Патологическая анатомия : учебник / А. И. Струков, В. В. Серов. - 5-е изд., стер. - М.: Литтерра, 2010. - 848 с. : ил.
|
|
|
|
ДИСТРОФИЯ
Общие сведения
Дистрофия (от греч. dys - нарушение и trophe - питаю) - сложный патологический процесс, в основе которого лежит нарушение тканевого (клеточного) метаболизма, ведущее к структурным изменениям. Поэтому дистрофии рассматриваются как один из видов повреждения.
Под трофикой понимают совокупность механизмов, определяющих метаболизм и структурную организацию ткани (клетки), которые необходимы для отправления специализированной функции. Среди этих механизмов выделяют клеточные и внеклеточные (рис. 26). Клеточные механизмы обеспечиваются структурной организацией клетки и ее ауторегуляцией. Это значит, что трофика клетки в значительной мере явля-
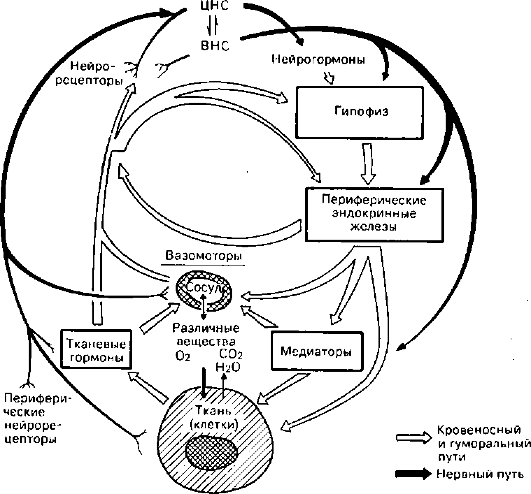 Рис. 26. Механизмы регуляции трофики (по М.Г. Балш)
Рис. 26. Механизмы регуляции трофики (по М.Г. Балш)
ется свойством самой клетки как сложной саморегулирующейся системы. Жизнедеятельность клетки обеспечивается «окружающей средой» и регулируется с помощью ряда систем организма. Поэтому внеклеточные механизмы трофики располагают транспортными (кровь, лимфа, микроциркуляторное русло) и интегративными (нейро-эндокринные, нейрогуморальные) системами ее регуляции. Из сказанного следует, что непосредственной причиной развития дистрофий могут служить нарушения как клеточных, так и внеклеточных механизмов, обеспечивающих трофику.
1. Расстройства ауторегуляции клетки могут быть вызваны различными факторами (гиперфункция, токсические вещества, радиация, наследственная недостаточность или отсутствие фермента и т.д.). Большую роль придают полому генов - рецепторов, осуществляющих «координированное торможение» функций различных ультраструктур. Нарушение ауторегуляции клетки ведет к энергетическому ее дефициту и к нарушению ферментативных процессов в клетке. Ферментопатия, или энзимопатия (приобретенная или наследственная), становится основным патогенетическим звеном и выражением дистрофии при нарушениях клеточных механизмов трофики.
2. Нарушения функции транспортных систем, обеспечивающих метаболизм и структурную сохранность тканей (клеток), вызывают гипоксию, которая является ведущей в патогенезе дисциркуляторных дистрофий.
3. При расстройствах эндокринной регуляции трофики (тиреотоксикоз, диабет, гиперпаратиреоз и т.д.) можно говорить об эндокринных, а при нарушении нервной регуляции трофики (нарушенная иннервация, опухоль головного мозга и т.д.) - о нервных или церебральных дистрофиях.
Особенности патогенеза внутриутробных дистрофий определяются непосредственной связью их с болезнями матери. В исходе при гибели части зачатка органа или ткани может развиться необратимый порок развития.
При дистрофиях в клетке и (или) межклеточном веществе накапливаются различные продукты обмена (белки, жиры, углеводы, минералы, вода), которые характеризуются количественными или качественными изменениями в результате нарушения ферментативных процессов.
Морфогенез. Среди механизмов, ведущих к развитию характерных для дистрофий изменений, различают инфильтрацию, декомпозицию (фанероз), извращенный синтез и трансформацию.
Инфильтрация - избыточное проникновение продуктов обмена из крови и лимфы в клетки или межклеточное вещество с последующим их накоплением в связи с недостаточностью ферментных систем, метаболизирующих эти продукты. Таковы, например, инфильтрация грубодисперсным белком эпителия проксимальных канальцев почек при нефротическом синдроме, инфильтрация холестерином и липопротеидами интимы аорты и крупных артерии при атеросклерозе.
Декомпозиция (фанероз) - распад ультраструктур клеток и межклеточного вещества, ведущий к нарушению тканевого (клеточного) метаболизма и накоплению продуктов нарушенного обмена в ткани (клетке). Таковы жи-
ровая дистрофия кардиомиоцитов при дифтерийной интоксикации, фибриноидное набухание соединительной ткани при ревматических болезнях.
Извращенный синтез - это синтез в клетках или в тканях веществ, не встречающихся в них в норме. К ним относятся: синтез аномального белка амилоида в клетке и аномальных белково-полисахаридных комплексов амилоида в межклеточном веществе; синтез белка алкогольного гиалина гепатоцитом; синтез гликогена в эпителии узкого сегмента нефрона при сахарном диабете.
Трансформация - образование продуктов одного вида обмена из общих исходных продуктов, которые идут на построение белков, жиров и углеводов. Такова, например, трансформация компонентов жиров и углеводов в белки, усиленная полимеризация глюкозы в гликоген и др.
Инфильтрация и декомпозиция - ведущие морфогенетические механизмы дистрофий - часто являются последовательными стадиями в их развитии. Однако в некоторых органах и тканях в связи со структурнофункциональными их особенностями преобладает какой-либо один из морфогенетических механизмов (инфильтрация - в эпителии почечных канальцев, декомпозиция - в клетках миокарда), что позволяет говорить об ортологии (от греч. orthos - прямой, типичный) дистрофий.
Морфологическая специфика. При изучении дистрофий на разных уровнях - ультраструктурном, клеточном, тканевом, органном - морфологическая специфика проявляется неоднозначно. Ультраструктурная морфология дистрофий обычно не имеет какой-либо специфики. Она отражает не только повреждение органелл, но и их репарацию (внутриклеточная регенерация). Вместе с тем возможность выявления в органеллах ряда продуктов обмена (липиды, гликоген, ферритин) позволяет говорить об ультраструктурных изменениях, характерных для того или иного вида дистрофий.
Характерная морфология дистрофий выявляется, как правило, на тканевом и клеточном уровнях, причем для доказательства связи дистрофии с нарушениями того или иного вида обмена требуется применение гистохимических методов. Без установления качества продукта нарушенного обмена нельзя верифицировать тканевую дистрофию, т.е. отнести ее к белковым, жировым, углеводным или другим дистрофиям. Изменения органа при дистрофии (размер, цвет, консистенция, структура на разрезе) в одних случаях представлены исключительно ярко, в других - отсутствуют, и лишь микроскопическое исследование позволяет выявить их специфичность. В ряде случаев можно говорить о системном характере изменений при дистрофии (системный гемосидероз, системный мезенхимальный амилоидоз, системный липоидоз).
В классификации дистрофий придерживаются нескольких принципов. Выделяют дистрофии.
I. В зависимости от преобладания морфологических изменений в специализированных элементах паренхимы или строме и сосудах: 1) паренхиматозные; 2) стромально-сосудистые; 3) смешанные.
II. По преобладанию нарушений того или иного вида обмена: 1) белковые; 2) жировые; 3) углеводные; 4) минеральные.
III. В зависимости от влияния генетических факторов: 1) приобретенные; 2) наследственные.
IV. По распространенности процесса: 1) общие; 2) местные.
Паренхиматозные дистрофии
Паренхиматозные дистрофии - проявления нарушений обмена в высокоспециализированных в функциональном отношении клетках. Поэтому при паренхиматозных дистрофиях преобладают нарушения клеточных механизмов трофики. Различные виды паренхиматозных дистрофий отражают недостаточность определенного физиологического (ферментативного) механизма, служащего выполнению специализированной функции клеткой (гепатоцит, нефроцит, кардиомиоцит и т.д.). В связи с этим в разных органах (печень, почки, сердце и т.д.) при развитии одного и того же вида дистрофии участвуют различные пато- и морфогенетические механизмы. Из этого следует, что переход одного вида паренхиматозной дистрофии в другой вид исключается, возможно лишь сочетание разных видов этой дистрофии.
В зависимости от нарушений того или иного вида обмена паренхиматозные дистрофии делят на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Паренхиматозные белковые дистрофии (диспротеинозы)
Большая часть белков цитоплазмы (простых и сложных) находится в соединении с липидами, образуя липопротеидные комплексы. Эти комплексы составляют основу мембран митохондрий, эндоплазматической сети, пластинчатого комплекса и других структур. Помимо связанных белков, в цитоплазме содержатся и свободные. Многие из последних обладают функцией ферментов.
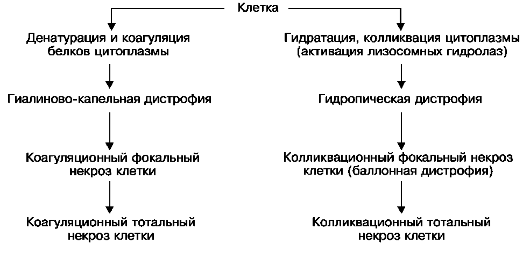
Сущность паренхиматозных диспротеинозов состоит в изменении физико-химических и морфологических свойств белков клетки: они подвергаются денатурации и коагуляции или, наоборот, колликвации, что ведет к гидратации цитоплазмы; в тех случаях, когда нарушаются связи белков с липидами, возникает деструкция мембранных структур клетки. В исходе этих нарушений может развиться коагуляционный (сухой) или колликвационный (влажный) некроз (схема I).
К паренхиматозным диспротеинозам относят гиалиново-капельную, гидропическую и роговую дистрофии.
К паренхиматозным белковым дистрофиям со времен Р. Вирхова причисляли и многие патологи продолжают причислять так называемую зернистую дистрофию, при которой в клетках паренхиматозных органов появляются белковые зерна. Сами органы увеличиваются в размерах, становятся дряблыми и тусклыми на разрезе, что послужило причиной называть также зернистую дистрофию тусклым (мутным) набуханием. Однако электронно-микроскопическое и гистоферменто-
Схема I. Морфогенез паренхиматозных диспротеинозов
 химическое
изучение «зернистой дистрофии» показало, что в ее основе лежит не
накопление белка в цитоплазме, а гиперплазия ультраструктур клеток
паренхиматозных органов как выражение функционального напряжения этих
органов в ответ на различные воздействия; гиперплазированные
ультраструктуры клетки выявляются при светооптическом исследовании как
белковые гранулы.
химическое
изучение «зернистой дистрофии» показало, что в ее основе лежит не
накопление белка в цитоплазме, а гиперплазия ультраструктур клеток
паренхиматозных органов как выражение функционального напряжения этих
органов в ответ на различные воздействия; гиперплазированные
ультраструктуры клетки выявляются при светооптическом исследовании как
белковые гранулы.
Гиалиново-капельная дистрофия
При гиалиново-капельной дистрофии в цитоплазме появляются крупные гиалиноподобные белковые капли, сливающиеся между собой и заполняющие тело клетки; при этом происходит деструкция ультраструктурных элементов клетки. В ряде случаев гиалиново-капельная дистрофия завершается фокальным коагуляционным некрозом клетки.
Этот вид диспротеиноза часто встречается в почках, редко - в печени и совсем редко - в миокарде.
В почках при микроскопическом исследовании накопление гиалиновых капель находят в нефроцитах. При этом наблюдается деструкция митохондрий, эндоплазматической сети, щеточной каемки (рис. 27). В основе гиалиново-капельной дистрофии нефроцитов лежит недостаточность вакуолярно-лизосомального аппарата эпителия проксимальных канальцев, в норме реабсорбирующего белки. Поэтому этот вид дистрофии нефроцитов очень часто встречается при нефротическом синдроме. Этот синдром является одним из проявлений многих заболеваний почек, при которых первично поражается гломерулярный фильтр (гломерулонефрит, амилоидоз почек, парапротеинемическая нефропатия и др.).
Внешний вид почек при этой дистрофии не имеет каких-либо характерных черт, он определяется прежде всего особенностями основного заболевания (гломерулонефрит, амилоидоз).
В печени при микроскопическом исследовании в гепатоцитах находят гиалиноподобные тельца (тельца Мэллори), которые состоят из фибрилл
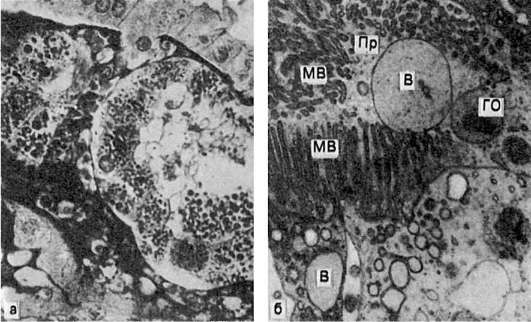 Рис. 27. Гиалиново-капельная дистрофия эпителия почечных канальцев:
Рис. 27. Гиалиново-капельная дистрофия эпителия почечных канальцев:
а - в цитоплазме эпителия крупные белковые капли (микроскопическая картина); б - в цитоплазме клетки много белковых (гиалиновых) образований (ГО) овальной формы и вакуолей (В); отмечаются десквамация микроворсинок (МВ) щеточной каемки и выход в просвет (Пр) канальца вакуолей и белковых образований. Электронограмма. х18 000
особого белка - алкогольного гиалина (см. рис. 22). Образование этого белка и телец Мэллори служит проявлением извращенной белковосинтетической функции гепатоцита, что встречается постоянно при алкогольном гепатите и сравнительно редко при первичном билиарном и индийском детском циррозах, гепатоцеребральной дистрофии (болезни Вильсона-Коновалова).
Внешний вид печени различен; изменения характерны для тех ее заболеваний, при которых встречается гиалиново-капельная дистрофия.
Исход гиалиново-капельной дистрофии неблагоприятен: она завершается необратимым процессом, ведущим к некрозу клетки.
Функциональное значение этой дистрофии очень велико. С гиалиновокапельной дистрофией эпителия почечных канальцев связаны появление в моче белка (протеинурия) и цилиндров (цилиндрурия), потеря белков плазмы (гипопротеинемия), нарушение ее электролитного баланса. Гиалиново-капельная дистрофия гепатоцитов нередко является морфологической основой нарушений многих функций печени.
Гидропическая дистрофия
Гидропическая, или водяночная, дистрофия характеризуется появлением в клетке вакуолей, наполненных цитоплазматической жидкостью. Она наблюдается чаще в эпителии кожи и почечных канальцев, в гепа-
тоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников.
Микроскопическая картина: паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражением фокального колликвационного некроза называют баллонной дистрофией.
Внешний вид органов и тканей мало изменяется при гидропическои дистрофии, она обнаруживается обычно под микроскопом.
Механизм развития гидропическои дистрофии сложен и отражает нарушения водно-электролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Причины развития гидропической дистрофии в разных органах неоднозначны. В почках - это повреждение гломерулярного фильтра (гломерулонефрит, амилоидоз, сахарный диабет), что ведет к гиперфильтрации и недостаточности ферментной системы базального лабиринта нефроцитов, в норме обеспечивающей реабсорбцию воды; поэтому гидропическая дистрофия нефроцитов так характерна для нефротического синдрома. В печени гидропическая дистрофия возникает при вирусном и токсическом гепатитах (рис. 28) и нередко является причиной печеночной недостаточности. Причиной гидропическои дистрофии эпидермиса может быть инфекция (оспа), отек кожи различного механизма. Вакуолизация цитоплазмы может быть проявлением физиологической деятельности клетки, что отмечается, например, в ганглиозных клетках центральной и периферической нервной системы.
Исход гидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает.
Роговая дистрофия
Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным.
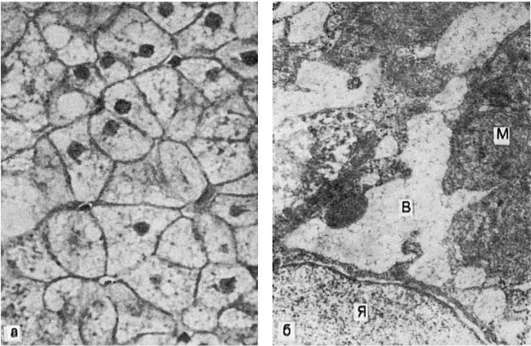 Рис. 28. Гидропическая дистрофия печени (биопсия):
Рис. 28. Гидропическая дистрофия печени (биопсия):
а - микроскопическая картина; вакуолизация гепатоцитов; б - электронограмма: расширение канальцев эндоплазматической сети и образование вакуолей (В), заполненных хлопьевидным содержимым. Мембраны, ограничивающие вакуоли, почти полностью лишены рибосом. Вакуоли сдавливают расположенные между ними митохондрии (М), часть которых подвергается деструкции; Я - ядро гепатоцита. х18 000
Причины роговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др.
Исход может быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток.
Значение роговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью.
К группе паренхиматозных диспротеинозов примыкает ряд дистрофий, в основе которых лежат нарушения внутриклеточного метаболизма ряда аминокислот в результате наследственной недостаточности метаболизирующих их ферментов, т.е. в результате наследственной ферментопатии. Эти дистрофии относятся к так называемым болезням накопления.
Наиболее яркими примерами наследственных дистрофий, связанных с нарушением внутриклеточного метаболизма аминокислот, являются цистиноз, тирозиноз, фенилпировиноградная олигофрения (фенилкетонурия). Их характеристика представлена в табл. 1.
Таблица 1. Наследственные дистрофии, связанные с нарушением обмена аминокислот
Название | Дефицит фермента | Локализация накоплений аминокислоты |
Цистиноз | Неизвестен | Печень, почки, селезенка, глаза, костный мозг, лимфатические узлы, кожа |
Тирозиноз | Тирозинаминотрансфераза или оксидаза пара-оксифенилпировиноградной кислоты | Печень, почки, кости |
Фенилпирови- ноградная олигофрения | Фенилаланин-4-гидроксилаза | Нервная система, мышцы, кожа, кровь, моча |
Паренхиматозные жировые дистрофии (липидозы)
В цитоплазме клеток содержатся в основном липиды, которые образуют с белками сложные лабильные жиробелковые комплексы - липопротеиды. Эти комплексы составляют основу мембран клетки. Липиды вместе с белками являются составной частью и клеточных ультраструктур. Помимо липопротеидов, в цитоплазме встречаются и нейтральные жиры, которые представляют собой сложные эфиры глицерина и жирных кислот.
Для выявления жиров используют срезы нефиксированных замороженных или фиксированных в формалине тканей. Гистохимически жиры выявляются с помощью ряда методов: судан III и шарлах окрашивают их в красный цвет, судан IV и осмиевая кислота - в черный, сульфат нильского голубого окрашивает жирные кислоты в темно-синий цвет, а нейтральные жиры - в красный.
С помощью поляризационного микроскопа можно дифференцировать изотропные и анизотропные липиды, последние дают характерное двойное лучепреломление.
Нарушения обмена цитоплазматических липидов могут проявляться в увеличении их содержания в клетках, где они обнаруживаются и в норме, в появлении липидов там, где они обычно не встречаются, и в образовании жиров необычного химического состава. Обычно в клетках накапливаются нейтральные жиры.
Паренхиматозная жировая дистрофия встречается наиболее часто там же, где и белковая, - в миокарде, печени, почках.
В миокарде жировая дистрофия характеризуется появлением в мышечных клетках мельчайших жировых капель (пылевидное ожирение). При нарастании изменений эти капли (мелкокапельное ожирение) полностью замещают цитоплазму (рис. 29). Большинство митохондрий при этом распадается, поперечная исчерченность волокон исчезает. Процесс имеет очаговый характер и наблюдается в группах мышечных клеток, расположенных по ходу венозного колена капилляров и мелких вен.
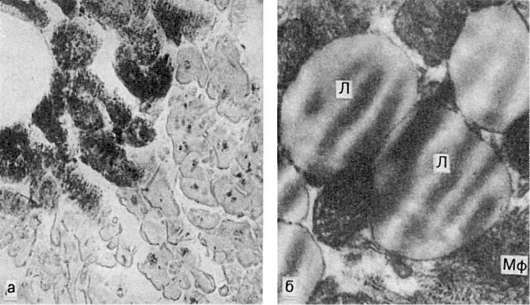 Рис. 29. Жировая дистрофия миокарда:
Рис. 29. Жировая дистрофия миокарда:
а - капли жира (на рисунке черного цвета) в цитоплазме мышечных волокон (микроскопическая картина); б - включения липидов (Л), имеющие характерную исчерченность; Мф - миофибриллы. Электронограмма. х21 000
Внешний вид сердца зависит от степени жировой дистрофии. Если процесс выражен слабо, его можно распознать лишь под микроскопом, применяя специальные окраски на липиды; если он выражен сильно, сердце выглядит увеличенным в объеме, камеры его растянуты, оно дряблой консистенции, миокард на разрезе тусклый, глинисто-желтый. Со стороны эндокарда видна желто-белая исчерченность, особенно хорошо выраженная в сосочковых мышцах и трабекулах желудочков сердца («тигровое сердце»). Эта исчерченность миокарда связана с очаговым характером дистрофии, преимущественным поражением мышечных клеток вокруг венул и вен. Жировая дистрофия миокарда рассматривается как морфологический эквивалент его декомпенсации.
Развитие жировой дистрофии миокарда связывают с тремя механизмами: повышенным поступлением жирных кислот в кардиомиоциты, нарушением обмена жиров в этих клетках и распадом липопротеидных комплексов внутриклеточных структур. Чаще всего эти механизмы реализуются путем инфильтрации и декомпозиции (фанероза) при энергетическом дефиците миокарда, связанном с гипоксией и интоксикацией (дифтерия). При этом основное значение декомпозиции не в высвобождении липидов из липопротеидных комплексов клеточных мембран, а в деструкции митохондрий, что ведет к нарушению окисления жирных кислот в клетке.
В печени жировая дистрофия (ожирение) проявляется резким увеличением содержания жиров в гепатоцитах и изменением их состава. В клетках печени вначале появляются гранулы липидов (пылевидное ожирение), затем мелкие капли их (мелкокапельное ожирение), которые в дальнейшем
сливаются в крупные капли (крупнокапельное ожирение) или в одну жировую вакуоль, которая заполняет всю цитоплазму и отодвигает ядро на периферию. Измененные таким образом печеночные клетки напоминают жировые. Чаще отложение жиров в печени начинается на периферии, реже - в центре долек; при значительно выраженной дистрофии ожирение клеток печени имеет диффузный характер.
Внешний вид печени достаточно характерен: она увеличена, дряблая, охряно-желтого или желто-коричневого цвета. При разрезе на лезвии ножа и поверхности разреза виден налет жира.
Среди механизмов развития жировой дистрофии печени различают: чрезмерное поступление в гепатоциты жирных кислот или повышенный их синтез этими клетками; воздействие токсических веществ, блокирующих окисление жирных кислот и синтез липопротеидов в гепатоцитах; недостаточное поступление в печеночные клетки аминокислот, необходимых для синтеза фосфолипидов и липопротеидов. Из этого следует, что жировая дистрофия печени развивается при липопротеидемии (алкоголизм, сахарный диабет, общее ожирение, гормональные расстройства), гепатотропных интоксикациях (этанол, фосфор, хлороформ и др.), нарушениях питания (недостаток белка в пище - алипотропная жировая дистрофия печени, авитаминозы, болезни пищеварительной системы).
В почках при жировой дистрофии жиры появляются в эпителии проксимальных и дистальных канальцев. Обычно это нейтральные жиры, фосфолипиды или холестерин, который обнаруживают не только в эпителии канальцев, но и в строме. Нейтральные жиры в эпителии узкого сегмента и собирательных трубок встречаются как физиологическое явление.
Внешний вид почек: они увеличены, дряблые (при сочетании с амилоидозом плотные), корковое вещество набухшее, серое с желтым крапом, заметным на поверхности и разрезе.
Механизм развития жировой дистрофии почек связан с инфильтрацией эпителия почечных канальцев жиром при липемии и гиперхолестеринемии (нефротический синдром), что ведет к гибели нефроцитов.
Причины жировой дистрофии разнообразны. Чаще всего она связана с кислородным голоданием (тканевая гипоксия), поэтому жировая дистрофия так часто встречается при заболеваниях сердечно-сосудистой системы, хронических заболеваниях легких, анемиях, хроническом алкоголизме и т.д. В условиях гипоксии страдают в первую очередь отделы органа, находящиеся в функциональном напряжении. Вторая причина - инфекции (дифтерия, туберкулез, сепсис) и интоксикации (фосфор, мышьяк, хлороформ), ведущие к нарушениям обмена (диспротеиноз, гипопротеинемия, гиперхолестеринемия), третья - авитаминозы и одностороннее (с недостаточным содержанием белков) питание, сопровождающееся дефицитом ферментов и липотропных факторов, которые необходимы для нормального жирового обмена клетки.
Исход жировой дистрофии зависит от ее степени. Если она не сопровождается грубым поломом клеточных структур, то, как правило, оказывается обратимой. Глубокое нарушение обмена клеточных липидов в
большинстве случаев заканчивается гибелью клетки, функция органов при этом резко нарушается, а в ряде случаев и выпадает.
Группу наследственных липидозов составляют так называемые системные липидозы, возникающие вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов. Поэтому системные липидозы относят к наследственным ферментопатиям (болезни накопления), поскольку дефицит фермента определяет накопление субстрата, т.е. липидов, в клетках.
В зависимости от вида накапливающихся в клетках липидов различают: цереброзидлипидоз, или глюкозилцерамидлипидоз (болезнь Гоше), сфингомиелинлипидоз (болезнь Ниманна-Пика), ганглиозидлипидоз (болезнь Тея-Сакса, или амавротическая идиотия), генерализованный ганглиозидоз (болезнь Нормана-Ландинга) и др. Чаще всего липиды накапливаются в печени, селезенке, костном мозге, центральной нервной системе (ЦНС), нервных сплетениях. При этом появляются характерные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика), что имеет диагностическое значение при изучении биоптатов (табл. 2).
Таблица 2. Системные липидозы (наследственные ферментопатии, болезни накопления, лизосомные болезни)
Название | Дефицит фермента | Локализация накоплений липида | Диагностический критерий при биопсии |
Болезнь Гоше - цереброзидлипидоз или глюкозидцерамидлипидоз | Глюкоцереброзидаза | Печень, селезенка, костный мозг, ЦНС (у детей) | Клетки Гоше |
Болезнь Ниманна- Пика - сфингомиелинлипидоз | Сфингомиелиназа | Печень, селезенка, костный мозг, ЦНС | Клетки Пика |
Амавротическая идиотия, болезнь Тея-Сакса - ганглиозидлипидоз | Гексозаминидаза | ЦНС, сетчатка глаз, нервные сплетения, селезенка, печень | Изменения мейсснеровского сплетения (ректобиопсия) |
Болезнь Нормана- Ландинга - генерализованный ганглиозидоз | β-Галактозидаза | ЦНС, нервные сплетения, печень, селезенка, костный мозг, почки и др. | Отсутствует |
Многие ферменты, дефицит которых определяет развитие системных липидозов, относятся, как видно из табл. 2, к лизосомным. На этом основании ряд липидозов рассматривают как лизосомные болезни.
Паренхиматозные углеводные дистрофии
Углеводы, которые определяются в клетках и тканях и могут быть идентифицированы гистохимически, делят на полисахариды, из которых в животных тканях выявляются лишь гликоген, гликозаминогликаны (му-
кополисахариды) и гликопротеиды. Среди гликозаминогликанов различают нейтральные, прочно связанные с белками, и кислые, к которым относятся гиалуроновая, хондроитинсерная кислоты и гепарин. Кислые гликозаминогликаны как биополимеры способны вступать в непрочные соединения с рядом метаболитов и осуществлять их транспорт. Главными представителями гликопротеидов являются муцины и мукоиды. Муцины составляют основу слизи, продуцируемой эпителием слизистых оболочек и железами, мукоиды входят в состав многих тканей.
Полисахариды, гликозаминогликаны и гликопротеиды выявляются ШИКреакцией или реакцией Хочкиса-Мак-Мануса. Сущность реакции заключается в том, что после окисления йодной кислотой (или реакции с перйодатом) образующиеся альдегиды дают с фуксином Шиффа красное окрашивание. Для выявления гликогена ШИК-реакцию дополняют ферментативным контролем - обработкой срезов амилазой. Гликоген окрашивается кармином Беста в красный цвет. Гликозаминогликаны и гликопротеиды определяют с помощью ряда методов, из которых наиболее часто применяют окраски толуидиновым синим или метиленовым синим. Эти окраски позволяют выявлять хромотропные вещества, дающие реакцию метахромазии. Обработка срезов ткани гиалуронидазами (бактериальной, тестикулярной) с последующей окраской теми же красителями позволяет дифференцировать различные гликозаминогликаны.
Паренхиматозная углеводная дистрофия может быть связана с нарушением обмена гликогена или гликопротеидов.
Углеводные дистрофии, связанные с нарушением обмена гликогена
Основные запасы гликогена находятся в печени и скелетных мышцах. Гликоген печени и мышц расходуется в зависимости от потребностей организма (лабильный гликоген). Гликоген нервных клеток, проводящей системы сердца, аорты, эндотелия, эпителиальных покровов, слизистой оболочки матки, соединительной ткани, эмбриональных тканей, хряща и лейкоцитов является необходимым компонентом клеток, и его содержание не подвергается заметным колебаниям (стабильный гликоген). Однако деление гликогена на лабильный и стабильный условно.
Регуляция обмена углеводов осуществляется нейроэндокринным путем. Основная роль принадлежит гипоталамической области, гипофизу (АКТГ, тиреотропный, соматотропный гормоны), (β-клеткам (В-клеткам) поджелудочной железы (инсулин), надпочечникам (глюкокортикоиды, адреналин) и щитовидной железе.
Нарушения содержания гликогена проявляются в уменьшении или увеличении количества его в тканях и появлении там, где он обычно не выявляется. Эти нарушения наиболее ярко выражены при сахарном диабете и при наследственных углеводных дистрофиях - гликогенозах.
При сахарном диабете, развитие которого связывают с патологией β-клеток островков поджелудочной железы, происходят недостаточное использование глюкозы тканями, увеличение ее содержания в крови (гипергликемия) и выведение с мочой (глюкозурия). Тканевые запасы гликогена резко уменьшаются. Это в первую очередь касается печени,
в которой нарушается синтез гликогена, что ведет к инфильтрации ее жирами - развивается жировая дистрофия печени; при этом в ядрах гепатоцитов появляются включения гликогена, они становятся светлыми («дырчатые», «пустые», ядра).
С глюкозурией связаны характерные изменения почек при диабете. Они выражаются в гликогенной инфильтрации эпителия канальцев, главным образом узкого и дистального сегментов. Эпителий становится высоким, со светлой пенистой цитоплазмой; зерна гликогена видны и в просвете канальцев. Эти изменения отражают состояние синтеза гликогена (полимеризация глюкозы) в канальцевом эпителии при резорбции богатого глюкозой ультрафильтрата плазмы.
При диабете страдают не только почечные канальцы, но и клубочки, их капиллярные петли, базальная мембрана которых становится значительно более проницаемой для сахаров и белков плазмы. Возникает одно из проявлений диабетической микроангиопатии - интеркапиллярный (диабетический) гломерулосклероз.
Наследственные углеводные дистрофии, в основе которых лежат нарушения обмена гликогена, называются гликогенозами. Гликогенозы обусловлены отсутствием или недостаточностью фермента, участвующего в расщеплении депонированного гликогена, и относятся поэтому к наследственным ферментопатиям, или болезням накопления. В настоящее время хорошо изучены 6 типов гликогенозов, обусловленных наследственной недостаточностью 6 различных ферментов. Это болезни Гирке (I тип), Помпе (II тип), Мак-Ардля (V тип) и Герса (VI тип), при которых структура накапливаемого в тканях гликогена не нарушена, и болезни Форбса-Кори (III тип) и Андерсена (IV тип), при которых она резко изменена (табл. 3).
Таблица 3. Гликогенозы (наследственные ферментопатии, болезни накопления)
Название болезни | Дефицит фермента | Локализация накоплений гликогена |
Без нарушения структуры гликогена | ||
Гирке (I тип) | Глюкозо-6-фосфатаза | Печень, почки |
Помпе (II тип) | Кислая α-клюкозидаза | Гладкие и скелетные мышцы, миокард |
Мак-Ардля (V тип) | Система фосфорилаз мышц | Скелетные мышцы |
Герса (VI тип) | Фосфорилаза печени | Печень |
С нарушением структуры гликогена | ||
Форбса-Кори, лимитдекстриноз (III тип) | Амило-1,6-глюкозидаза | Печень, мышцы, сердце |
Андерсена, амилопектиноз (IV тип) | Амило-(1,4-1,6)-трансглюкозидаза | Печень, селезенка, лимфатические узлы |
Морфологическая диагностика гликогеноза того или иного типа возможна при биопсии с помощью гистоферментативных методов.
Углеводные дистрофии, связанные с нарушением обмена гликопротеидов
При нарушении обмена гликопротеидов в клетках или в межклеточном веществе происходит накопление муцинов и мукоидов, называемых также слизистыми или слизеподобными веществами. В связи с этим при нарушении обмена гликопротеидов говорят о слизистой дистрофии.
Микроскопическое исследование. Оно позволяет выявить не только усиленное слизеобразование, но и изменения физико-химических свойств слизи. Многие секретирующие клетки погибают и десквамируются, выводные протоки желез обтурируются слизью, что ведет к развитию кист. Нередко в этих случаях присоединяется воспаление. Слизь может закрывать просветы бронхов, следствием чего является возникновение ателектазов и очагов пневмонии.
Иногда в железистых структурах накапливается не истинная слизь, а слизеподобные вещества (псевдомуцины). Эти вещества могут уплотняться и принимать характер коллоида. Тогда говорят о коллоидной дистрофии, которая наблюдается, например, при коллоидном зобе.
Причины слизистой дистрофии разнообразны, но чаще всего это воспаление слизистых оболочек в результате действия различных патогенных раздражителей (см. Катаральное воспаление).
Слизистая дистрофия лежит в основе наследственного системного заболевания, называемого муковисцидозом, для которого характерно изменение качества слизи, выделяемой эпителием слизистых желез: слизь становится густой и вязкой, она плохо выводится, что обусловливает развитие ретенционных кист и склероза (кистозный фиброз). Поражаются экзокринный аппарат поджелудочной железы, железы бронхиального дерева, пищеварительного и мочевого тракта, желчных путей, потовые и слезные железы (подробнее см. Пренатальная патология).
Исход в значительной мере определяется степенью и длительностью повышенного слизеобразования. В одних случаях регенерация эпителия приводит к полному восстановлению слизистой оболочки, в других - она атрофируется, подвергается склерозу, что, естественно, отражается на функции органа.
Стромально-сосудистые дистрофии
Стромально-сосудистые (мезенхимальные) дистрофии развиваются в результате нарушений обмена в соединительной ткани и выявляются в строме органов и стенках сосудов. Они развиваются на территории гистиона, который, как известно, образован отрезком микроциркуляторного русла с окружающими его элементами соединительной ткани (основное вещество, волокнистые структуры, клетки) и нервными волокнами. Понятными становятся в связи с этим преобладание среди механизмов развития стромально-сосудистых дистрофий нарушений транспортных систем трофики, общность морфогенеза, возможность не только сочетания различных видов дистрофии, но и перехода одного вида в другой.
При нарушениях обмена в соединительной ткани, преимущественно в ее межклеточном веществе, накапливаются продукты метаболизма, которые могут приноситься с кровью и лимфой, быть результатом извращенного синтеза или появляться в результате дезорганизации основного вещества и волокон соединительной ткани.
В зависимости от вида нарушенного обмена мезенхимальные дистрофии делят на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Стромально-сосудистые белковые дистрофии (диспротеинозы)
Среди белков соединительной ткани основное значение имеет коллаген, из макромолекул которого строятся коллагеновые и ретикулярные волокна. Коллаген является неотъемлемой частью базальных мембран (эндотелия, эпителия) и эластических волокон, в состав которых, помимо коллагена, входит эластин. Коллаген синтезируется клетками соединительной ткани, среди которых главную роль играют фибробласты. Кроме коллагена, эти клетки синтезируют гликозаминогликаны основного вещества соединительной ткани, которое содержит также белки и полисахариды плазмы крови.
Волокна соединительной ткани имеют характерную ультраструктуру. Они хорошо выявляются с помощью ряда гистологических методов: коллагеновые - окраской пикрофуксиновой смесью (по ван Гизону), эластические - окраской фукселином или орсеином, ретикулярные - импрегнацией солями серебра (ретикулярные волокна являются аргирофильными).
В соединительной ткани, помимо ее клеток, синтезирующих коллаген и гликозаминогликаны (фибробласт, ретикулярная клетка), а также ряд биологически активных веществ (лаброцит, или тучная клетка), находятся клетки гематогенного происхождения, осуществляющие фагоцитоз (полиморфно-ядерные лейкоциты, гистиоциты, макрофаги) и иммунные реакции (плазмобласты и плазмоциты, лимфоциты, макрофаги).
К стромально-сосудистым диспротеинозам относят мукоидное набухание, фибриноидное набухание (фибриноид), гиалиноз, амилоидоз.
Нередко мукоидное набухание, фибриноидное набухание и гиалиноз являются последовательными стадиями дезорганизации соединительной ткани; в основе этого процесса лежат накопление продуктов плазмы крови в основном веществе в результате повышения тканево-сосудистой проницаемости (плазморрагия), деструкция элементов соединительной ткани и образование белковых (белково-полисахаридных) комплексов. Амилоидоз отличается от этих процессов тем, что в состав образующихся белково-полисахаридных комплексов входит не встречающийся обычно фибриллярный белок, синтезируемый клетками - амилоидобластами (схема II).
Схема II. Морфогенез стромально-сосудистых диспротеинозов
 Мукоидное набухание
Мукоидное набухание
Мукоидное набухание - поверхностная и обратимая дезорганизация соединительной ткани. При этом в основном веществе происходят накопление и перераспределение гликозаминогликанов за счет увеличения содержания прежде всего гиалуроновой кислоты. Гликозаминогликаны обладают гидрофильными свойствами, накопление их обусловливает повышение тканевой и сосудистой проницаемости. В результате этого к гликозаминогликанам примешиваются белки плазмы (главным образом глобулины) и гликопротеиды. Развиваются гидратация и набухание основного межуточного вещества.
Микроскопическое исследование. Основное вещество базофильное, при окраске толуидиновым синим - сиреневое или красное (рис. 30, см. на цв. вкл.). Возникает феномен метахромазии, в основе которого лежит изменение состояния основного межуточного вещества с накоплением хромотропных веществ. Коллагеновые волокна обычно сохраняют пучковое строение, но набухают и подвергаются фибриллярному разволокнению. Они становятся малоустойчивыми к действию коллагеназы и при окраске пикрофуксином выглядят желто-оранжевыми, а не кирпично-красными. Изменения основного вещества и коллагеновых волокон при мукоидном набухании могут сопровождаться клеточными реакциями - появлением лимфоцитарных, плазмоклеточных и гистиоцитарных инфильтратов.
Мукоидное набухание встречается в различных органах и тканях, но чаще в стенках артерий, клапанах сердца, эндокарде и эпикарде, т.е. там, где хромотропные вещества встречаются и в норме; при этом количество хромотропных веществ резко возрастает. Наиболее часто оно наблюдается при инфекционных и аллергических заболеваниях, ревматических болезнях, атеросклерозе, эндокринопатиях и пр.
Внешний вид. При мукоидном набухании ткань или орган сохранены, характерные изменения устанавливаются с помощью гистохимических реакций при микроскопическом исследовании.
Причины. Большое значение в его развитии имеют гипоксия, инфекция, особенно стрептококковая, иммунопатологические реакции (реакции гиперчувствительности).
Исход может быть двояким: полное восстановление ткани или переход в фибриноидное набухание. Функция органа при этом страдает (например, нарушения функции сердца в связи с развитием ревматического эндокардита - вальвулита).
Фибриноидное набухание (фибриноид)
Фибриноидное набухание - глубокая и необратимая дезорганизация соединительной ткани, в основе которой лежит деструкция ее основного вещества и волокон, сопровождающаяся резким повышением сосудистой проницаемости и образованием фибриноида.
Фибриноид представляет собой сложное вещество, в состав которого входят белки и полисахариды распадающихся коллагеновых волокон, основного вещества и плазмы крови, а также клеточные нуклеопротеиды. Гистохимически при различных заболеваниях фибриноид различен, но обязательным компонентом его является фибрин (рис. 31) (отсюда и термины «фибриноидное набухание», «фибриноид»).
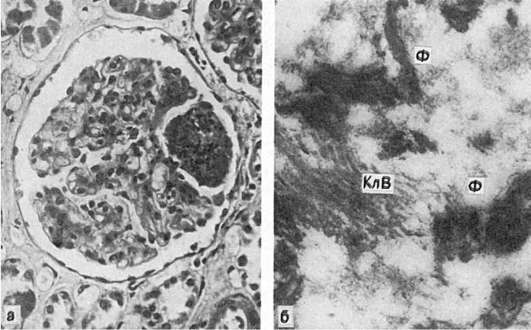 Рис. 31. Фибриноидное набухание:
Рис. 31. Фибриноидное набухание:
а - фибриноидное набухание и фибриноидный некроз капилляров почечных клубочков (системная красная волчанка); б - в фибриноиде среди набухших, потерявших поперечную исчерченность коллагеновых волокон (КлВ), массы фибрина (Ф). Электронограмма. х35 000 (по Гизекингу)
Микроскопическая картина. При фибриноидном набухании пучки коллагеновых волокон, пропитанные белками плазмы, становятся гомогенными, образуя с фибрином нерастворимые прочные соединения; они эозинофильны, пирофуксином окрашиваются в желтый цвет, резко ШИК-положительны и пиронинофильны при реакции Браше, а также аргирофильны при импрегнации солями серебра. Метахромазия соединительной ткани при этом не выражена или выражена слабо, что объясняется деполимеризацией гликозаминогликанов основного вещества.
В исходе фибриноидного набухания иногда развивается фибриноидный некроз, характеризующийся полной деструкцией соединительной ткани. Вокруг очагов некроза обычно выражена реакция макрофагов.
Внешний вид. Различные органы и ткани, где встречается фибриноидное набухание, внешне мало изменяются, характерные изменения обнаруживаются обычно лишь при микроскопическом исследовании.
Причины. Чаще всего это проявление инфекционно-аллергических (например, фибриноид сосудов при туберкулезе с гиперергическими реакциями), аллергических и аутоиммунных (фибриноидные изменения соединительной ткани при ревматических болезнях, капилляров почечных клубочков при гломерулонефрите) и ангионевротических (фибриноид артериол при гипертонической болезни и артериальных гипертензиях) реакций. В таких случаях фибриноидное набухание имеет распространенный (системный) характер. Местно фибриноидное набухание может возникать при воспалении, особенно хроническом (фибриноид в червеобразном отростке при аппендиците, в дне хронической язвы желудка, трофических язв кожи и т.д.).
Исход фибриноидных изменений характеризуется развитием некроза, замещением очага деструкции соединительной тканью (склероз) или гиалинозом. Фибриноидное набухание ведет к нарушению, а нередко и прекращению функции органа (например, острая почечная недостаточность при злокачественной гипертонии, характеризующейся фибриноидным некрозом и изменениями артериол клубочков).
Гиалиноз
При гиалинозе (от греч. hyalos - прозрачный, стекловидный), или гиалиновой дистрофии, в соединительной ткани образуются однородные полупрозрачные плотные массы (гиалин), напоминающие гиалиновый хрящ. Ткань уплотняется, поэтому гиалиноз рассматривается и как разновидность склероза.
Гиалин - это фибриллярный белок. При иммуногистохимическом исследовании в нем обнаруживают не только белки плазмы, фибрин, но и компоненты иммунных комплексов (иммуноглобулины, фракции комплемента), а также липиды. Гиалиновые массы устойчивы по отношению к кислотам, щелочам, ферментам, ШИК-положительны, хорошо воспринимают кислые красители (эозин, кислый фуксин), пикрофуксином окрашиваются в желтый или красный цвет.
Механизм гиалиноза сложен. Ведущими в его развитии являются деструкция волокнистых структур и повышение тканево-сосудистой проницаемости (плазморрагия) в связи с ангионевротическими (дисциркуляторными), метаболическими и иммунопатологическими процессами. С плазморрагией связаны пропитывание ткани белками плазмы и адсорбция их на измененных волокнистых структурах с последующей преципитацией и образованием белка - гиалина. В образовании сосудистого гиалина принимают участие гладкомышечные клетки. Гиалиноз может развиваться в исходе разных процессов: плазматического пропитывания, фибриноидного набухания (фибриноида), воспаления, некроза, склероза.
Классификация. Различают гиалиноз сосудов и гиалиноз собственно соединительной ткани. Каждый из них может быть распространенным (системным) и местным.
Гиалиноз сосудов. Гиалинозу подвергаются преимущественно мелкие артерии и артериолы. Ему предшествуют повреждение эндотелия, его мембраны и гладкомышечных клеток стенки и пропитывание ее плазмой крови.
Микроскопическое исследование. Гиалин обнаруживают в субэндотелиальном пространстве, он оттесняет кнаружи и разрушает эластическую пластинку, средняя оболочка истончается, в финале артериолы превращаются в утолщенные стекловидные трубочки с резко суженным или полностью закрытым просветом (рис. 32).
Гиалиноз мелких артерий и артериол носит системный характер, но наиболее выражен в почках, головном мозге, сетчатке глаза, поджелудочной железе, коже. Он особенно характерен для гипертонической болезни и гипертонических состояний (гипертонический артериологиалиноз), диабетической микроангиопатии (диабетический артериологиалиноз) и заболеваний с нарушениями иммунитета. Как физиологическое явление местный гиалиноз артерий наблюдается в селезенке взрослых и пожилых людей, отражая функционально-морфологические особенности селезенки как органа депонирования крови.
Сосудистый гиалин - вещество преимущественно гематогенной природы. В его образовании играют роль не только гемодинамические и метаболические, но и иммунные механизмы. Руководствуясь особенностями патогенеза гиалиноза сосудов, выделяют 3 вида сосудистого гиалина: 1) простой, возникающий вследствие инсудации неизмененных или малоизмененных компонентов плазмы крови (встречается чаще при гипертонической болезни доброкачественного течения, атеросклерозе и у здоровых людей); 2) липогиалин, содержащий липиды и β-липопротеиды (обнаруживается чаще всего при сахарном диабете); 3) сложный гиалин, строящийся из иммунных комплексов, фибрина и разрушающихся структур сосудистой стенки (см. рис. 32) (характерен для болезней с иммунопатологическими нарушениями, например для ревматических заболеваний).
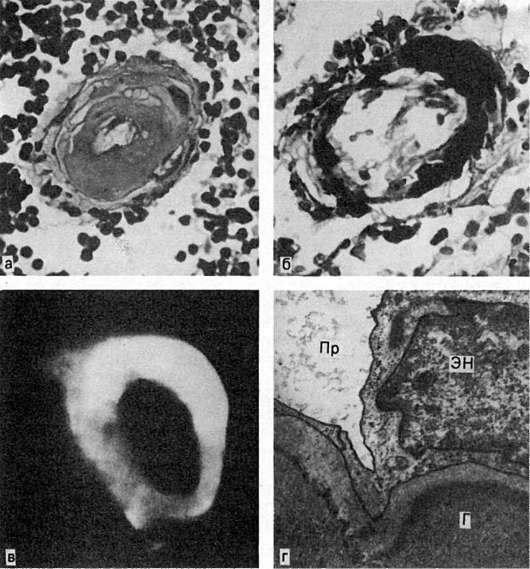 Рис. 32. Гиалиноз сосудов селезенки:
Рис. 32. Гиалиноз сосудов селезенки:
а - стенка центральной артерии фолликула селезенки представлена гомогенными массами гиалина; б - фибрин среди гиалиновых масс при окраске по методу Вейгерта; в - фиксация в гиалине IgG иммунных комплексов (люминесцентная микроскопия); г - массы гиалина (Г) в стенке артериолы; Эн - эндотелий; Пр - просвет артериолы. Электронограмма.
х15 000
Гиалиноз собственно соединительной ткани. Развивается обычно в исходе фибриноидного набухания, ведущего к деструкции коллагена и пропитыванию ткани белками плазмы и полисахаридами.
Микроскопическое исследование. Находят набухание соединительнотканных пучков, они теряют фибриллярность и сливаются в однородную плотную хрящеподобную массу; клеточные элементы сдавливаются и подвергаются атрофии. Этот механизм развития системного гиалиноза соединительной ткани особенно часто встречается при заболеваниях с иммунными нарушениями (ревматические болезни). Гиалиноз может завершать фибриноидные изменения в дне хронической язвы желудка, в
червеобразном отростке при аппендиците; он подобен механизму местного гиалиноза в очаге хронического воспаления.
Гиалиноз как исход склероза имеет в основном также местный характер: он развивается в рубцах, фиброзных спайках серозных полостей, сосудистой стенке при атеросклерозе, инволюционном склерозе артерий, при организации тромба, в капсулах, строме опухоли и т.д. В основе гиалиноза в этих случаях лежат нарушения обмена соединительной ткани. Подобный механизм имеет гиалиноз некротизированных тканей и фибринозных наложений.
Внешний вид. При выраженном гиалинозе внешний вид органов изменяется. Гиалиноз мелких артерий и артериол ведет к атрофии, деформации и сморщиванию органа (например, развитие артериолосклеротического нефроцирроза).
При гиалинозе собственно соединительной ткани она становится плотной, белесоватой, полупрозрачной (например, гиалиноз клапанов сердца при ревматическом пороке).
Исход. В большинстве случаев неблагоприятный, но возможно и рассасывание гиалиновых масс. Так, гиалин в рубцах - так называемых келоидах - может подвергаться разрыхлению и рассасыванию. Обратим гиалиноз молочной железы, причем рассасывание гиалиновых масс происходит в условиях гиперфункции желез. Иногда гиалинизированная ткань ослизняется.
Функциональное значение. Различно в зависимости от локализации, степени и распространенности гиалиноза. Распространенный гиалиноз артериол может вести к функциональной недостаточности органа (почечная недостаточность при артериолосклеротическом нефроциррозе). Местный гиалиноз (например, клапанов сердца при его пороке) также может быть причиной функциональной недостаточности органа. Но в рубцах он может не причинять особых расстройств.
Амилоидоз
Амилоидоз (от лат. amylum - крахмал), или амилоидная дистрофия, - стромально-сосудистый диспротеиноз, сопровождающийся глубоким нарушением белкового обмена, появлением аномального фибриллярного белка и образованием в межуточной ткани и стенках сосудов сложного вещества - амилоида.
В 1844 г. венский патолог К. Рокитанский описал своеобразные изменения паренхиматозных органов, которые, помимо резкого уплотнения, приобретали восковой, сальный, вид. Заболевание, при котором возникали подобные изменения органов, он назвал «сальной болезнью». Спустя несколько лет Р. Вирхов показал, что изменения эти связаны с появлением в органах особого вещества, которое под действием йода и серной кислоты окрашивается в синий цвет. Поэтому он назвал его амилоидом, а «сальную болезнь» - амилоидозом. Белковая природа амилоида была установлена М.М. Рудневым вместе с Кюне в 1865 г.
Химический состав и физические свойства амилоида. Амилоид представляет собой гликопротеид, основным компонентом которого являются фибриллярные белки (F-компонент). Они образуют фибриллы, имеющие характерную ультрамикроскопическую структуру (рис. 33). Фибриллярные белки амилоида неоднородны. Выделяют 4 типа этих белков, характерных для определенных форм амилоидоза: 1) АА-белок (неассоциированный с иммуноглобулинами), образующийся из своего сывороточного аналога - белка SAA; 2) AL-белок (ассоциированный с иммуноглобулинами), предшественником его являются L-цепи (легкие цепи) иммуноглобулинов; 3) AF-белок, в образовании которого участвует главным образом преальбумин; 4) ASC^-белок, предшественник которого также преальбумин.
Белки фибрилл амилоида можно идентифицировать с помощью специфических сывороток при иммуногистохимическом исследовании, а также ряде химических (реакции с перманганатом калия, щелочным гуанидином) и физических (автоклавирование) реакций.
Фибриллярные белки амилоида, которые продуцируют клетки - амилоидобласты, входят в сложные соединения с глюкопротеидами плазмы крови. Этот плазменный компонент (Р-компонент) амилоида представлен палочковидными структурами («периодические палочки» - см. рис. 33). Фибриллярный и плазменный компоненты амилоида обладают антигенными свойствами. Фибриллы амилоида и плазменный компонент вступают в соединения с хондроитинсульфатами ткани и к образующемуся комплексу присоединяются так называемые гематогенные добавки, среди которых основное значение имеют фибрин и иммунные комплексы. Связи белков и полисахаридов в амилоидном веществе чрезвычайно прочные, чем объясняется отсутствие эффекта при действии на амилоид различных ферментов организма.
 Рис. 33. Ультраструктура амилоида:
Рис. 33. Ультраструктура амилоида:
а - фибриллы амилоида (Ам), х35 000; б - палочковидные образования, состоящие из пентагональных структур (ПСт), х300 000 (по Гленнер и др.)
Характерным для амилоида является его красное окрашивание конго красным, метиловым (илигенциановым) фиолетовым; характерна специфическая люминесценция с тиофлавинами S или Т. Амилоид выявляют также с помощью поляризационного микроскопа. Ему свойственны дихроизм и анизотропия (спектр двойного лучепреломления лежит в пределах 540-560 нм). Эти свойства позволяют отличать амилоид от других фибриллярных белков. Для макроскопической диагностики амилоидоза пользуются воздействием на ткань люголевским раствором, а затем 10% раствором серной кислоты; амилоид становится синефиолетовым или грязно-зеленым.
Красочные реакции амилоида, связанные с особенностями его химического состава, могут быть различными в зависимости от формы, вида и типа амилоидоза. В ряде случаев они отсутствуют, тогда говорят об ахроматическом амилоиде, или ахроамилоиде.
Классификация амилоидоза учитывает следующие признаки: 1) возможную причину; 2) специфику белка фибрилл амилоида; 3) распространенность амилоидоза; 4) своеобразие клинических проявлений в связи с преимущественным поражением определенных органов и систем.
1. Руководствуясь причиной, выделяют первичный (идиопатический), наследственный (генетический, семейный), вторичный (приобретенный) и старческий амилоидоз. Первичный, наследственный, старческий амилоидозы рассматривают в качестве нозологических форм. Вторичный амилоидоз, встречающийся при тех или иных заболеваниях, является осложнением этих заболеваний, «второй болезнью».
Для первичного (идиопатического) амилоидоза характерно: отсутствие предшествующего или сопутствующего «причинного» заболевания; поражение преимущественно мезодермальных тканей - сердечно-сосудистой системы, поперечно-полосатых и гладких мышц, нервов и кожи (генерализованный амилоидоз); склонность к образованию узловатых отложений, непостоянство красочных реакций амилоидного вещества (часты отрицательные результаты при окраске конго красным).
Наследственный (генетический, семейный) амилоидоз. Значение генетических факторов в развитии амилоидоза подтверждается своеобразием его географической патологии и особой предрасположенностью к нему определенных этнических групп населения. Наиболее часто встречающийся тип наследственного амилоидоза с преимущественным поражением почек характерен для периодической болезни (семейная средиземноморская лихорадка), которая чаще наблюдается у представителей древних народов (евреи, армяне, арабы).
Встречаются и другие типы наследственного амилоидоза. Так, известен семейный нефропатический амилоидоз, протекающий с лихорадкой, крапивницей и глухотой, описанный в английских семьях (форма Маккла и Уэллса). Наследственный нефропатический амилоидоз имеет несколько вариантов. Для наследственной нейропатии I типа (португальский амилоидоз) характерно поражение периферических нервов ног, а для нейропатии II типа, встречающейся в американских семьях, - поражение периферических нервов рук. При нейропатии III типа, которая описана также у американцев, встречается сочетание ее с не-
фропатией, а при нейропатии IV типа, описанной в финских семьях, отмечается сочетание не только с нефропатией, но и сетчатой дистрофией роговицы. Наследственный кардиопатический амилоидоз, встречающийся у датчан, мало чем отличается от генерализованного первичного амилоидоза.
Вторичный (приобретенный) амилоидоз в отличие от других форм развивается как осложнение ряда заболеваний («вторая болезнь»). Это хронические инфекции (особенно туберкулез), болезни, характеризующиеся гнойно-деструктивными процессами (хронические неспецифические воспалительные заболевания легких, остеомиелит, нагноение ран), злокачественные новообразования (парапротеинемические лейкозы, лимфогранулематоз, рак), ревматические болезни (особенно ревматоидный артрит). Вторичный амилоидоз, при котором, как правило, поражаются многие органы и ткани (генерализованный амилоидоз), встречается по сравнению с другими формами амилоидоза наиболее часто.
При старческом амилоидозе типичны поражения сердца, артерий, головного мозга и островков поджелудочной железы. Эти изменения, как и атеросклероз, обусловливают старческую физическую и психическую деградацию. У старых людей имеется несомненная связь между амилоидозом, атеросклерозом и диабетом, которая объединяет возрастные нарушения обмена. При старческом амилоидозе наиболее часты локальные формы (амилоидоз предсердий, головного мозга, аорты, островков поджелудочной железы), хотя встречается и генерализованный старческий амилоидоз с преимущественным поражением сердца и сосудов, который клинически мало чем отличается от генерализованного первичного амилоидоза.
2. Специфика белка фибрилл амилоида позволяет выделить AL-, АА-, AF- и ASC1-амилоидоз.
AL-амилоидоз включает первичный (идиопатический) амилоидоз и амилоидоз при «плазмоклеточной дискразии», которая объединяет парапротеинемические лейкозы (миеломная болезнь, болезнь Вальденстрема, болезнь тяжелых цепей Франклина), злокачественные лимфомы и др. AL-амилоидоз всегда генерализованный с поражением сердца, легких и сосудов. АА-амилоидоз охватывает вторичный амилоидоз и две формы наследственного - периодическую болезнь и болезнь Маккла и Уэллса. Это также генерализованный амилоидоз, но с преимущественным поражением почек. AF-амилоидоз - наследственный, представлен семейной амилоидной нейропатией (FAP); поражаются прежде всего периферические нервы. ASC-амилоидоз - старческий генерализованный или системный (SSA) с преимущественным поражением сердца и сосудов.
3. Учитывая распространенность амилоидоза, различают генерализованную и локальную формы. К генерализованному амилоидозу, как это видно уже из сказанного, относят первичный амилоидоз и амилоидоз при «плазмоклеточной дискразии» (формы AL-амилоидоза), вторичный амилоидоз и некоторые типы наследственного (формы АА-амилоидоза), а также старческий системный амилоидоз (ASC^-амилоидоз). Локальный амилоидоз
объединяет ряд форм наследственного и старческого амилоидоза, а также локальный опухолевидный амилоидоз («амилоидная опухоль»).
4. Своеобразие клинических проявлений в связи с преимущественным поражением органов и систем позволит выделять кардиопатический, нефропатический, нейропатический, гепатопатический, эпинефропатический, смешанный типы амилоидоза и APUD-амилоидоз. Кардиопатический тип, как говорилось ранее, чаще встречается при первичном и старческом системном амилоидозе, нефропатический - при вторичном амилоидозе, периодической болезни и болезни Маккла и Уэллса; для вторичного амилоидоза характерны и смешанные типы (сочетание поражения почек, печени, надпочечников, желудочно-кишечного тракта). Нейропатический амилоидоз, как правило, имеет наследственный характер. APUD-амилоид развивается в органах APUD-системы при развитии в них опухолей (апудом), а также в островках поджелудочной железы при старческом амилоидозе.
Морфо- и патогенез амилоидоза. Функцию амилоидобластов, продуцирующих белок фибрилл амилоида (рис. 34), при различных формах амилоидоза выполняют разные клетки. При генерализованных формах амилоидоза - это главным образом макрофаги, плазматические и миеломные клетки; однако не исключается роль фибробластов, ретикулярных клеток и эндотелиоцитов. При локальных формах в роли амилоидобластов могут выступать кардиомиоциты (амилоидоз сердца), гладкие мышечные клетки (амилоидоз аорты), кератиноциты (амилоидоз кожи), В-клетки островков поджелудочной железы (инсулярный амилоидоз), С-клетки щитовидной железы и другие эпителиальные клетки APUD-системы.
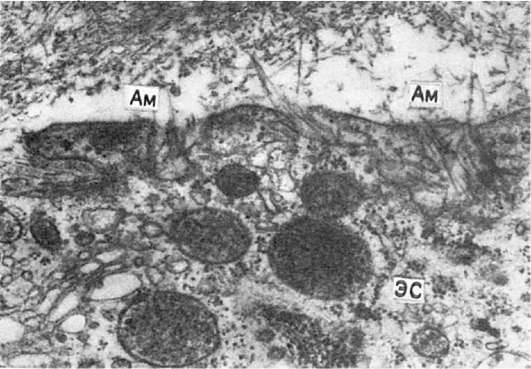 Рис. 34. Амилоидобласт.
Фибриллы амилоида (Ам) в инвагинатах плазмолеммы звездчатого
ретикулоэндотелиоцита с гиперплазией гранулярной эндоплазматической сети
(ЭС), свидетельствующей о его высокой синтетической активности. х30 000
Рис. 34. Амилоидобласт.
Фибриллы амилоида (Ам) в инвагинатах плазмолеммы звездчатого
ретикулоэндотелиоцита с гиперплазией гранулярной эндоплазматической сети
(ЭС), свидетельствующей о его высокой синтетической активности. х30 000
Появление клона амилоидобластов объясняет мутационная теория амилоидоза (Серов В.В., Шамов И.А., 1977). При вторичном амилоидозе (исключая амилоидоз при «плазмоклеточной дискразии») мутации и появление амилоидобластов можно связать с длительной антигенной стимуляцией. Клеточные мутации при «плазмоклеточной дискразии» и амилоидозе опухолей, а возможно, и при опухолевидном локальном амилоидозе обусловлены опухолевыми мутагенами. При генетическом (семейном) амилоидозе речь идет о мутации гена, которая может произойти в различных локусах, чем и определяются различия в составе амилоидного белка у разных людей и животных. При старческом амилоидозе, вероятнее всего, имеют место подобные механизмы, так как эту разновидность амилоидоза рассматривают как фенокопию генетического. Поскольку антигены белка амилоидных фибрилл являются чрезвычайно слабыми иммуногенами, мутирующиеся клетки не распознаются иммунокомпетентной системой и не элиминируются. Развивается иммунологическая толерантность к белкам амилоида, что обусловливает прогрессирование амилоидоза, чрезвычайно редкое рассасывание амилоида - амилоидоклазия - с помощью макрофагов (гигантские клетки инородных тел).
Образование амилоидного белка может быть связано с ретикулярными (периретикулярныи амилоидоз) или коллагеновыми (периколлагеновыи амилоидоз) волокнами. Для периретикулярного амилоидоза, при котором амилоид выпадает по ходу мембран сосудов и желез, а также ретикулярной стромы паренхиматозных органов характерно преимущественное поражение селезенки, печени, почек, надпочечников, кишечника, интимы сосудов мелкого и среднего калибра (паренхиматозный амилоидоз). Для периколлагенового амилоидоза, при котором амилоид выпадает по ходу коллагеновых волокон, свойственно преимущественное поражение адвентиции сосудов среднего и крупного калибра, миокарда, поперечнополосатой и гладкой мускулатуры, нервов, кожи (мезенхимальный амилоидоз). Таким образом, амилоидные отложения имеют довольно типичную локализацию: в стенках кровеносных и лимфатических капилляров и сосудов в интиме или адвентиции; в строме органов по ходу ретикулярных и коллагеновых волокон; в собственной оболочке железистых структур. Амилоидные массы вытесняют и замещают паренхиматозные элементы органов, что ведет к развитию их хронической функциональной недостаточности.
Патогенез амилоидоза сложен и неоднозначен у различных его форм и типов. Лучше других форм изучен патогенез АА- и AL-амилоидоза.
При АА-амилоидозе фибриллы амилоида образуются из поступающего в макрофаг - амилоидобласт плазменного предшественника фибриллярного белка амилоида - белка SAA, который усиленно синтезируется в печени (схема III). Усиленный синтез SAA гепатоцитами стимулирует макрофагальный медиатор интерлейкин-1, что приводит к резкому увеличению содержания SAA в крови (предамилоидная стадия). В этих условиях макрофаги не в состоянии осуществить полную деградацию SAA, и из
Схема III. Патогенез AA-амилоидоза
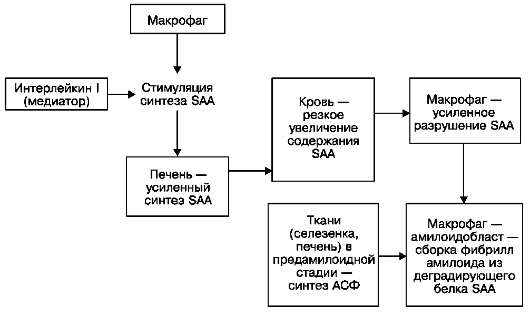 его
фрагментов в инвагинатах плазматической мембраны амилоидобласта
происходит сборка фибрилл амилоида (см. рис. 34). Стимулирует эту сборку
амилоидстимулирующий фактор (АСФ), который обнаруживается в
тканях (селезенка, печень) в предамилоидной стадии. Таким образом,
ведущую роль в патогенезе АА-амилоидоза играет макрофагальная система:
она стимулирует усиленный синтез белка предшественника - SAA печенью,
она же участвует и в образовании фибрилл амилоида из деградирующих
фрагментов этого белка.
его
фрагментов в инвагинатах плазматической мембраны амилоидобласта
происходит сборка фибрилл амилоида (см. рис. 34). Стимулирует эту сборку
амилоидстимулирующий фактор (АСФ), который обнаруживается в
тканях (селезенка, печень) в предамилоидной стадии. Таким образом,
ведущую роль в патогенезе АА-амилоидоза играет макрофагальная система:
она стимулирует усиленный синтез белка предшественника - SAA печенью,
она же участвует и в образовании фибрилл амилоида из деградирующих
фрагментов этого белка.
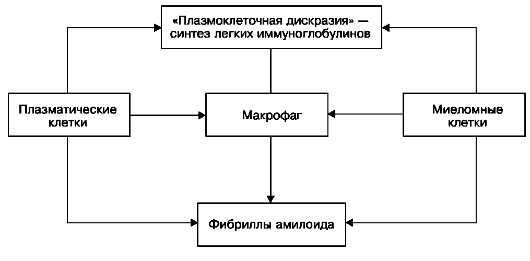
При AL-амилоидозе сывороточным предшественником белка амилоидных фибрилл являются L-цепи иммуноглобулинов. Считают, что возможны два механизма образования AL-амилоидных фибрилл: 1) нарушение деградации моноклоновых легких цепей с образованием фрагментов, способных к агрегации в амилоидные фибриллы; 2) появление L-цепей с особыми вторичными и третичными структурами при аминокислотных заменах. Синтез амилоидных фибрилл из L-цепей иммуноглобулинов может происходить не только в макрофагах, но и в плазматических и миеломных клетках, синтезирующих парапротеины (схема IV). Таким образом, к патогенезу AL-амилоидоза причастна прежде всего лимфоидная система; с ее извращенной функцией связано появление «амилоидогенных» легких цепей иммуноглобулинов - предшественника амилоидных фибрилл. Роль макрофагальной системы при этом вторичная, соподчиненная.
Макро- и микроскопическая характеристика амилоидоза. Внешний вид органов при амилоидозе зависит от степени процесса. Если отложения амилоида небольшие, внешний вид органа изменяется мало и амилоидоз
Схема IV. Патогенез AL-амилоидоза
 обнаруживается
лишь при микроскопическом исследовании. При выраженном амилоидозе орган
увеличивается в объеме, становится очень плотным и ломким, а на разрезе
имеет своеобразный восковидный, или сальный, вид.
обнаруживается
лишь при микроскопическом исследовании. При выраженном амилоидозе орган
увеличивается в объеме, становится очень плотным и ломким, а на разрезе
имеет своеобразный восковидный, или сальный, вид.
В селезенке амилоид откладывается в лимфатических фолликулах (рис. 35) или же равномерно по всей пульпе. В первом случае амилоидноизмененные фолликулы увеличенной и плотной селезенки на разрезе имеют вид полупрозрачных зерен, напоминающих зерна саго (саговая селезенка). Во втором случае селезенка увеличена, плотная, коричнево-красная, гладкая, имеет сальный блеск на разрезе (сальная селезенка). Саговая и сальная селезенка представляют последовательные стадии процесса.
В почках амилоид откладывается в стенке сосудов, в капиллярных петлях и мезангии клубочков, в базальных мембранах канальцев и в строме. Почки становятся плотными, большими и «сальными». По мере нарастания процесса клубочки и пирамиды полностью замещаются амилоидом (см. рис. 35), разрастается соединительная ткань и развивается амилоидное сморщивание почек.
В печени отложение амилоида наблюдается между звездчатыми ретикулоэндотелиоцитами синусоидов, по ходу ретикулярной стромы долек, в стенках сосудов, протоков и в соединительной ткани портальных трактов. По мере накопления амилоида печеночные клетки атрофируются и погибают. При этом печень увеличена, плотная, выглядит «сальной».
В кишечнике амилоид выпадает по ходу ретикулярной стромы слизистой оболочки, а также в стенках сосудов как слизистой оболочки, так и подслизистого слоя. При резко выраженном амилоидозе железистый аппарат кишечника атрофируется.
Амилоидоз надпочечников, как правило, двусторонний, отложение амилоида встречается в корковом веществе по ходу сосудов и капилляров.
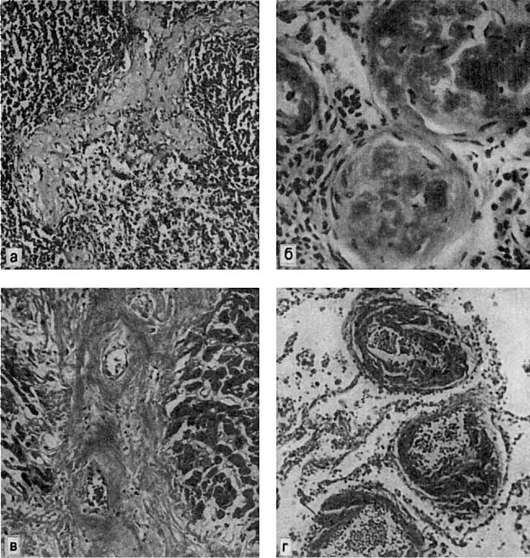 Рис. 35. Амилоидоз:
Рис. 35. Амилоидоз:
а - амилоид в фолликулах селезенки (саговая селезенка); б - амилоид в сосудистых клубочках почек; в - амилоид между мышечными волокнами сердца; г - амилоид в стенках сосудов легких
В сердце амилоид обнаруживается под эндокардом, в строме и сосудах миокарда (см. рис. 35), а также в эпикарде по ходу вен. Отложение амилоида в сердце ведет к резкому его увеличению (амилоидная кардиомегалия). Оно становится очень плотным, миокард приобретает сальный вид.
В скелетных мышцах, как и в миокарде, амилоид выпадает по ходу межмышечной соединительной ткани, в стенках сосудов и в нервах.
Периваскулярно и периневрально нередко образуются массивные отложения амилоидного вещества. Мышцы становятся плотными, полупрозрачными.
В легких отложения амилоида появляются сначала в стенках разветвлений легочных артерии и вены (см. рис. 35), а также в перибронхиальной соединительной ткани. Позже амилоид появляется в межальвеолярных перегородках.
В головном мозге при старческом амилоидозе амилоид находят в сенильных бляшках коры, сосудах и оболочках.
Амилоидоз кожи характеризуется диффузным отложением амилоида в сосочках кожи и ее ретикулярном слое, в стенках сосудов и по периферии сальных и потовых желез, что сопровождается деструкцией эластических волокон и резкой атрофией эпидермиса.
Амилоидоз поджелудочной железы имеет некоторое своеобразие. Помимо артерий железы, встречается и амилоидоз островков, что наблюдается в глубокой старости.
Амилоидоз щитовидной железы также своеобразен. Отложения амилоида в строме и сосудах железы могут быть проявлением не только генерализованного амилоидоза, но и медуллярного рака железы (медуллярный рак щитовидной железы с амилоидозом стромы). Амилоидоз стромы часто встречается в опухолях эндокринных органов и APUD-системы (медуллярный рак щитовидной железы, инсулома, карциноид, феохромоцитома, опухоли каротидных телец, хромофобная аденома гипофиза, гипернефроидный рак), причем в образовании APUD-амилоида доказано участие эпителиальных опухолевых клеток.
Исход. Неблагоприятный. Амилоидоклазия - исключительно редкое явление при локальных формах амилоидоза.
Функциональное значение определяется степенью развития амилоидоза. Выраженный амилоидоз ведет к атрофии паренхимы и склерозу органов, к их функциональной недостаточности. При выраженном амилоидозе возможна хроническая почечная, печеночная, сердечная, легочная, надпочечниковая, кишечная (синдром нарушенного всасывания) недостаточность.
Стромально-сосудистые жировые дистрофии (липидозы)
Стромально-сосудистые жировые дистрофии возникают при нарушениях обмена нейтральных жиров или холестерина и его эфиров.
Нарушения обмена нейтральных жиров
Нарушения обмена нейтральных жиров проявляются в увеличении их запасов в жировой ткани, которое может иметь общий или местный характер.
Нейтральные жиры - это лабильные жиры, обеспечивающие энергетические запасы организма. Они сосредоточены в жировых депо (подкожная клетчатка, брыжейка, сальник, эпикард, костный мозг). Жировая ткань выполняет не только обменную, но и опорную, механическую, функцию, поэтому она способна замещать атрофирующиеся ткани.
Ожирение, или тучность, - увеличение количества нейтральных жиров в жировых депо, имеющее общий характер. Оно выражается в обильном отложении жиров в подкожной клетчатке, сальнике, брыжейке, средостении, эпикарде. Жировая ткань появляется также там, где она обычно отсутствует или имеется лишь в небольшом количестве, например в строме миокарда, поджелудочной железе (рис. 36, а). Большое клиническое зна-
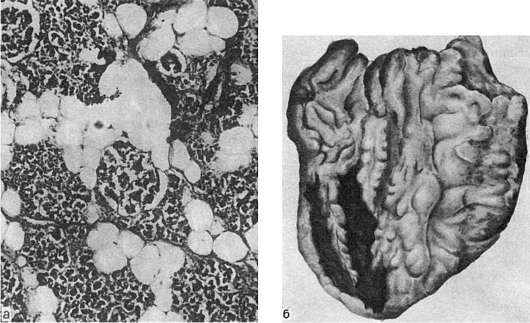 Рис. 36. Ожирение:
Рис. 36. Ожирение:
а - разрастание жировой ткани в строме поджелудочной железы (сахарный диабет); б - ожирение сердца, под эпикардом толстый слой жира
чение имеет ожирение сердца при тучности. Жировая ткань, разрастаясь под эпикардом, окутывает сердце, как футляром (рис. 36, б). Она прорастает строму миокарда, особенно в субэпикардиальных отделах, что ведет к атрофии мышечных клеток. Ожирение обычно резче выражено в правой половине сердца. Иногда вся толща миокарда правого желудочка замещается жировой тканью, в связи с чем может произойти разрыв сердца.
Классификация. Она основывается на различных принципах и учитывает причину, внешние проявления (типы ожирения), степень превышения «идеальной» массы тела, морфологические изменения жировой ткани (варианты ожирения).
По этиологическому принципу выделяют первичную и вторичную формы ожирения. Причина первичного ожирения неизвестна, поэтому его называют также идиопатическим. Вторичное ожирение представлено следующими его видами: 1) алиментарное, причиной которого является несбалансированное питание и гиподинамия; 2) церебральное, развивающееся при травме, опухолях мозга, ряде нейротропных инфекций; 3) эндокринное, представленное рядом синдромов (синдромы Фрелиха и Иценко-Кушинга, адипозогенитальная дистрофия, гипогонадизм, гипотиреоз); 4) наследственное в виде синдрома Лоренса-Муна-Бидля и болезни Гирке.
По внешним проявлениям различают симметричный (универсальный), верхний, средний и нижний типы ожирения. При симметричном типе
жиры относительно равномерно откладываются в разных частях тела. Верхний тип характеризуется накоплением жира преимущественно в области подкожной клетчатки лица, затылка, шеи, верхнего плечевого пояса, молочных желез. При среднем типе жир откладывается в подкожной клетчатке живота в виде фартука, при нижнем типе - в области бедер и голеней.
По превышению массы тела больного выделяют несколько степеней ожирения. При I степени ожирения избыточная масса тела составляет 20-29%, при II - 30-49%, при III - 50-99% и при IV - до 100% и более.
При характеристике морфологических изменений жировой ткани при ожирении учитывают число адипозоцитов и их размер. На этом основании выделяют гипертрофический и гиперпластический варианты общего ожирения. При гипертрофическом варианте жировые клетки увеличены и содержат в несколько раз больше триглицеридов, чем обычные; при этом число адипозоцитов не меняется. Адипозоциты малочувствительны к инсулину, но высокочувствительны к липолитическим гормонам; течение болезни злокачественное. При гиперпластическом варианте число адипозоцитов увеличено (известно, что число жировых клеток достигает максимума в пубертатном периоде и в дальнейшем не меняется). Однако функция адипозоцитов не нарушена, метаболические изменения их отсутствуют; течение болезни доброкачественное.
Причины и механизмы развития. Среди причин общего ожирения, как уже говорилось, большое значение имеют несбалансированное питание и гиподинамия, нарушение нервной (ЦНС) и эндокринной регуляции жирового обмена, наследственные (семейно-конституциональные) факторы. Непосредственный механизм ожирения лежит в нарушении равновесия липогенеза и липолиза в жировой клетке в пользу липогенеза (схема V). Как видно из схемы V, усиление липогенеза, как и ослабление липолиза,
Схема V. Липогенез и липолиз в жировой клетке
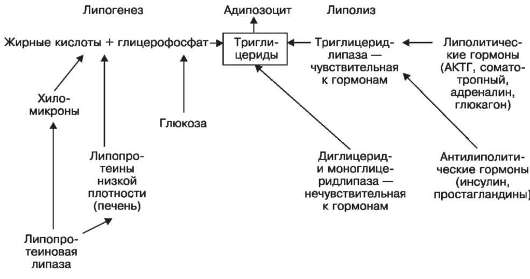 связано
не только с активацией липопротеиновой липазы и угнетением
липолитических липаз, но и нарушением гормональной регуляции в пользу
антилиполитических гормонов, состоянием жирового обмена в кишечнике и
печени.
связано
не только с активацией липопротеиновой липазы и угнетением
липолитических липаз, но и нарушением гормональной регуляции в пользу
антилиполитических гормонов, состоянием жирового обмена в кишечнике и
печени.
Значение. Будучи проявлением ряда заболеваний, общее ожирение определяет развитие тяжелых осложнений. Избыточная масса тела, например, является одним из факторов риска при ишемической болезни сердца.
Исход общего ожирения редко бывает благоприятным.
Антиподом общего ожирения является истощение, в основе которого лежит атрофия. Истощение наблюдается также в терминальной стадии кахексии (от греч. kakos - плохой, hexis - состояние).
При увеличении количества жировой клетчатки, имеющем местный характер, говорят о липоматозах. Среди них наибольший интерес представляет болезнь Деркума (lipomatosis dolorosa), при которой в подкожной клетчатке конечностей и туловища появляются узловатые болезненные отложения жира, похожие на липомы. В основе заболевания лежит полигландулярная эндокринопатия. Местное увеличение количества жировой ткани нередко является выражением вакатного ожирения (жировое замещение) при атрофии ткани или органа (например, жировое замещение почки или вилочковой железы при их атрофии).
Антиподом липоматозов служат регионарные липодистрофии, сущность которых состоит в очаговой деструкции жировой ткани и распаде жиров, нередко с воспалительной реакцией и образованием липогранулем (например, липогранулематоз при рецидивирующем ненагнаивающемся панникулите, или болезни Вебера-Крисчена).
Нарушения обмена холестерина и его эфиров
Нарушения обмена холестерина и его эфиров лежат в основе тяжелого заболевания - атеросклероза. При этом в интиме артерий накапливаются не только холестерин и его эфиры, но и β-липопротеиды низкой плотности и белки плазмы крови, чему способствует повышение сосудистой проницаемости. Накапливающиеся высокомолекулярные вещества ведут к деструкции интимы, распадаются и омыляются. В результате этого в интиме образуется жиробелковый детрит (athere - кашицеобразная масса), разрастается соединительная ткань (sclerosis - уплотнение) и формируется фиброзная бляшка, нередко суживающая просвет сосуда (см. Атеросклероз).
Наследственной дистрофией, развивающейся в связи с нарушением обмена холестерина, является семейный гиперхолестеринемический ксантоматоз. Его относят к болезням накопления, хотя характер ферментопатии не установлен. Холестерин откладывается в коже, стенках крупных сосудов (развивается атеросклероз), клапанах сердца и других органах.
Стромально-сосудистые углеводные дистрофии
Стромально-сосудистые углеводные дистрофии могут быть связаны с нарушением баланса гликопротеидов и гликозаминогликанов. Стромальнососудистую дистрофию, связанную с нарушением обмена гликопроте-
идов, называют ослизнением тканей. Сущность его состоит в том, что хромотропные вещества высвобождаются из связей с белками и накапливаются главным образом в межуточном веществе. В отличие от мукоидного набухания при этом происходит замещение коллагеновых волокон слизеподобной массой. Собственно соединительная ткань, строма органов, жировая ткань, хрящ становятся набухшими, полупрозрачными, слизеподобными, а клетки их - звездчатыми или причудливыми отростчатыми.
Причина. Ослизнение тканей происходит чаще всего вследствие дисфункции эндокринных желез, истощения (например, слизистый отек, или микседема, при недостаточности щитовидной железы; ослизнение соединительнотканных образований при кахексии любого генеза).
Исход. Процесс может быть обратимым, однако прогрессирование его приводит к колликвации и некрозу ткани с образованием полостей, заполненных слизью.
Функциональное значение определяется тяжестью процесса, его продолжительностью и характером ткани, подвергшейся дистрофии.
Наследственные нарушения обмена гликозаминогликанов (мукополисахаридов) представлены большой группой болезней накопления - мукополисахаридозами. Среди них основное клиническое значение имеет гаргоилизм, или болезнь Пфаундлера-Гурлера, для которой характерны непропорциональный рост, деформация черепа («массивный череп»), других костей скелета, наличие пороков сердца, паховой и пупочной грыж, помутнение роговицы, гепато- и спленомегалии. Считают, что в основе мукополисахаридозов лежит недостаточность специфического фактора, определяющего обмен гликозаминогликанов.
Смешанные дистрофии
О смешанных дистрофиях говорят в тех случаях, когда морфологические проявления нарушенного метаболизма выявляются как в паренхиме, так и в строме, стенке сосудов органов и тканей. Они возникают при нарушениях обмена сложных белков - хромопротеидов, нуклеопротеидов и липопротеидов1, а также минералов.
Нарушения обмена хромопротеидов (эндогенные пигментации)2
Хромопротеиды - окрашенные белки, или эндогенные пигменты, играют важную роль в жизни организма. С помощью хромопротеидов осуществляются дыхание (гемоглобин, цитохромы), выработка секретов (желчь) и инкретов (серотонин), защита организма от воздействия лучевой энергии (меланин), пополнение запасов железа (ферритин), баланс витаминов (липохромы) и т.д. Обмен пигментов регулируется вегетативной нервной системой, эндокринными железами, он тесно связан с функцией органов кроветворения и системы моноцитарных фагоцитов.
1 Нарушения обмена липопртеидов приведены в разделах о липидогенных пигментах, жировых и белковых дистрофиях.
2 Помимо эндогенных, существуют экзогенные пигментации (см. Профессиональныне болезни).
Классификация. Эндогенные пигменты принято делить на 3 группы: гемоглобиногенные, представляющие собой различные производные гемоглобина, протеиногенные, или тирозиногенные, связанные с обменом тирозина, и липидогенные, или липопигменты, образующиеся при обмене жиров.
Нарушения обмена гемоглобиногенных пигментов
В норме гемоглобин проходит ряд циклических превращений, обеспечивающих его ресинтез и образование необходимых для организма продуктов. Эти превращения связаны со старением и разрушением эритроцитов (гемолиз, эритрофагия), постоянным обновлением эритроцитной массы. В результате физиологического распада эритроцитов и гемоглобина образуются пигменты ферритин, гемосидерин и билирубин. В патологических условиях вследствие многих причин гемолиз может быть резко усилен и осуществляться как в циркулирующей крови (интраваскулярно), так и в очагах кровоизлияний (экстраваскулярно). В этих условиях, помимо увеличения образующихся в норме гемоглобиногенных пигментов, может появляться ряд новых пигментов - гематоидин, гематины и порфирин.
В связи с накоплением гемоглобиногенных пигментов в тканях могут возникать различные виды эндогенных пигментации, которые становятся проявлением ряда заболеваний и патологических состояний.
Ферритин - железопротеид, содержащий до 23% железа. Железо ферритина связано с белком, который носит название апоферритина. В норме ферритин обладает дисульфидной группой. Это неактивная (окисленная) форма ферритина - SS-ферритин. При недостаточности кислорода происходит восстановление ферритина в активную форму - SH-ферритин, который обладает вазопаралитическими и гипотензивными свойствами. В зависимости от происхождения различают анаболический и катаболический ферритин. Анаболический ферритин образуется из железа, всасывающегося в кишечнике, катаболический - из железа гемолизированных эритроцитов. Ферритин (апоферритин) обладает антигенными свойствами. Ферритин образует берлинскую лазурь (железосинеродистое железо) под действием железосинеродистого калия и соляной, или хлористоводородной, кислоты (реакция Перлса) и может быть идентифицирован с помощью специфической антисыворотки при иммунофлюоресцентном исследовании. Большое количество ферритина содержится в печени (депо ферритина), селезенке, костном мозге и лимфатических узлах, где обмен его связан с синтезом гемосидерина, гемоглобина и цитохромов.
В условиях патологии количество ферритина может увеличиваться как в тканях, так и в крови. Повышение содержания ферритина в тканях наблюдается при гемосидерозе, так как полимеризация ферритина ведет к образованию гемосидерина. Ферритинемией объясняют необратимость шока, сопровождающегося сосудистым коллапсом, так как SH-ферритин выступает в роли антагониста адреналина.
Гемосидерин образуется при расщеплении гема и является полимером ферритина. Он представляет собой коллоидную гидроокись железа, связанную с белками, гликозаминогликанами и липидами клетки. Клетки, в которых образуется гемосидерин, называются сидеробластами. В их сидеросомах происходит синтез гранул гемосидерина (рис. 37). Сидеробласты могут быть как мезенхимальной,
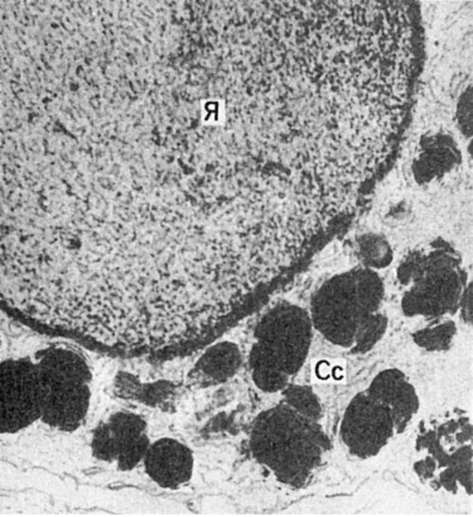 Рис. 37. Сидеробласт. Крупное ядро (Я), узкий ободок цитоплазмы с большим числом сидеросом (Сс). Электронограмма. х20 000
Рис. 37. Сидеробласт. Крупное ядро (Я), узкий ободок цитоплазмы с большим числом сидеросом (Сс). Электронограмма. х20 000
так и эпителиальной природы. Гемосидерин постоянно обнаруживается в ретикулярных и эндотелиальных клетках селезенки, печени, костного мозга, лимфатических узлах. В межклеточном веществе он подвергается фагоцитозу сидерофагами.
Присутствие в гемосидерине железа позволяет выявлять его с помощью характерных реакций: образование берлинской лазури (реакция Перлса), турнбулевой сини (обработка срезов сульфидом аммония, а затем железосинеродистым калием и хлористоводородной кислотой). Положительные реакции на железо отличают гемосидерин от сходных с ним пигментов (гемомеланин, липофусцин, меланин).
В условиях патологии наблюдается избыточное образование гемосидерина - гемосидероз. Он может иметь как общий, так и местный характер.
Общий, или распространенный, гемосидероз наблюдается при внутрисосудистом разрушении эритроцитов (интраваскулярный гемолиз) и встречается при болезнях системы кроветворения (анемии, гемобластозы), интоксикациях гемолитическими ядами, некоторых инфекционных заболеваниях (возвратный тиф, бруцеллез, малярия и др.), переливаниях иногруппной крови, резус-конфликте и т.д. Разрушенные эритроциты, их обломки, гемоглобин идут на построение гемосидерина. Сидеробластами становятся ретикулярные, эндотелиальные и гистиоцитарные элементы селезенки, печени, костного мозга, лимфатических узлов, а также эпителиальные клетки печени, почек, легких, потовых и слюнных желез. Появляется большое число сидерофагов, которые не успевают поглощать гемосидерин, загружающий межклеточное вещество. В результате этого коллагеновые и эластические волокна пропитываются железом. При этом селезенка, печень, костный мозг и лимфатические узлы становятся ржаво-коричневыми.
Близко к общему гемосидерозу своеобразное заболевание - гемохроматоз, который может быть первичным (наследственный гемохроматоз) или вторичным.
Первичный гемохроматоз - самостоятельное заболевание из группы болезней накопления. Передается доминантно-аутосомным путем и связано с наследственным дефектом ферментов тонкой кишки, что ведет к повышенному всасыванию пищевого железа, которое в виде гемосидерина откладывается в большом количестве в органах. Обмен железа эритроцитов при этом не нарушен. Количество железа в организме увеличивается
в десятки раз, достигая 50-60 г. Развивается гемосидероз печени, поджелудочной железы, эндокринных органов, сердца, слюнных и потовых желез, слизистой оболочки кишечника, сетчатки глаза и даже синовиальных оболочек; одновременно в органах увеличивается содержание ферритина. В коже и сетчатке глаз увеличивается содержание меланина, что связано с поражением эндокринной системы и нарушением регуляции меланинобразования. Основными симптомами болезни являются бронзовая окраска кожи, сахарный диабет (бронзовый диабет) и пигментный цирроз печени. Возможно развитие и пигментной кардиомиопатии с нарастающей сердечной недостаточностью.
Вторичный гемохроматоз - заболевание, развивающееся при приобретенной недостаточности ферментных систем, обеспечивающих обмен пищевого железа, что ведет к распространенному гемосидерозу. Причиной этой недостаточности могут быть избыточное поступление железа с пищей (железосодержащие препараты), резекция желудка, хронический алкоголизм, повторные переливания крови, гемоглобинопатии (наследственные,заболевания, в основе которых лежит нарушение синтеза гема или глобина). При вторичном гемохроматозе содержание железа повышено не только в тканях, но и в сыворотке крови. Накопление гемосидерина и ферритина, наиболее выраженное в печени, поджелудочной железе и сердце, приводит к циррозу печени, сахарному диабету и кардиомиопатии.
Местный гемосидероз - состояние, развивающееся при внесосудистом разрушении эритроцитов (экстраваскулярный гемолиз), т.е. в очагах кровоизлияний. Оказавшиеся вне сосудов эритроциты теряют гемоглобин и превращаются в бледные круглые тельца («тени» эритроцитов), свободный гемоглобин и обломки эритроцитов идут на построение пигмента. Сидеробластами и сидерофагами становятся лейкоциты, гистиоциты, ретикулярные клетки, эндотелий, эпителий. Сидерофаги могут долго сохраняться на месте бывшего кровоизлияния, нередко они переносятся током лимфы в близлежащие лимфатические узлы, где задерживаются, и узлы становятся ржавыми. Часть сидерофагов разрушается, пигмент высвобождается и в дальнейшем снова подвергается фагоцитозу.
Гемосидерин образуется при всех кровоизлияниях, как мелких, так и крупных. В небольших кровоизлияниях, которые чаще имеют характер диапедезных, обнаруживается только гемосидерин. При крупных кровоизлияниях по периферии, среди живой ткани образуется гемосидерин, а в центре - кровоизлияния, где аутолиз происходит без доступа кислорода и участия клеток, появляются кристаллы гематоидина.
В зависимости от условий развития местный гемосидероз может возникать в пределах не только участка ткани (гематома), но и целого органа. Таков гемосидероз легких, наблюдающийся при ревматическом митральном пороке сердца, кардиосклерозе и др. (рис. 38). Хронический венозный застой в легких ведет к множественным диапедезным кровоизлияниям, в связи с чем в межальвеолярных перегородках, альвеолах,
 Рис. 38. Гемосидероз легких. Цитоплазма гистиоцитов и альвеолярного эпителия (сидеробластов и сидерофагов) нагружена зернами пигмента
Рис. 38. Гемосидероз легких. Цитоплазма гистиоцитов и альвеолярного эпителия (сидеробластов и сидерофагов) нагружена зернами пигмента
лимфатических сосудах и узлах легких появляется большое число нагруженных гемосидерином клеток (см. Венозное полнокровие).
Билирубин - важнейший желчный пигмент. Его образование начинается в гистиоцитарно-макрофагальной системе при разрушении гемоглобина и отщеплении от него гема. Гем теряет железо и превращается в биливердин, при восстановлении которого образуется билирубин в комплексе с белком. Гепатоциты осуществляют захват пигмента, конъюгацию его с глюкуроновой кислотой и экскрецию в желчные капилляры. С желчью билирубин поступает в кишечник, где часть его всасывается и вновь попадает в печень, часть - выводится с калом в виде стер-кобилина и мочой в виде уробилина. В норме билирубин встречается в растворенном состоянии в желчи и в небольшом количестве в плазме крови.
Билирубин представлен красножелтыми кристаллами. Он не содержит железа. Для выявления его употребляют реакции, основанные на способности пигмента легко окисляться с образованием различно окрашенных продуктов. Такова, например, реакция Гмелина, при которой под воздействием концентрированной азотной кислоты билирубин дает сначала зеленое, а затем синее или пурпурное окрашивание.
Нарушение обмена билирубина связано с расстройством его образования и выделения. Это ведет к повышенному содержанию билирубина в плазме крови и желтому окрашиванию им кожи, склер, слизистых и серозных оболочек и внутренних органов - желтухе.
Механизм развития желтухи различен, что позволяет выделять три ее вида: надпеченочную (гемолитическую), печеночную (паренхиматозную) и подпеченочную (механическую).
Надпеченочная (гемолитическая) желтуха характеризуется повышенным образованием билирубина в связи с увеличенным распадом эритроцитов. Печень в этих условиях образует большее, чем в норме, количество пигмента, однако вследствие недостаточности захвата билирубина гепатоцитами уровень его в крови остается повышенным. Гемолитическая желтуха наблюдается при инфекциях (сепсис, малярия, возвратный тиф) и интоксикациях (гемолитические яды), при изоиммунных (гемолитическая болезнь новорожденных, переливание несовместимой крови) и аутоиммунных (гемобластозы, системные заболевания соединительной ткани) конфликтах. Она может развиваться и при массивных кровоизли-
яниях, геморрагических инфарктах в связи с избыточным поступлением билирубина в кровь из очага распада эритроцитов, где желчный пигмент выявляется в виде кристаллов. С образованием в гематомах билирубина связано изменение их окраски.
Гемолитическая желтуха может быть обусловлена дефектом эритроцитов. Таковы наследственные ферментопатии (микросфероцитоз, овалоцитоз), гемоглобинопатии, или гемоглобинозы (талассемия, или гемоглобиноз F; серповидноклеточная анемия, или гемоглобиноз S), пароксизмальная ночная гемоглобинурия, так называемые шунтовые желтухи (при дефиците витамина В12, некоторых гипопластических анемиях и др.).
Печеночная (паренхиматозная) желтуха возникает при поражении гепатоцитов, в результате чего нарушаются захват ими билирубина, конъюгация его с глюкуроновой кислотой и экскреция. Такая желтуха наблюдается при остром и хроническом гепатитах, циррозах печени, медикаментозных ее повреждениях и аутоинтоксикации, например при беременности, ведущих к внутрипеченочному холестазу. Особую группу составляют ферментопатические печеночные желтухи, возникающие при наследственных пигментных гепатозах, при которых нарушена одна из фаз внутрипеченочного обмена билирубина.
Подпеченочная (механическая) желтуха связана с нарушением проходимости желчных протоков, что затрудняет экскрецию и определяет регургитацию желчи. Эта желтуха развивается при наличии препятствий оттоку желчи из печени, лежащих внутри или вне желчных протоков, что наблюдается при желчнокаменной болезни, раке желчных путей, головки поджелудочной железы и сосочка двенадцатиперстной кишки, атрезии (гипоплазии) желчных путей, метастазах рака в перипортальные лимфатические узлы и печень. При застое желчи в печени возникают очаги некроза с последующим замещением их соединительной тканью и развитием цирроза (вторичный билиарный цирроз). Застой желчи приводит к расширению желчных протоков и разрыву желчных капилляров. Развивается холемия, которая вызывает не только интенсивную окраску кожи, но и явления общей интоксикации, главным образом от воздействия на организм циркулирующих в крови желчных кислот (холалемия). В связи с интоксикацией понижается способность крови к свертыванию, появляются множественные кровоизлияния (геморрагический синдром). С аутоинтоксикацией связано поражение почек, развитие печеночно-почечной недостаточности.
Гематоидин - не содержащий железа пигмент, кристаллы которого имеют вид ярко-оранжевых ромбических пластинок или иголок, реже - зерен. Он возникает при распаде эритроцитов и гемоглобина внутриклеточно, но в отличие от гемосидерина в клетках не остается и при гибели их оказывается свободно лежащим среди некротических масс. Химически он идентичен билирубину.
Скопления гематоидина находят в старых гематомах, рубцующихся инфарктах, причем в центральных участках кровоизлияний - вдали от живых тканей.
Гематины представляют собой окисленную форму гема и образуются при гидролизе оксигемоглобина. Они имеют вид темно-коричневых или черных ромбовидных кристаллов или зерен, дают двойное лучепреломление в поляризованном свете (анизотропны), содержат железо, но в связанном состоянии.
К выявляемым в тканях гематинам относят: гемомеланин (малярийный пигмент), солянокислый гематин (гемин) и формалиновый пигмент. Гистохимические свойства этих пигментов идентичны.
Гемомеланин (малярийный пигмент) возникает из простетической части гемоглобина под влиянием плазмодиев малярии, паразитирующих в эритроцитах. При малярии развивается гемомеланоз, так как при разрушении эритроцитов малярийный пигмент попадает в кровь и подвергается фагоцитозу макрофагами селезенки, печени, костного мозга, лимфатических узлов, головного мозга (при малярийной коме). Эти органы приобретают аспидно-серую окраску. В них наряду с малярийным пигментом наблюдается отложение гемосидерина.
Солянокислый гематин (гемин) находят в эрозиях и язвах желудка, где он возникает под воздействием на гемоглобин ферментов желудочного сока и хлористоводородной кислоты. Область дефекта слизистой оболочки желудка приобретает буро-черный цвет.
Формалиновый пигмент в виде темно-коричневых игл или гранул встречается в тканях при фиксации их в кислом формалине (этот пигмент не образуется, если формалин имеет рН >6,0). Его считают производным гематина.
Порфирины - предшественники простетической части гемоглобина, имеющие, как и гем, то же тетрапиррольное кольцо, но лишенное железа. По химической природе порфирины близки билирубину: они растворимы в хлороформе, эфире, пиридине. Метод выявления порфиринов основан на способности растворов этих пигментов давать красную или оранжевую флюоресценцию в ультрафиолетовом свете (флюоресцирующие пигменты). В норме порфирины обнаруживаются в крови, моче, тканях. Они обладают свойством повышать чувствительность организма, прежде всего кожи, к свету и являются поэтому антагонистами меланина.
При нарушениях обмена порфиринов возникают порфирии, для которых характерно увеличение содержания пигментов в крови (порфиринемия) и моче (порфиринурия), резкое повышение чувствительности к ультрафиолетовым лучам (светобоязнь, эритема, дерматит). Различают приобретенную и врожденную порфирии.
Приобретенная порфирия наблюдается при интоксикациях (свинец, сульфазол, барбитураты), авитаминозах (пеллагра), пернициозной анемии, некоторых заболеваниях печени. Отмечаются нарушения функции нервной системы, повышенная чувствительность к свету, нередко развиваются желтуха, пигментация кожи, в моче обнаруживают большое количество порфиринов.
Врожденная порфирия - редкое наследственное заболевание. При нарушении синтеза порфирина в эритробластах (недостаточность уропорфириногена III - косинтетазы) развивается эритропоэтическая форма,
а при нарушении синтеза порфирина в клетках печени (недостаточность уропорфирина III - косинтетазы) - печеночная форма порфирии. При эритропоэтической форме порфирии развивается гемолитическая анемия, поражаются нервная система и желудочно-кишечный тракт (рвота, диарея). Порфирины накапливаются в селезенке, костях и зубах, которые приобретают коричневый цвет; моча, содержащая большое количество порфиринов, становится желто-красной. При печеночной форме порфирии печень увеличивается, становится серо-коричневой, в ожиревших гепатоцитах, помимо отложений порфиринов, находят гемосидерин.
Нарушения обмена протеиногенных (тирозиногенных) пигментов
К протеиногенным (тирозиногенным) пигментам относят меланин, пигмент гранул энтерохромаффинных клеток и адренохром. Накопление этих пигментов в тканях служит проявлением ряда заболеваний.
Меланин (от греч. melas - черный) - широко распространенный буро-черный пигмент, с которым у человека связана окраска кожи, волос, глаз. Он дает положительную аргентаффинную реакцию, т.е. обладает способностью восстанавливать аммиачный раствор нитрата серебра до металлического серебра. Эти реакции позволяют гистохимически отличить его в тканях от других пигментов.
Синтез меланина происходит из тирозина в клетках меланинобразующей ткани - меланоцитах, имеющих нейроэктодермальное происхождение. Их предшественниками являются меланобласты. Под действием тирозиназы в меланосомах меланоцитов (рис. 39) из тирозина образуется диоксифенилаланин (ДОФА), или промеланин, который полимеризуется в меланин. Клетки, фагоцитирующие меланин, называют меланофагами.
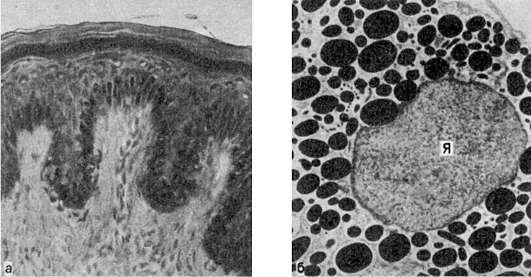 Рис. 39. Кожа при аддисоновой болезни:
Рис. 39. Кожа при аддисоновой болезни:
а - в базальном слое эпидермиса скопления меланоцитов; в дерме много меланофагов; б - меланоцит кожи. В цитоплазме много меланосом. Я - ядро. Электронограмма. х10 000
Меланоциты и меланофаги содержатся в эпидермисе, дерме, радужной и сетчатой оболочках глаз, в мягкой мозговой оболочке. Содержание меланина в коже, сетчатке и радужке зависит от индивидуальных и расовых особенностей и подвергается колебаниям в различные периоды жизни. Регуляция меланогенеза осуществляется нервной системой и эндокринными железами. Стимулируют синтез меланина меланостимулирующий гормон гипофиза, АКТГ, половые гормоны, медиаторы симпатической нервной системы, тормозят - мелатонин и медиаторы парасимпатической нервной системы. Образование меланина стимулируется ультрафиолетовыми лучами, что объясняет возникновение загара как адаптивной защитной биологической реакции.
Нарушения обмена меланина выражаются в усиленном его образовании или исчезновении. Эти нарушения имеют распространенный или местный характер и могут быть приобретенными или врожденными.
Распространенный приобретенный гипермеланоз (меланодермия) особенно часто и резко выражен при аддисоновой болезни (см. рис. 39), обусловленной поражением надпочечников, чаще туберкулезной или опухолевой природы. Гиперпигментация кожи при этой болезни объясняется не столько тем, что при разрушении надпочечников вместо адреналина из тирозина и ДОФА синтезируется меланин, сколько усилением продукции АКТГ в ответ на уменьшение адреналина в крови. АКТГ стимулирует синтез меланина, в меланоцитах увеличивается количество меланосом. Меланодермия встречается также при эндокринных расстройствах (гипогонадизм, гипопитуитаризм), авитаминозах (пеллагра, цинга), кахексии, интоксикации углеводородами.
Распространенный врожденный гипермеланоз (пигментная ксеродерма) связан с повышенной чувствительностью кожи к ультрафиолетовым лучам и выражается в пятнистой пигментации кожи с явлениями гиперкератоза и отека.
К местному приобретенному меланозу относят меланоз толстой кишки, который встречается у людей, страдающих хроническим запором, гиперпигментированные участки кожи (черный акантоз) при аденомах гипофиза, гипертиреоидизме, сахарном диабете. Очаговое усиленное образование меланина наблюдается в пигментных пятнах (веснушки, лентиго) и в пигментных невусах. Из пигментных невусов могут возникать злокачественные опухоли - меланомы.
Распространенный гипомеланоз, или альбинизм (от лат. albus - белый), связан с наследственной недостаточностью тирозиназы. Альбинизм проявляется отсутствием меланина в волосяных луковицах, эпидермисе и дерме, в сетчатке и радужке.
Очаговый гипомеланоз (лейкодерма, или витилиго) возникает при нарушении нейроэндокринной регуляции меланогенеза (лепра, гиперпаратиреоидизм, сахарный диабет), образовании антител к меланину (зоб Хасимото), воспалительных и некротических поражениях кожи (сифилис).
Пигмент гранул энтерохромаффинных клеток, разбросанных в различных отделах желудочно-кишечного тракта, является производным триптофана. Он может быть выявлен с помощью ряда гистохимических реакций - аргентаффинной, хромаффинной реакции Фалька, образование пигмента связано с синтезом серотонина и мелатонина.
Накопление гранул, содержащих пигмент энтерохромаффинных клеток, постоянно обнаруживают в опухолях из этих клеток, называемых карциноидами.
Адренохром - продукт окисления адреналина - встречается в виде гранул в клетках мозгового вещества надпочечников. Дает характерную хромаффинную реакцию, в основе которой лежит способность окрашиваться хромовой кислотой в темно-коричневый цвет и восстанавливать бихромат. Природа пигмента изучена мало.
Патология нарушений обмена адренохрома не изучена.
Нарушения обмена липидогенных пигментов (липопигментов)
В эту группу входят жиробелковые пигменты - липофусцин, пигмент недостаточности витамина Е, цероид и липохромы. Липофусцин, пигмент недостаточности витамина Е и цероид имеют одинаковые физические и химические (гистохимические) свойства, что дает право считать их разновидностями одного пигмента - липофусцина. Однако в настоящее время липофусцином считают липопигмент лишь паренхиматозных и нервных клеток; пигмент недостаточности витамина Е - разновидность липофусцина. Цероидом называют липопигмент мезенхимальных клеток, главным образом макрофагов.
Патология обмена липопигментов разнообразна.
Липофусцин представляет собой гликолипопротеид. Он представлен зернами золотистого или коричневого цвета, электронно-микроскопически выявляется в виде электронно-плотных гранул (рис. 40), окруженных трехконтурной мембраной, которая содержит миелиноподобные структуры.
Образование липофусцина происходит путем аутофагии и проходит несколько стадий. Первичные гранулы, или пропигмент-гранулы, появляются перинуклеарно в зоне наиболее активно протекающих обменных процессов. Они содержат ферменты митохондрий и рибосом (металлофлавопротеиды, цитохромы), связанные с липопротеидами их мембран. Пропигмент-гранулы поступают в пластинчатый комплекс, где происходит синтез гранул незрелого липофусцина, который суданофилен, ШИК-положителен, содержит железо, иногда медь, обладает светло-желтой аутофлюоресценцией в ультрафиолетовом свете. Гранулы незрелого пигмента перемещаются в периферическую зону клетки и абсорбируются там лизосомами; появляется зрелый липофусцин, обладающий высокой активностью лизосомных, а не дыхательных ферментов. Гранулы его становятся коричневыми, они стойко суданофильны, ШИК-положительны, железо в них не выявляется, аутофлюоресценция становится красно-коричневой. Накапливающийся в лизосомах липофусцин превращается в остаточные тельца - телолизосомы.
В условиях патологии содержание липофусцина в клетках может резко увеличиваться. Это нарушение обмена называется липофусцинозом. Он может быть вторичным и первичным (наследственным).
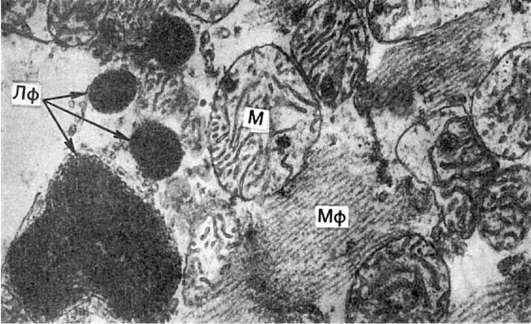 Рис. 40. Липофусцин (Лф) в мышечной клетке сердца, тесно связанный с митохондриями (М). Мф - миофибриллы. Электронограмма. х21 000
Рис. 40. Липофусцин (Лф) в мышечной клетке сердца, тесно связанный с митохондриями (М). Мф - миофибриллы. Электронограмма. х21 000
Вторичный липофусциноз развивается в старости, при истощающих заболеваниях, ведущих к кахексии (бурая атрофия миокарда, печени), при повышении функциональной нагрузки (липофусциноз миокарда при пороке сердца, печени - при язвенной болезни желудка и двенадцатиперстной кишки), при злоупотреблении некоторыми лекарствами (анальгетики), при недостаточности витамина Е (пигмент недостаточности витамина Е).
Первичный (наследственный) липофусциноз характеризуется избирательным накоплением пигмента в клетках определенного органа или системы. Он проявляется в виде наследственного гепатоза, или доброкачественной гипербилирубинемии (синдромы Дабина-Джонсона, Жильбера, Кригера-Найяра) с избирательным липофусцинозом гепатоцитов, а также нейронального липофусциноза (синдром Бильшовского-Янского, Шпильмейера-Шегрена, Кафа), когда пигмент накапливается в нервных клетках, что сопровождается снижением интеллекта, судорогами, нарушением зрения.
Цероид образуется в макрофагах путем гетерофагии при резорбции липидов или липидсодержащего материала; основу цероида составляют липиды, к которым вторично присоединяются белки. К образованию гетерофагических вакуолей (липофагосом) приводит эндоцитоз. Липофагосомы трансформируются во вторичные лизосомы (липофаголизосомы). Липиды не перевариваются лизосомальными ферментами и остаются в лизосомах, появляются остаточные тельца, т.е. телолизосомы.
В условиях патологии образование цероида чаще всего отмечается при некрозе тканей, особенно если окисление липидов усиливается кровоизлиянием (поэтому раньше цероид называли гемофусцином, что принци-
пиально неверно) или если липиды присутствуют в таком количестве, что их аутоокисление начинается раньше, чем переваривание.
Липохромы представлены липидами, в которых присутствуют каротиноиды, являющиеся источником образования витамина А. Липохромы придают желтую окраску жировой клетчатке, коре надпочечников, сыворотке крови, желтому телу яичников. Выявление их основано на обнаружении каротиноидов (цветные реакции с кислотами, зеленая флюоресценция в ультрафиолетовом свете).
В условиях патологии может наблюдаться избыточное накопление липохромов.
Например, при сахарном диабете пигмент накапливается не только в жировой клетчатке, но и в коже, костях, что связано с резким нарушением липидно-витаминного обмена. При резком и быстром похудании происходит конденсация липохромов в жировой клетчатке, которая становится охряно-желтой.
Нарушения обмена нуклеопротеидов
Нуклеопротеиды построены из белка и нуклеиновых кислот - дезоксирибонуклеиновой (ДНК) и рибонуклеиновой (РНК). ДНК выявляется с помощью метода Фельгена, РНК - метода Браше. Эндогенная продукция и поступление нуклеопротеидов с пищей (пуриновый обмен) уравновешиваются их распадом и выведением в основном почками конечных продуктов нуклеинового обмена - мочевой кислоты и ее солей.
При нарушениях обмена нуклеопротеидов и избыточном образовании мочевой кислоты ее соли могут выпадать в тканях, что наблюдается при подагре, мочекаменной болезни и мочекислом инфаркте.
Подагра (от греч. podos - нога и agra - охота) характеризуется периодическим выпадением в суставах мочекислого натрия, что сопровождается болевым приступом. У больных обнаруживается повышенное содержание солей мочевой кислоты в крови (гиперурикемия) и моче (гиперурикурия). Соли обычно откладываются в синовии и хрящах мелких суставов ног и рук, голеностопных и коленных суставов, в сухожилиях и суставных сумках, в хряще ушных раковин. Ткани, в которых выпадают соли в виде кристаллов или аморфных масс, некротизируются. Вокруг отложений солей, как и очагов некроза, развивается воспалительная гранулематозная реакция со скоплением гигантских клеток (рис. 41). По мере увеличения отложений солей и разрастания вокруг них соединительной ткани образуются подагрические шишки (tophi urici), суставы деформируются. Изменения почек при подагре заключаются в скоплениях мочевой кислоты и солей мочекислого натрия в канальцах и собирательных трубках с обтурацией их просветов, развитии вторичных воспалительных и атрофических изменений (подагрические почки).
В большинстве случаев развитие подагры обусловлено врожденными нарушениями обмена веществ (первичная подагра), о чем свидетельствует семейный ее характер; при этом велика роль особенностей питания, употребления больших количеств животных белков. Реже подагра является
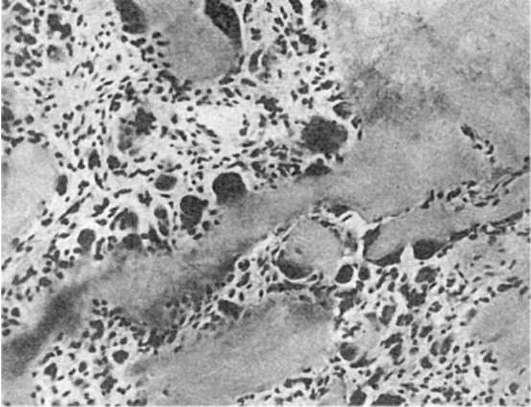 Рис. 41. Подагра. Отложения солей мочевой кислоты с выраженной воспалительной гигантоклеточной реакцией вокруг них
Рис. 41. Подагра. Отложения солей мочевой кислоты с выраженной воспалительной гигантоклеточной реакцией вокруг них
осложнением других заболеваний, нефроцирроза, болезней крови (вторичная подагра).
Мочекаменная болезнь, как и подагра, может быть связана прежде всего с нарушением пуринового обмена, т.е. быть проявлением так называемого мочекислого диатеза. При этом в почках и мочевыводящих путях образуются преимущественно или исключительно ураты (см. Почечнокаменная болезнь).
Мочекислый инфаркт встречается у новорожденных, проживших не менее 2 сут, и проявляется выпадением в канальцах и собирательных трубках почек аморфных масс мочекислых натрия и аммония. Отложения солей мочевой кислоты выглядят на разрезе почки в виде желто-красных полос, сходящихся у сосочков мозгового слоя почки. Возникновение мочекислого инфаркта связано с интенсивным обменом в первые дни жизни новорожденного и отражает адаптацию почек к новым условиям существования.
Нарушения минерального обмена (минеральные дистрофии)
Минералы участвуют в построении структурных элементов клеток и тканей и входят в состав ферментов, гормонов, витаминов, пигментов, белковых комплексов. Они являются биокатализаторами, участвуют во многих обменных процессах, играют важную роль в поддержании кислотно-основного состояния и в значительной мере определяют нормальную жизнедеятельность организма.
Минеральные вещества в тканях определяют методом микросжигания в сочетании с гистоспектрографией. С помощью радиоавтографии можно изучить локализацию в тканях элементов, вводимых в организм в форме изотопов. Кроме того, для выявления ряда элементов, высвобождающихся из связей с белками и выпадающих в тканях, применяются обычные гистохимические методы.
Наибольшее практическое значение имеют нарушения обмена кальция, меди, калия и железа.
Нарушения обмена кальция
Кальций связан с процессами проницаемости клеточных мембран, возбудимости нервно-мышечных приборов, свертывания крови, регуляции кислотно-основного состояния, формирования скелета и т.д.
Кальций абсорбируется с пищей в виде фосфатов в верхнем отрезке тонкой кишки, кислая среда которой обеспечивает всасывание. Большое значение для абсорбции кальция в кишечнике имеет витамин D, который катализирует образование растворимых фосфорных солей кальция. В утилизации кальция (кровь, ткани) большое значение имеют белковые коллоиды и рН крови. В высвобожденной концентрации (0,25-0,3 ммоль/л) кальций удерживается в крови и тканевой жидкости. Основная масса кальция находится в костях (депо кальция), где соли кальция связаны с органической основой костной ткани. В компактном веществе костей кальций является относительно стабильным, а в губчатом веществе эпифизов и метафизов - лабильным. Растворение кости и «вымывание» кальция проявляются в одних случаях лакунарным рассасыванием, в других - так называемым пазушным рассасыванием, или гладкой резорбцией. Лакунарное рассасывание кости осуществляется с помощью клеток - остеокластов; при пазушном рассасывании, как и при гладкой резорбции, происходит растворение кости без участия клеток, образуется «жидкая кость». В тканях кальций выявляют методом серебрения Косса. Поступление кальция с пищей и из депо уравновешивается экскрецией его толстой кишкой, почками, печенью (с желчью) и некоторыми железами.
Регуляция обмена кальция осуществляется нейрогуморальным путем. Наибольшее значение имеют околощитовидные железы (паратгормон) и щитовидная железа (кальцитонин). При гипофункции околощитовидных желез (паратгормон стимулирует вымывание кальция из костей), как и при гиперпродукции кальцитонина (кальцитонин способствует переходу кальция из крови в костную ткань), содержание кальция в крови снижается; гиперфункция околощитовидных желез, как и недостаточная продукция кальцитонина, наоборот, сопровождается вымыванием кальция из костей и гиперкальциемией.
Нарушения обмена кальция называют кальцинозом, известковой дистрофией, или обызвествлением. В его основе лежит выпадение солей кальция из растворенного состояния и отложение их в клетках или межклеточном веществе. Матрицей обызвествления могут быть митохондрии и лизосомы клеток, гликозаминогликаны основного вещества, коллагеновые или эластические волокна. В связи с этим различают внутриклеточное и внеклеточное обызвествление. Кальциноз может быть системным (распространенным) или местным.
Механизм развития. В зависимости от преобладания общих или местных факторов в развитии кальциноза различают три формы обызвествления: метастатическое, дистрофическое и метаболическое.
Метастатическое обызвествление (известковые метастазы) имеет распространенный характер. Основной причиной его возникновения является гиперкальциемия, связанная с усиленным выходом солей кальция из депо, пониженным их выведением из организма, нарушением эндокринной регуляции обмена кальция (гиперпродукция паратгормона, недо-
статок кальцитонина). Поэтому возникновение известковых метастазов отмечают при разрушении костей (множественные переломы, миеломная болезнь, метастазы опухоли), остеомаляции и гиперпаратиреоидной остеодистрофии, поражениях толстой кишки (отравление сулемой, хроническая дизентерия) и почек (поликистоз, хронический нефрит), избыточном введении в организм витамина D и др.
Соли кальция при метастическом обызвествлении выпадают в разных органах и тканях, но наиболее часто - в легких, слизистой оболочке желудка, почках, миокарде и стенке артерий. Это объясняется тем, что легкие, желудок и почки выделяют кислые продукты и их ткани вследствие большей щелочности менее способны удерживать соли кальция в растворе, чем ткани других органов. В миокарде и стенке артерий известь откладывается в связи с тем, что их ткани омываются артериальной кровью и относительно бедны углекислотой.
Внешний вид органов и тканей мало изменяется, иногда на поверхности разреза видны беловатые плотные частицы. При известковых метастазах соли кальция инкрустируют как клетки паренхимы, так и волокна и основное вещество соединительной ткани. В миокарде (рис. 42) и почках первичные отложения извести находят в митохондриях и фаголизосомах, обладающих высокой активностью фосфатаз (образование фосфата кальция). В стенке артерий и в соединительной ткани известь первично выпадает по ходу мембран и волокнистых структур. Вокруг отложений извести наблюдается воспалительная реакция, иногда отмечают скопление макрофагов, гигантских клеток, образование гранулемы.
При дистрофическом обызвествлении, или петрификации, отложения солей кальция имеют местный характер и обычно обнаруживаются в тка-
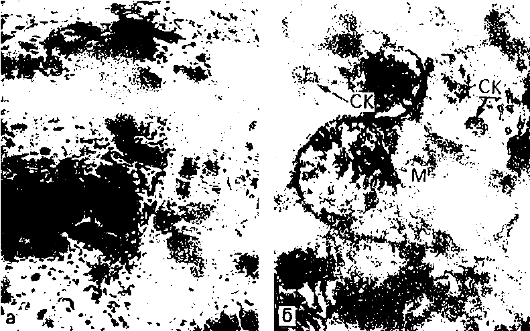 Рис. 42. Известковые метастазы в миокарде:
Рис. 42. Известковые метастазы в миокарде:
а - обызвествленные мышечные волокна (черного цвета) (микроскопическая картина); б - соли кальция (СК) фиксированы на кристах митохондрий (М). Электронограмма. х40 000
нях, омертвевших или находящихся в состоянии глубокой дистрофии; гиперкальциемия отсутствует. Основная причина дистрофического обызвествления - физико-химические изменения тканей, обеспечивающие абсорбцию извести из крови и тканей жидкости. Наибольшее значение придается ощелачиванию среды и усилению активности фосфатаз, высвобождающихся из некротизированных тканей.
При дистрофическом обызвествлении в тканях образуются разных размеров известковые сростки каменной плотности - петрификаты; в ряде случаев в петрификатах появляется костная ткань (оссификация). Петрификаты образуются в казеозных очагах при туберкулезе, гуммах, инфарктах, фокусах хронического воспаления и т.д. Дистрофическому обызвествлению подвергаются также рубцовая ткань (например, клапанов сердца при его пороке, атеросклеротических бляшек - рис. 43), хрящи (хондрокальциноз), погибшие паразиты (эхинококк, трихины), мертвый плод при внематочной беременности (литопедион) и др.
Механизм метаболического обызвествления (известковая подагра, интерстициальный кальциноз) не выяснен: общие (гиперкальциемия) и местные (дистрофия, некроз, склероз) предпосылки отсутствуют. В развитии метаболического обызвествления главное значение придают нестойкости буферных систем (рН и белковые коллоиды), в связи с чем кальций не удерживается в крови и тканевой жидкости даже при невысокой его концентрации, а также наследственно обусловленной повышенной чувствительности тканей к кальцию - кальцергии, или кальцифилаксии (Селье Г., 1970).
Различают системный и ограниченный интерстициальный кальциноз. При интерстициальном системном (универсальном) кальцинозе известь выпадает в коже, подкожной клетчатке, по ходу сухожилий, фасций и
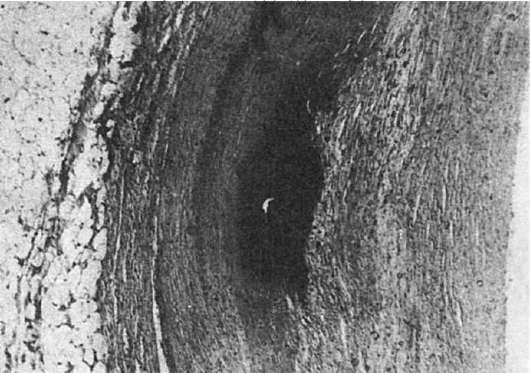 Рис. 43. Дистрофическое обызвествление стенки артерии. В толще атеросклеротической бляшки видны отложения извести
Рис. 43. Дистрофическое обызвествление стенки артерии. В толще атеросклеротической бляшки видны отложения извести
апоневрозов, в мышцах, нервах и сосудах; иногда локализация отложений извести бывает такой же, как при известковых метастазах. Интерстициальный ограниченный (местный) кальциноз, или известковая подагра, характеризуется отложением извести в виде пластинок в коже пальцев рук, реже ног.
Исход. Неблагоприятен: выпавшая известь обычно не рассасывается или рассасывается с трудом.
Значение. Имеют значение распространенность, локализация и характер обызвествлений. Так, отложение извести в стенке сосуда ведет к функциональным нарушениям и может явиться причиной ряда осложнений (например, тромбоза). Наряду с этим отложение извести в казеозном туберкулезном очаге свидетельствует о его заживлении, т.е. имеет репаративный характер.
Нарушения обмена меди
Медь - обязательный компонент цитоплазмы, где она участвует в ферментативных реакциях.
В тканях медь находится в очень небольших количествах, лишь в печени новорожденного ее относительно много. Для выявления меди наиболее точным является метод Окамото, основанный на применении рубеановодородной кислоты (дитиооксамид).
Нарушение обмена меди наиболее ярко проявляется при гепатоцеребральной дистрофии (гепатолентикулярная дегенерация), или болезни Вильсона-Коновалова. При этом наследственном заболевании медь депонируется в печени, мозге, почках, роговице (патогномонично кольцо Кайзера-Флейшера - зеленовато-бурое кольцо по периферии роговицы), поджелудочной железе, яичках и других органах. Развиваются цирроз печени и дистрофические симметричные изменения ткани головного мозга в области чечевичных ядер, хвостатого тела, бледного шара, коры. Содержание меди в плазме крови понижено, а в моче - повышено. Различают печеночную, лентикулярную и гепатолентикулярную формы болезни. Депонирование меди обусловлено пониженным образованием в печени церулоплазмина, который принадлежит к а2-глобулинам и способен связывать в крови медь. В результате она высвобождается из непрочных связей с белками плазмы и выпадает в ткани. Не исключено, что при болезни Вильсона-Коновалова повышено сродство некоторых тканевых белков к меди.
Нарушения обмена калия
Калий - важнейший элемент, принимающий участие в построении клеточной цитоплазмы.
Баланс калия обеспечивает нормальный белково-липидный обмен, нейроэндокринную регуляцию. Калий может быть выявлен с помощью метода МакКаллума.
Увеличение количества калия в крови (гиперкалиемия) и тканях отмечается при аддисоновой болезни и связано с поражением коры надпочеч-
ников, гормоны которых контролируют баланс электролитов. Дефицитом калия и нарушением его обмена объясняют возникновение периодического паралича - наследственного заболевания, проявляющегося приступами слабости и развитием двигательного паралича.
Нарушения обмена железа
Железо в основном содержится в гемоглобине, и морфологические проявления нарушений его обмена связаны с гемоглобиногенными пигментами (см. Нарушения обмена гемоглобиногенных пигментов).
Образование камней
Камни, или конкременты (от лат. concrementum - сросток), представляют собой очень плотные образования, свободно лежащие в полостных органах или выводных протоках желез.
Вид камней (форма, величина, цвет, структура на распиле) различен в зависимости от их локализации в той или иной полости, химического состава, механизма образования. Встречаются огромные камни и микролиты. Форма камня нередко повторяет полость, которую он заполняет: круглые или овальные камни находятся в мочевом и желчном пузырях, отростчатые - в лоханках и чашечках почек, цилиндрические - в протоках желез. Камни могут быть одиночными и множественными. В последнем случае они нередко имеют граненые притертые друг к другу поверхности (фасетированные камни). Поверхность камней бывает не только гладкой, но и шероховатой (оксалаты, например, напоминают тутовую ягоду), что травмирует слизистую оболочку, вызывает ее воспаление. Цвет камней различный, что определяется их разным химическим составом: белый (фосфаты), желтый (ураты), темно-коричневый или темно-зеленый (пигментные). В одних случаях на распиле камни имеют радиарное строение (кристаллоидные), в других - слоистое (коллоидные), в третьих - слоисторадиарное (коллоидно-кристаллоидные). Химический состав камней также различен. Желчные камни могут быть холестериновыми, пигментными, известковыми или холестериново-пигментно-известковыми (сложные, или комбинированные, камни). Мочевые камни могут состоять из мочевой кислоты и ее солей (ураты), фосфата кальция (фосфаты), оксалата кальция (оксалаты), цистина и ксантина. Бронхиальные камни состоят обычно из инкрустированной известью слизи.
Наиболее часто камни образуются в желчных и мочевых путях, являясь причиной развития желчнокаменной и мочекаменной болезней. Они встречаются также в других полостях и протоках: в выводных протоках поджелудочной железы и слюнных желез, в бронхах и бронхоэктазах (бронхиальные камни), в криптах миндалин. Особым видом камней являются так называемые венные камни (флеболиты), представляющие собой отделившиеся от стенки петрифицированные тромбы, и кишечные камни (копролиты), возникающие при инкрустации уплотнившегося содержимого кишечника.
Механизм развития. Патогенез камнеобразования сложен и определяется как общими, так и местными факторами. К общим факторам, которые имеют основное значение для образования камней, следует отнести нарушения обмена веществ приобретенного или наследственного характера. Особое значение имеют нарушения обмена жиров (холестерин), нуклеопротеидов, ряда углеводов, минералов. Хорошо известна, например, связь желчнокаменной болезни с общим ожирением и атеросклерозом, мочекаменной болезни - с подагрой, оксалурией и т.д. Среди местных факторов велико значение нарушений секреции, застоя секрета и воспалительных процессов в органах, где образуются камни. Нарушения секреции, как и застой секрета, ведут к увеличению концентрации веществ, из которых строятся камни, и осаждению их из раствора, чему способствует усиление реабсорбции и сгущение секрета. При воспалении в секрете появляются белковые вещества, что создает органическую (коллоидную) матрицу, в которую откладываются соли и на которой строится камень. Впоследствии камень и воспаление нередко становятся дополняющими друг друга факторами, определяющими прогрессирование камнеобразования.
Непосредственный механизм образования камня складывается из двух процессов: образования органической матрицы и кристаллизации солей, причем каждый из этих процессов в определенных ситуациях может быть первичным.
Значение и последствия образования камней. Они могут быть очень серьезными. В результате давления камней на ткань может возникнуть ее омертвение (почечные лоханки, мочеточники, желчный пузырь и желчные протоки, червеобразный отросток), что приводит к образованию пролежней, перфорации, спаек, свищей. Камни часто бывают причиной воспаления полостных органов (пиелоцистит, холецистит) и протоков (холангит, холангиолит). Нарушая отделение секрета, они ведут к тяжелым осложнениям общего (например, желтуха при закупорке общего желчного протока) или местного (например, гидронефроз при обтурации мочеточника) характера.