Патологическая анатомия : учебник / А. И. Струков, В. В. Серов. - 5-е изд., стер. - М.: Литтерра, 2010. - 848 с. : ил.
|
|
|
|
ОБЩАЯ ПАТОЛОГИЧЕСКАЯ АНАТОМИЯ ПОВРЕЖДЕНИЕ
В патологии под повреждением, или альтерацией (от лат. alteratio - изменение), понимают изменения структуры клеток, межклеточного вещества, тканей и органов, которые сопровождаются нарушением их жизнедеятельности. Альтеративные изменения в органах и тканях как филогенетически наиболее древний вид реактивных процессов встречаются на самых ранних этапах развития человеческого зародыша.
Повреждение способны вызывать самые разнообразные причины. Они могут действовать на клеточные и тканевые структуры непосредственно или опосредовано (через гуморальные и рефлекторные влияния), причем характер и степень повреждения зависят от силы и природы патогенного фактора, структурно-функциональных особенностей органа или ткани, а также от реактивности организма. В одних случаях возникают поверхностные и обратимые изменения, касающиеся обычно лишь ультраструктур, в других - глубокие и необратимые, которые могут завершиться гибелью не только клеток и тканей, но и целых органов. Повреждение имеет различное морфологическое выражение на клеточном и тканевом уровнях. На клеточном уровне оно представлено разнообразными ультраструктурными изменениями клетки, что составляет содержание большого раздела общей патологии - патологии клетки. На тканевом уровне повреждение представлено двумя общепатологическими процессами - дистрофией и некрозом, которые нередко являются последовательными стадиями альтерации.
Патология клетки
Клетка - элементарная живая система, обладающая способностью к обмену с окружающей средой. Строение клеток организма человека обеспечивает выполнение ими специализированной функции и «сохранение себя», т.е. поддержание клеточного пула. Органоиды клетки, обладая определенными морфологическими особенностями, обеспечивают основные проявления жизнедеятельности клетки (рис. 1). С ними связаны дыхание и энергетические запасы (митохондрии), синтез белков (рибосомы, гранулярная цитоплазматическая сеть), накопление и транспорт липидов и гликогена, детоксикационная функция (гладкая цитоплазматическая сеть), синтез продуктов и их секреция (пластинчатый комплекс), внутриклеточное пищеварение и защитная функция (лизосомы). Деятельность ультраструктур клетки строго координирована, причем координация в
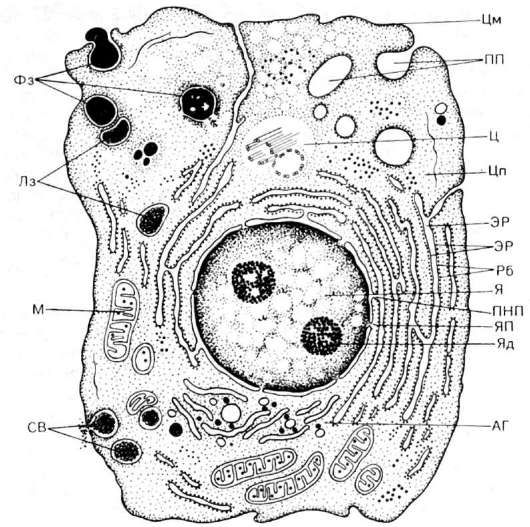 Рис. 1. Строение клетки (схема):
Рис. 1. Строение клетки (схема):
Я - ядро, ЯП - ядерные поры, Яд - ядрышко, ПНП - перинуклеарное пространство, Цп - цитоплазма (гиалоплазма), Цм - оболочка клетки (цитомембрана), ЭР - эндоплазматический ретикулум (эндоплазматическая сеть), Рб - рибосомы, М - митохондрии, АГ - пластинчатый комплекс (комплекс Гольджи), Лз - лизосомы, Ц - центросома, СВ - секреторные вакуоли, ПП - пиноцитозные пузырьки, Фз - стадии фагоцитоза
выработке специфического продукта клеткой подчинена закону «внутриклеточного конвейера». По принципу ауторегуляции он осуществляет взаимосвязь между структурными компонентами клетки и протекающими в ней процессами обмена.
Функции органоидов не строго детерминированы, так как они могут участвовать в различных внутриклеточных процессах. Более специализированы метаплазматические образования клетки, выполняющие частные функции: тонофибриллы, выполняющие опорную функцию клетки; миофибриллы, осуществляющие сокращение клетки и способствующие ее движению; микроворсинки, щеточная каемка, участвующие в процессах всасывания; десмосомы, обеспечивающие клеточные контакты, и т.д. Однако ни одна функция клетки не является результатом деятельности одного органоида или одного метаплазматического образования. Каждое функциональное проявление клетки - это результат совместной работы
всех взаимосвязанных компонентов. Понятно поэтому, что структурные изменения клетки, отражающие нарушения ее функции, не могут быть поняты без учета возможных изменений каждой из ее двух основных частей - ядра и цитоплазмы, ее органелл, метаплазматических образований и включений. От нарушений элементарных структур клетки и их функций к патологии клетки как элементарной саморегулирующейся живой системе и к патологии клеточных коопераций, объединенных конечной функцией, - таков путь познания патологии клетки - структурной основы патологии человека.
Поэтому патология клетки - понятие неоднозначное. Во-первых, это патология специализированных ультраструктур клетки, она представлена не только достаточно стереотипными изменениями той или иной ультраструктуры в ответ на различные воздействия, но и настолько специфичными изменениями ультраструктур, что можно говорить о хромосомных болезнях и «болезнях» рецепторов, лизосомных, митохондриальных, пероксисомных и других «болезнях» клетки. Во-вторых, патология клетки - это изменения ее компонентов и ультраструктур в причинно-следственных связях. При этом речь идет о выявлении общих закономерностей повреждения клетки и ее реакции на повреждение. Сюда могут быть отнесены: рецепция патогенной информации клеткой и реакция на повреждение, нарушения проницаемости клеточных мембран и циркуляции внутриклеточной жидкости; нарушения метаболизма клетки, смерть клетки (некроз), клеточная дисплазия и метаплазия, гипертрофия и атрофия, патология движения клетки, ее ядра и генетического аппарата и др.
Патология клеточного ядра
Морфологически она проявляется в изменении структуры, размеров, формы и количества ядер и ядрышек, в появлении разнообразных ядерных включений и изменений ядерной оболочки. Особую форму ядерной патологии представляет патология митоза; с патологией хромосом ядра связано развитие хромосомных синдромов и хромосомных болезней.
Структура и размеры ядер
Структура и размеры ядра (речь идет об интерфазном, интермитозном, ядре) зависят в первую очередь от плоидности, в частности от содержания в ядре ДНК, и от функционального состояния ядра. Тетраплоидные ядра имеют диаметр больше, чем диплоидные, октоплоидные - больше, чем тетраплоидные.
Большая часть клеток содержит диплоидные ядра. В пролиферирующих клетках в период синтеза ДНК (S-фаза) содержание ДНК в ядре удваивается, в постмитотический период, напротив, снижается. Если после синтеза ДНК в диплоидной клетке не происходит нормального митоза, то появляются тетраплоидные ядра. Возникает полиплоидия - кратное увеличение числа наборов хромосом в ядрах клеток, или состояние плоидности от тетраплоидии и выше.
Полиплоидные клетки выявляют различными способами: по размеру ядра, по увеличенному количеству ДНК в интерфазном ядре или по увеличению числа хромосом в митотической клетке. Они встречаются в нормально функционирующих тканях человека. Увеличение числа полиплоидных ядер во многих органах отмечается в старости. Особенно ярко полиплоидия представлена при репаративной регенерации (печень), компенсаторной (регенерационной) гипертрофии (миокард), при опухолевом росте.
Другой вид изменений структуры и размеров ядра клетки встречается при анеуплоидии, под которой понимают изменения в виде неполного набора хромосом. Анеуплоидия связана с хромосомными мутациями. Ее проявления (гипертетраплоидные, псевдоплоидные, «приблизительно» диплоидные или триплоидные ядра) часто обнаруживаются в злокачественных опухолях.
Размеры ядер и ядерных структур независимо от плоидии определяются в значительной мере функциональным состоянием клетки. В связи с этим следует помнить, что процессы, постоянно совершающиеся в интерфазном ядре, разнонаправленны: во-первых, это репликация генетического материала в S-периоде («полуконсервативный» синтез ДНК); во-вторых, образование РНК в процессе транскрипции, транспортировка РНК из ядра в цитоплазму через ядерные поры для осуществления специфической функции клетки и для репликации ДНК.
Функциональное состояние ядра находит отражение в характере и распределении его хроматина. В наружных отделах диплоидных ядер нормальных тканей находят конденсированный (компактный) хроматин - гетерохроматин, в остальных ее отделах - неконденсированный (рыхлый) хроматин - эухроматин. Гетеро- и эухроматин отражают различные состояния активности ядра; первый из них считается «малоактивным» или «неактивным», второй - «достаточно активным». Поскольку ядро может переходить из состояния относительно функционального покоя в состояние высокой функциональной активности и обратно, морфологическая картина распределения хроматина, представленная гетеро- и эухроматином, не может считаться статичной. Возможна «гетерохроматинизация» или «эухроматинизация» ядер (рис. 2), механизмы которой изучены недостаточно. Неоднозначна и трактовка характера и распределения хроматина в ядре.
Например, маргинация хроматина, т.е. расположение его под ядерной оболочкой, трактуется и как признак активности ядра, и как проявление его повреждения. Однако конденсация эухроматиновых структур (гиперхроматоз стенки ядра), отражающая инактивацию активных участков транскрипции, рассматривается как патологическое явление, как предвестник гибели клетки. К патологическим изменениям ядра относят также его дисфункциональное (токсическое) набухание, встречающееся при различных повреждениях клетки. При этом происходит изменение коллоидно-осмотического состояния ядра и цитоплазмы вследствие торможения транспорта веществ через оболочку клетки.
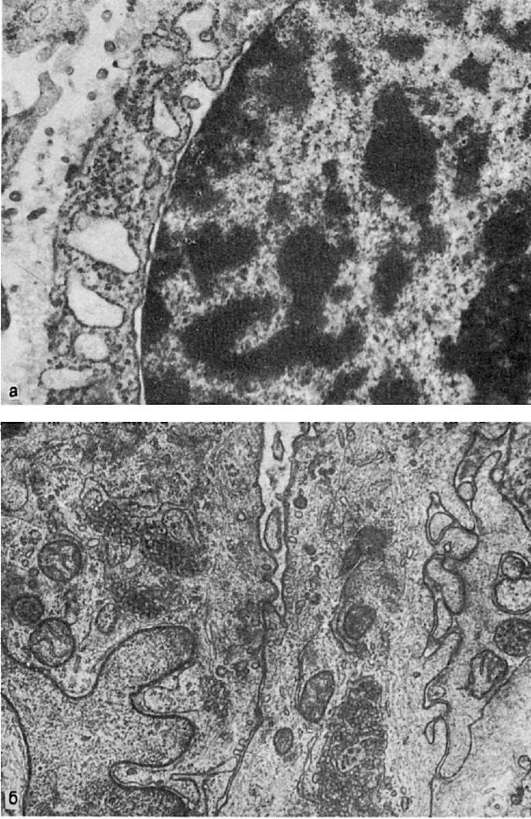 Рис. 2. Гетеро- и эухроматизация ядер:
Рис. 2. Гетеро- и эухроматизация ядер:
а - гетерохроматин ядра опухолей клетки. х25 000; б - эухроматизация хроматина ядра эндотелиоцита. Многочисленные инвагинаты ядерной оболочки; в цитоплазме - тубулярные включения и скопления промежуточных филаментов. х30 000
Форма ядер и их количество
Изменения формы ядра - существенный диагностический признак: деформация ядер цитоплазматическими включениями при дистрофических процессах, полиморфизм ядер при воспалении (гранулематоз) и опухолевом росте (клеточный атипизм).
Форма ядра может меняться также в связи с образованием множественных выпячиваний ядра в цитоплазму (рис. 3), которое обусловлено увеличением ядерной поверхности и свидетельствует о синтетической активности ядра в отношении нуклеиновых кислот и белка.
Изменения количества ядер в клетке могут быть представлены многоядерностью, появлением «спутника ядра» и безъядерностью. Многоядерность возможна при слияний клеток. Таковы, например, гигантские многоядерные клетки инородных тел и Пирогова-Лангханса, образующиеся при слиянии эпителиоидных клеток (см. рис. 72). Но возможно образование многоядерных клеток и при нарушениях митоза - деление ядра без последующего деления цитоплазмы, что наблюдается после облучения или введения цитостатиков, а также при злокачественном росте.
«Спутниками ядра», кариомерами (маленькими ядрами) называют мелкие подобные ядру образования с соответствующей структурой и собственной оболочкой, которые расположены в цитоплазме около неизмененного ядра. Причиной их образования считают хромосомные мутации. Таковы кариомеры в клетках злокачественной опухоли при наличии большого числа фигур патологических митозов.
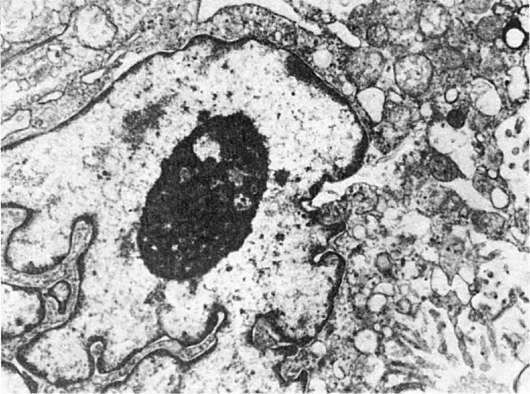 Рис. 3. Атипизм ядер клетки опухоли. Множественные выпячивания ядерной оболочки. х15 500
Рис. 3. Атипизм ядер клетки опухоли. Множественные выпячивания ядерной оболочки. х15 500
Безъядерность в отношении функциональной оценки клетки неоднозначна. Известны безъядерные клеточные структуры, которые являются вполне жизнеспособными (эритроциты, тромбоциты). При патологических состояниях можно наблюдать жизнеспособность частей цитоплазмы, отделенных от клетки. Но безъядерность может свидетельствовать и о гибели ядра, которая проявляется кариопикнозом, кариорексисом (рис. 4) и кариолизисом (см. Некроз).
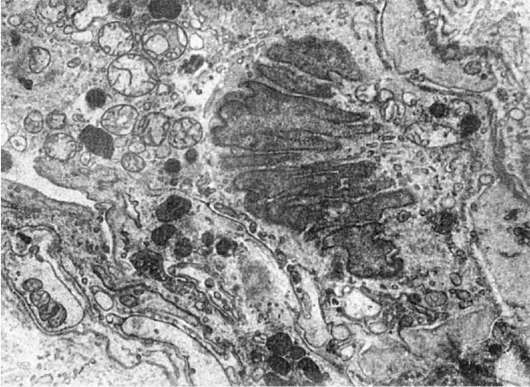 Рис. 4. Распад пикнотического ядра (кариорексис)
Рис. 4. Распад пикнотического ядра (кариорексис)
Структура и размеры ядрышек
Изменения ядрышек имеют существенное значение в морфофункциональной оценке состояния клетки, так как с ядрышками связаны процессы транскрипции и трансформации рибосомальной РНК (р-РНК). Размеры и структура ядрышек в большинстве случаев коррелируют с объемом клеточного белкового синтеза, выявляемого биохимическими методами. Размеры ядрышек зависят также от функции и типа клеток.
Увеличение размеров и количества ядрышек (рис. 5) свидетельствует о повышении их функциональной активности. Вновь образованная в ядрышке рибосомальная РНК транспортируется в цитоплазму и, вероятно, через поры внутренней ядерной мембраны. Интенсивный синтез белка в таких случаях подтверждается увеличением количества рибосом эндоплазматической сети.
Гипергранулированные ядрышки с преобладанием гранул над фибриллярной субстанцией могут отражать различное функциональное состояние как ядрышек, так и клетки. Наличие таких ядрышек с хорошо выраженной лакунарной системой и резкой базофилией цитоплазмы
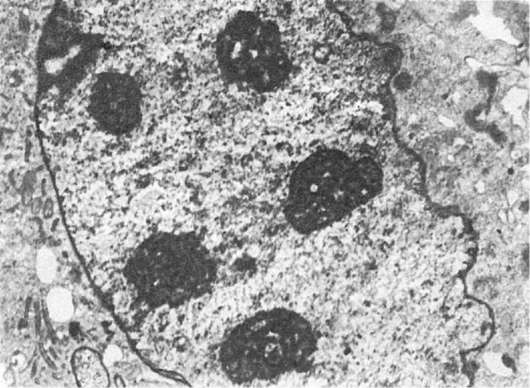 Рис. 5. Увеличение количества и размеров ядрышек. х12 500
Рис. 5. Увеличение количества и размеров ядрышек. х12 500
свидетельствует как о повышенном синтезе р-РНК, так и о трансмиссии. Такие «гиперфункциональные ядрышки» встречаются в молодых плазматических клетках, активных фибробластах, гепатоцитах, во многих опухолевых клетках. Те же гипергранулированные ядрышки со слабовыраженной базофилией цитоплазмы могут отражать нарушение трансмиссии (транспортировки гранул) при продолжающемся синтезе р-РНК. Они обнаруживаются в опухолевых клетках, отличающихся большим ядром и незначительной цитоплазматической базофилией.
Разрыхление (диссоциация) ядрышек, отражающее их гипогрануляцию, может быть следствием «извержения» р-РНК в цитоплазму или торможения ядрышковои транскрипции. Дезорганизация (сегрегация) ядрышек отражает, как правило, полное и быстрое прекращение ядрышковой транскрипции: ядро уменьшается в размерах, наблюдается выраженная конденсация ядрышкового хроматина, происходит разделение гранул и протеиновых нитей. Эти изменения встречаются при энергетическом дефиците клетки.
Ядерные включения
Ядерные включения делят на три группы: ядерные цитоплазматические, истинные ядерные и ядерные вирусобусловленные.
Ядерными цитоплазматическими включениями называют отграниченные оболочкой части цитоплазмы в ядре. Они могут содержать все составные части клетки (органеллы, пигмент, гликоген, капли жира и т.д.). Их появление в большинстве случаев связано с нарушением митотического деления.
Истинными ядерными включениями считают те, которые расположены внутри ядра (кариоплазмы) и соответствуют веществам, встречающимся
в цитоплазме - белок, гликоген (рис. 6, а), липиды и т.д. В большинстве случаев эти вещества проникают из цитоплазмы в ядро через неповрежденные или поврежденные поры ядерной оболочки или через разрушенную ядерную оболочку. Возможно также проникновение этих веществ в ядро при митозе. Таковы, например, включения гликогена в ядрах печени при сахарном диабете («ядерный гликоген», «дырчатые, пустые, ядра»).
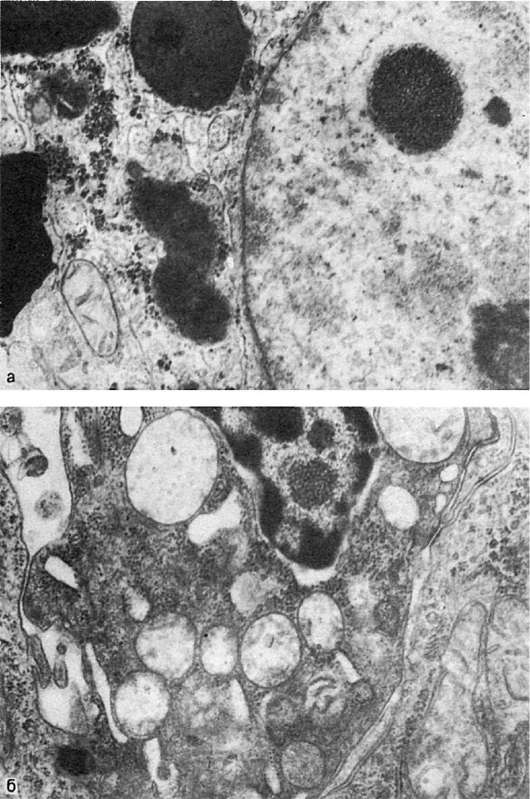 Рис. 6. Ядерные включения:
Рис. 6. Ядерные включения:
а - включения гликогена в ядре гепатоцита. х22 500; б - включения вируса в ядре опухолевой клетки
Вирусобусловленные ядерные включения (так называемые тельца ядерных включений) неоднозначны. Во-первых, это ядерные включения в кариоплазме кристаллической решетки вируса (рис. 6, б), во-вторых, включения белковых частиц, возникающих при внутриядерном размножении вируса; в-третьих, ядерные включения как проявление реакции на поражение вирусом цитоплазмы («реактивные включения»).
Ядерная оболочка
Ядерная оболочка выполняет ряд функций, нарушения которых могут служить основой для развития патологии клетки.
О роли ядерной оболочки в поддержании формы и размера ядра свидетельствует образование внутриядерных трубчатых систем, отходящих от внутренней ядерной мембраны, включений в перинуклеарной зоне - гипертрофия миокарда, легочный фиброз, системный васкулит, саркоидоз, опухоли печени, дерматомиозит (рис. 7).
О ядерной оболочке как месте прикрепления ДНК для облегчения репликации и транскрипции свидетельствует тот факт, что в ядерной оболочке имеются структуры, модулированные хроматином и в свою очередь ответственные за ориентацию и структуру хроматина. Показано, что функциональная активность ДНК связана с ее распределением при делении клетки и со степенью конденсации в интерфазе, причем повреждение оболочки может вызывать изменения таких участков распределения и быть причиной патологических изменений клетки.
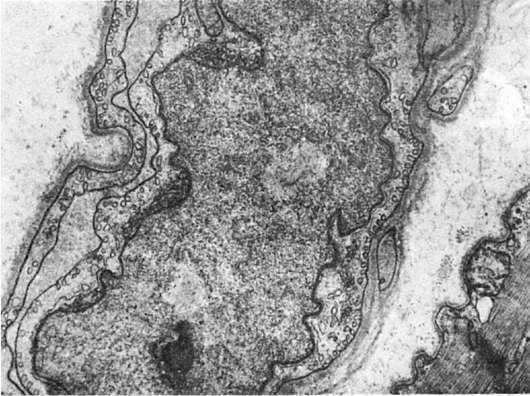 Рис. 7. Микротубулярные включения в перинуклеарной зоне эндотелиоцита при дерматомиозите. х15 500
Рис. 7. Микротубулярные включения в перинуклеарной зоне эндотелиоцита при дерматомиозите. х15 500
В пользу функции ядерной оболочки как физического барьера и модулятора нуклеоцитоплазматического обмена говорит установленная корреляция между изменениями структуры ядерной оболочки, модулем ее пор и выходом РНК в цитоплазму. Контроль ядерной оболочкой транспорта РНК в цитоплазму может оказывать существенное влияние на гомеостаз клетки при патологических состояниях. Участие ядерной оболочки в синтезе мембран не имеет достоверных доказательств, хотя и считают, что эта роль возможна, так как мембраны ядерной оболочки непосредственно переходят в эндоплазматическую сеть цитоплазмы. О возможном влиянии ферментов ядерной оболочки на функцию ядра свидетельствует наличие в ядерной оболочке различных ферментов детоксикации, а также веществ, обеспечивающих «гормональное управление» (аденилатциклаза, рецепторы инсулина и др.).
Патология митоза
В жизненном цикле клетки митоз занимает особое место. С его помощью осуществляется репродукция клеток, а значит, и передача их наследственных свойств. Подготовка клеток к митозу складывается из ряда последовательных процессов: репродукции ДНК, удвоения массы клетки, синтеза белковых компонентов хромосом и митотического аппарата, удвоения клеточного центра, накопления энергии для цитотомии. В процессе митотического деления, как известно, различают 4 основные фазы: профазу, метафазу, анафазу и телофазу.
При патологии митоза может страдать любая из этих фаз. Руководствуясь этим, создана классификация патологии митоза (Алов И.А., 1972), согласно которой выделяются следующие типы патологии митоза.
I. Повреждение хромосом: 1) задержка клеток в профазе; 2) нарушение спирализации и деспирализации хромосом; 3) фрагментация хромосом; 4) образование мостов между хромосомами в анафазе; 5) раннее разъединение сестринских хроматид; 6) повреждение кинетохора.
II. Повреждение митотического аппарата: 1) задержка развития митоза в метафазе; 2) рассредоточение хромосом в метафазе; 3) трехгрупповая метафаза; 4) полая метафаза; 5) многополюсные митозы; 6) асимметричные митозы; 7) моноцентрические митозы; 8) К-митозы.
III. Нарушение цитотомии: 1) преждевременная цитотомия; 2) задержка цитотомии; 3) отсутствие цитотомии.
Патологию митоза могут вызвать различные воздействия на клетку: ультрафиолетовое и ионизирующее излучение, высокая температура, химические вещества, в том числе канцерогены и митотические яды и др. Велико количество патологических митозов при малигнизации тканей (рис. 8).
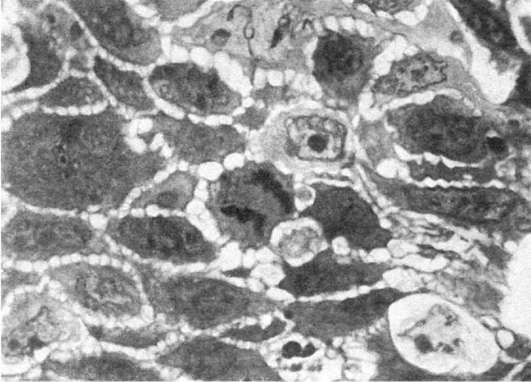 Рис. 8. Патология митоза. Полутонкий срез ткани опухоли. х1000
Рис. 8. Патология митоза. Полутонкий срез ткани опухоли. х1000
Хромосомные аберрации и хромосомные болезни
Хромосомные аберрации. Под хромосомными аберрациями понимают изменения структуры хромосом, вызванные их разрывами, с последующим перераспределением, утратой или удвоением генетического материала. Они отражают различные виды аномалий хромосом. У человека среди наиболее часто встречающихся хромосомных аберраций, проявляющихся развитием глубокой патологии, выделяют аномалии, касающиеся числа и структуры хромосом. Нарушения числа хромосом могут быть выражены отсутствием одной из пары гомологичных хромосом (моносомия) или появлением добавочной, третьей, хромосомы (трисомия). Общее количество хромосом в кариотипе в этих случаях отличается от модального числа и равняется 45 или 47. Полиплоидия и анеуплоидия имеют меньшее значение для развития хромосомных синдромов. К нарушениям структуры хромосом при общем нормальном их числе в кариотипе относят различные типы их «поломки»: транслокацию (обмен сегментами между двумя негомологичными хромосомами), делецию (выпадение части хромосомы), фрагментацию, кольцевые хромосомы и т.д.
Хромосомные аберрации, нарушая баланс наследственных факторов, являются причиной многообразных отклонений в строении и жизнедеятельности организма, проявляющихся в так называемых хромосомных болезнях.
Хромосомные болезни. Их делят на связанные с аномалиями соматических хромосом (аутосом) и с аномалиями половых хромосом (телец Барра). При этом учитывают характер хромосомной аномалии - нарушение числа отдельных хромосом, числа хромосомного набора или структуры
хромосом. Эти критерии позволяют выделять полные или мозаичные клинические формы хромосомных болезней.
Хромосомные болезни, обусловленные нарушениями числа отдельных хромосом (трисомиями и моносомиями), могут касаться как аутосом, так и половых хромосом.
Моносомии аутосом (любые хромосомы, кроме Х- и Y-хромосом) несовместимы с жизнью. Трисомии аутосом достаточно распространены в патологии человека. Наиболее часто они представлены синдромами Патау (13-я пара хромосом) и Эдвардса (18-я пара), а также болезнью Дауна (21-я пара). Хромосомные синдромы при трисомиях других пар аутосом встречаются значительно реже. Моносомия половой Х-хромосомы (генотип ХО) лежит в основе синдрома Шерешевского-Тернера, трисомия половых хромосом (генотип XXY) - в основе синдрома Клейнфелтера. Нарушения числа хромосом в виде тетраили триплоидии могут быть представлены как полными, так и мозаичными формами хромосомных болезней.
Нарушения структуры хромосом дают самую большую группу хромосомных синдромов (более 700 типов), которые, однако, могут быть связаны не только с хромосомными аномалиями, но и с другими этиологическими факторами.
Для всех форм хромосомных болезней характерна множественность проявлений в виде врожденных пороков развития, причем их формирование начинается на стадии гистогенеза и продолжается в органогенезе, что объясняет сходство клинических проявлений при различных формах хромосомных болезней.
Патология цитоплазмы
Изменения мембран и патология клетки
Клеточные мембраны, как известно, состоят из бислоя фосфолипидов, по обе стороны которого располагаются разнообразные мембранные белки. На внешней поверхности мембраны белковые молекулы несут полисахаридные компоненты (гликокаликс), которые содержат многочисленные поверхностные клеточные антигены. Они играют важную роль в клеточном узнавании, формировании клеточных стыков.
Изменения клеточных мембран. Среди них различают следующие (Авцын А.П., Шахламов В.А., 1979): чрезмерное везикулообразование («минусмембрана» - рис. 9); увеличение поверхности плазмолеммы клеток мембранами микропиноцитозных пузырьков («плюс-мембрана»); усиленный микроклазматоз и клазматоз («минус-мембрана» - см. рис. 9); образование цитоплазматических отростков из плазмолеммы клетки; образование пузырей на поверхности клетки; утолщение слоев мембраны; образование микропор; образование миелиноподобных структур из плазмолеммы и мембран органелл; слияние разнородных клеточных мембран; локальные разрушения мембран - «бреши» в плазмолемме; «штопка» локально разрушенной плазмолеммы мембранами микропиноцитозных везикул.
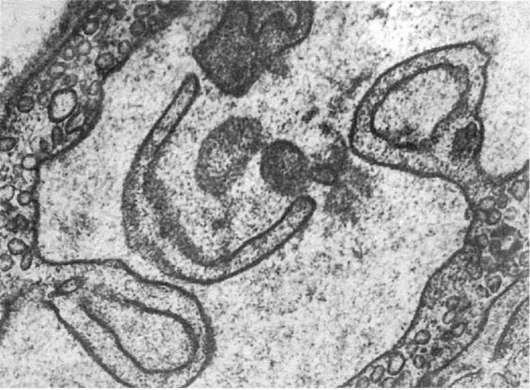 Рис. 9. Изменения мембран эндотелиоцитов. Усиленное везикулообразование и клазматоз. х25 500
Рис. 9. Изменения мембран эндотелиоцитов. Усиленное везикулообразование и клазматоз. х25 500
К патологии мембран клетки могут вести нарушения мембранного транспорта, изменения проницаемости мембран, изменения коммуникации клеток и их «узнавания», изменения подвижности мембран и формы клеток, нарушения синтеза и обмена мембран.
Нарушения мембранного транспорта. Процесс мембранного транспорта предполагает перенос ионов и других субстратов против градиента концентрации. Транспорт может быть активным, тогда он требует АТФ и «подвижности» транспортных белков в мембране, или пассивным посредством различных диффузионных и обменных процессов. Активный транспорт - это также функция эпителиальных барьеров. Нарушения мембранного транспорта, ведущие к патологии клетки, хорошо прослежены при ишемии, которая приводит к первичным изменениям митохондрий. В митохондриях резко падает эффективность окислительного фосфорилирования, они набухают, вначале увеличивается проницаемость их внутренней мембраны, в дальнейшем повреждение становится тотальным и необратимым (рис. 10).
Ишемическое повреждение митохондрий приводит к полому натрий-калиевого АТФ-насоса, постепенному накапливанию в клетке натрия и потере ею калия. Нарушение натрий-калиевого обмена ведет к вытеснению кальция из митохондрий. В результате в цитоплазме повышается уровень ионизированного кальция и увеличивается связывание его с кальмодулином. С повышением содержания кальцийкальмодулиновых комплексов связан ряд изменений клетки: рас-
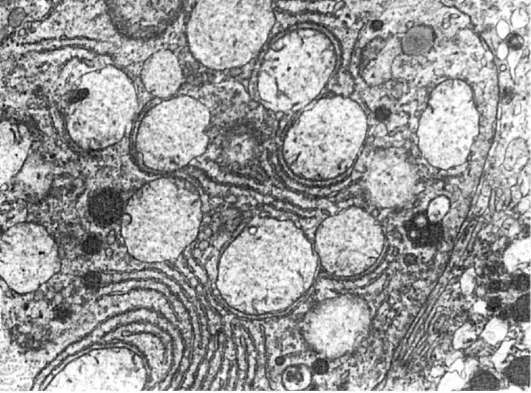 Рис. 10. Вакуолизация митохондрий в одном гепатоците (слева) и конденсация их в другом (справа). х16 000
Рис. 10. Вакуолизация митохондрий в одном гепатоците (слева) и конденсация их в другом (справа). х16 000
хождение клеточных стыков, поглощение кальция митохондриями, изменение микротрубочек и микрофиламентов, активация фосфолипаз. Эндоплазматическая сеть накапливает воду и ионы, следствием чего является расширение ее канальцев и цистерн, развитие гидропической дистрофии. Усиление гликолиза сопровождается истощением гликогена, накоплением лактата и снижением клеточного рН. С этими изменениями связано нарушение структуры хроматина и уменьшение синтеза РНК. Необратимые ишемические повреждения клетки связаны с гидролизом мембран, особенно мембранных липидов, под действием фосфолипаз. Возникают и нарушения лизосомальных мембран с высвобождением гидролаз.
Изменения проницаемости мембран. Контроль мембранной проницаемости предполагает поддержание структуры как фосфолипидного бислоя мембраны с необходимым обменом и ресинтезом, так и соответствующих белковых каналов. Важная роль в осуществлении этого контроля принадлежит гликокаликсу и взаимодействию мембранных белков с цитоскелетом, а также гормонам, взаимодействующим с мембранными рецепторами. Изменения проницаемости могут быть тяжелыми (необратимыми) или поверхностными. Наиболее изученной моделью изменения мембранной проницаемости является повреждение тяжелыми металлами (ртуть, уран). Тяжелые металлы, взаимодействуя с сульфгидрильными группами мембранных белков, изменяют их конформацию и резко увеличивают проницаемость мембраны для натрия,
калия, хлора, кальция и магния, что приводит к быстрому набуханию клеток, распаду их цитоскелета. Подобные изменения мембран отмечаются при повреждении их комплементом («болезни гиперчувствительности»). В мембранах образуются бреши, что снижает их сопротивление и резко увеличивает проницаемость.
Изменения коммуникации клеток и их «узнавания». Коммуникабельность клеток и опознавание «своих» и «чужих» - необходимое свойство клеточного кооперирования. Клеточное «общение» и «узнавание» подразумевают прежде всего различия во внешних поверхностях плазматической мембраны и мембран внутриклеточных органелл. Особый интерес в этом отношении представляет гликокаликс мембраны с поверхностными антигенами - маркерами определенного типа клеток.
Изменения клеточного «общения» и «узнавания» встречаются при тех патологических процессах (воспаление, регенерация, опухолевый рост), при которых поверхностные антигены могут изменяться, причем различия могут касаться как типа антигена, так и его «доступности» со стороны внеклеточного пространства. Показано, что при исчезновении характерных для данного типа клеток антигенов могут появляться «эмбриональные» и аномальные (например, карциноэмбриональный) антигены; изменения гликолипидов мембраны делают ее более доступной воздействию антител.
Коммуникабельность клеток определяется также состоянием клеточных стыков, которые могут повреждаться при различных патологических процессах и болезнях. В раковых клетках, например, найдена корреляция между изменениями клеточных стыков и нарушением межклеточных связей; в опухолях обнаружены аномальные клеточные соединения.
Изменения подвижности мембран и формы клеток. Различают два типа изменений, связанных с нарушением подвижности мембран: выпячивание мембраны наружу - экзотропия и внутрь цитоплазмы - эзотропия. При экзотропии мембрана, выпячивающаяся во внеклеточное пространство, образует окруженную мембраной цитоплазматическую структуру. При эзотропии появляется окруженная мембраной полость. Изменения формы клеток связаны не только с экзо- и эзотропией, но и с упрощением клеточной поверхности (потеря малых отростков подоцитов при нефротическом синдроме).
Нарушения синтеза и обмена мембран. Возможно усиление синтеза мембран (при воздействии ряда химических веществ на клетку) или его ослабление (снижение синтеза мембран щеточной каемки энтероцитов при угнетении мембранных ферментов). В равной мере возможно усиление обмена мембран (при стимуляции аутофагоцитоза) или его ослабление (при лизосомных болезнях).
Эндоплазматическая сеть
Однозначные изменения гранулярной и агранулярной эндоплазматической сети могут отражать нарушения различных функций цитоплазмы и клетки.
Изменения гранулярной эндоплазматической сети и рибосом
Функции гранулярной эндоплазматической сети и рибосом сопряжены достаточно жестко, поэтому морфологические проявления их нарушений касаются, как правило, обеих органелл.
Изменения гранулярной эндоплазматической сети и рибосом могут быть представлены гиперплазией и атрофией, упрощением структуры, дезагрегацией (диссоциацией) рибосом и полисом, образованием аномальных рибосомально-пластинчатых комплексов.
Гиперплазия гранулярной эндоплазматической сети и рибосом, т.е. увеличение их количества, светооптически проявляется повышенной базофилией цитоплазмы, которая отражает объемную плотность рибосом и является показателем интенсивности белкового синтеза в клетке. Электронно-микроскопически в таких случаях можно судить о сопряжении синтеза и экскреции белка или отсутствии такого сопряжения. В интенсивно секретирующих и экскретирующих белок клетках (например, в активных фибробластах) цистерны гранулярной эндоплазматической сети расширены и содержат мало электронно-плотного материала: отмечается гиперплазия как связанных с мембранами, так и свободных рибосом, образующих полисомы; пластинчатый комплекс (комплекс Гольджи), участвующий в экскреции синтезируемого белка, хорошо развит (рис. 11). В интенсивно секретирующих белок клетках с нарушенной его экскрецией в гиперплазированных расширенных цистернах эндоплазматической сети с обилием рибосом и полисом накапливается хлопьевидный электронноплотный материал (рис. 12), иногда происходит его кристаллизация; комплекс Гольджи в таких случаях развит плохо.
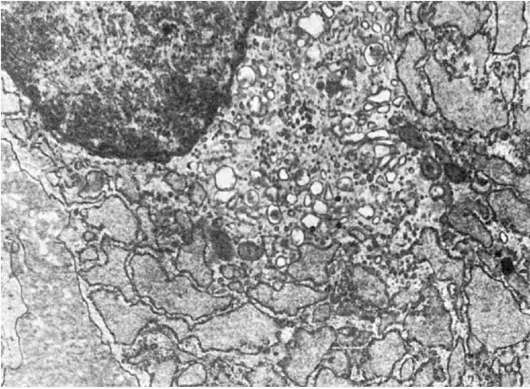 Рис. 11. Гиперплазия
гранулярной эндоплазматической сети, расширение ее цистерн, гиперплазия
пластинчатого комплекса (плазматическая клетка). х13 500
Рис. 11. Гиперплазия
гранулярной эндоплазматической сети, расширение ее цистерн, гиперплазия
пластинчатого комплекса (плазматическая клетка). х13 500
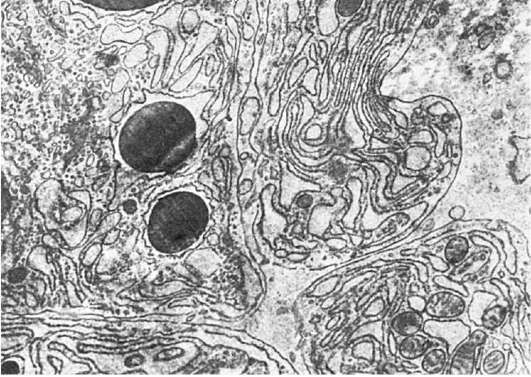 Рис. 12. Конденсированный белковый секрет в эндоплазматической сети (плазматическая клетка). х13 500
Рис. 12. Конденсированный белковый секрет в эндоплазматической сети (плазматическая клетка). х13 500
Атрофия гранулярной эндоплазматической сети, т.е. уменьшение ее размеров, светооптически представлена снижением или исчезновением базофилии цитоплазмы, а электронно-микроскопически - уменьшением размеров канальцев и объема сети, количества и размеров рибосом (рис. 13). Она отражает снижение белково-синтетической функции клетки (белковый дефицит при голодании, болезнях печени; старение).
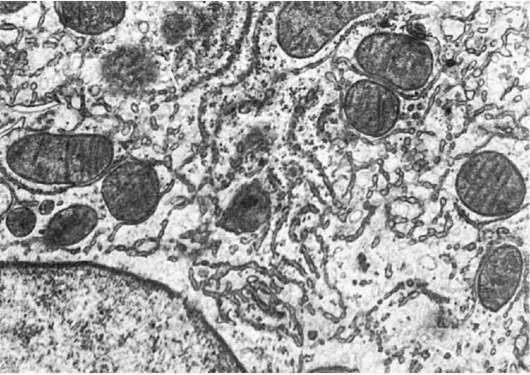 Рис. 13. Атрофия гранулярной и гиперплазия агранулярной эндоплазматической сети гепатоцитов. х16 500
Рис. 13. Атрофия гранулярной и гиперплазия агранулярной эндоплазматической сети гепатоцитов. х16 500
Упрощение структуры гранулярной эндоплазматической сети клеток свидетельствует о недостаточной их дифференцировке, нередко встречается в клетках злокачественных опухолей.
Дезагрегация (диссоциация) рибосом и полисом, выражающаяся в нарушениях рибосомально-мембранных взаимоотношений, «неорганизованной» ассоциации рибосом в полисомы, может быть выражением структурного упрощения эндоплазматической сети недифференцированной и опухолевой клетки. Но те же изменения наблюдаются и в дифференцированных клетках при кислородном голодании и дефиците белка в организме.
Образование аномальных рибосомально-пластинчатых комплексов является выражением субклеточной атипии и встречается при опухолях системы крови - гемобластозах (см. Опухоли системы крови).
Изменения агранулярной эндоплазматической сети
Агранулярная цитоплазматическая сеть может претерпевать ряд морфологических изменений, отражающих нарушения разнообразных функций этого органоида. Среди них главные - гиперплазия и атрофия.
Гиперплазия мембран эндоплазматической сети с расширением ее канальцев и систем (см. рис. 13) может отражать различные по интенсивности и разные по своей сути процессы. Во-первых, это усиление метаболической активности ряда веществ (белков, липидов, лекарственных средств). Во-вторых, это нарушенный внутриклеточный транспорт метаболизируемых продуктов, которые накапливаются в расширенных канальцах и цистернах сети, при этом пластинчатый комплекс редуцирован. В-третьих, это дефицит ферментов (ферментопатия), ведущий к недостаточности специфических функций этого органоида. При нарушении внутриклеточного транспорта метаболизируемых продуктов и ферментопатии в расширенных цистернах эндоплазматической сети накапливаются белки и вода (гидропическая дистрофия) или липиды и липопротеиды (жировая дистрофия).
Атрофия, а в дальнейшем и редукция гладкой эндоплазматической сети возникают при остром или хроническом воздействии на клетку различных ядов и токсических веществ (рис. 14), а также при белковом голодании.
Эндоплазматическая сеть и система оксигеназ со смешанной функцией
Ряд чужеродных веществ, подвергающихся метаболизму в эндоплазматической сети, способен взаимодействовать с макромолекулами клетки, что ведет к ее повреждению. Катализаторами таких метаболических процессов в эндоплазматической сети является группа родственных NADH- и 02-зависимых ферментов. Это - монооксигеназы (гидроксилазы) или оксигеназы со смешанной функцией (ОСФ); терминальной оксигеназой этой системы является цитохром-Р-450. Система ОСФ, связанная с цитохромом Р-450, найдена в эндоплазматической сети клеток многих органов (печень, легкие, кишечник, кора надпочечников, семенники, кожа). Эта система может, помимо гидроксилирования стероидов, утилизировать многие липофильные эндогенные (жирные кислоты) и экзогенные
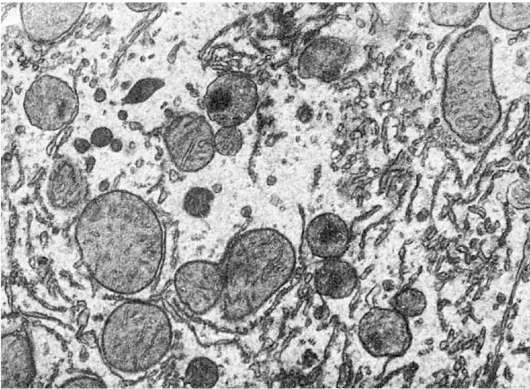 Рис. 14. Атрофия гладкой эндоплазматической сети гепатоцита. х18 000
Рис. 14. Атрофия гладкой эндоплазматической сети гепатоцита. х18 000
(лекарственные препараты, органические растворители, карциногены) вещества. Метаболизм чужеродных липофильных веществ требует сложного взаимодействия ряда ферментативных процессов, в которых система ОСФ - цитохром Р-450 занимает центральное место. Такой метаболизм не всегда ведет к инактивации метаболических веществ. Возможно образование реакционноспособных оксигенированных продуктов, которые могут взаимодействовать с нуклеиновыми кислотами и белками клетки, что ведет к ее повреждению. Основной механизм такого повреждения - это генерация супероксидных радикалов кислорода и перекиси водорода, индуцирующих переокисление липидов.
Пластинчатый комплекс (комплекс Гольджи), секреторные гранулы и вакуоли Синтетическая деятельность пластинчатого комплекса, тесно связанная с эндоплазматической сетью, завершается образованием секреторных гранул и вакуолей. Поэтому морфология нарушенной деятельности пластинчатого комплекса отражает и нарушения секреции, т.е. нарушения продукции клеточных включений - гранул и вакуолей. Можно говорить о двух основных морфологических проявлениях нарушенной деятельности пластинчатого комплекса и секретообразования: гипертрофии и атрофии.
Гипертрофия пластинчатого комплекса, т.е. его увеличение за счет гиперплазии его мембран, увеличения количества секреторных гранул, везикул и вакуолей, является проявлением повышенного синтеза и секреции белков, гликолипидов или полисахаридов (рис. 15). При этом увеличивается количество секреторных гранул и везикул в цитоплазме и за пределами пластинчатого комплекса. Гипертрофия пластинчатого
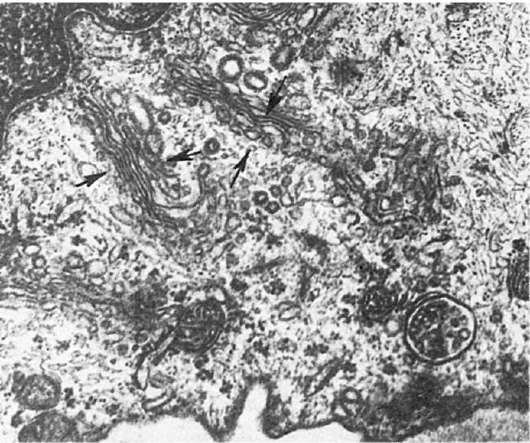 Рис. 15. Гиперплазия мембран пластинчатого комплекса в подоците. х20 500
Рис. 15. Гиперплазия мембран пластинчатого комплекса в подоците. х20 500
комплекса в таких случаях сочетается с гиперплазией эндоплазматической сети. В тех случаях, когда синтез тех или иных веществ опережает их секрецию и выведение, эти вещества избирательно накапливаются в гипертрофированном пластинчатом комплексе и могут повреждать его. Таково, например, скопление желчи в пластинчатом комплексе гепатоцитов при холестазе.
Атрофия пластинчатого комплекса, т.е. уменьшение его размеров с редукцией компонентов, потерей секреторных гранул и вакуолей, свидетельствует о снижении его функциональной активности. Одной из причин такого снижения может быть недостаточность белковых запасов организма (белковое голодание); при этом эндоплазматическая сеть также атрофична, в цитоплазме мало секреторных гранул. Другая причина снижения функциональной активности пластинчатого комплекса - это нарушение взаимодействия пластинчатого комплекса с эндоплазматической сетью, т.е. «повреждение» клеточного конвейера. В этих случаях эндоплазматическая сеть гиперплазирована, функционально активна, а цитоплазма заполнена множеством секреторных гранул и вакуолей.
Митохондрии
Митохондрии являются наиболее лабильными внутриклеточными структурами. Они первыми подвергаются изменениям при гиперфункции клетки и различных ее повреждениях. Изменения митохондрий, возникающие при многих патологических процессах и болезнях, достаточно стереотипны, хотя ряд патологических состояний и болезней имеет специфические признаки повреждения митохондрий.
Изменения структуры, размеров, формы и числа митохондрий
Среди изменений структуры митохондрий наибольшее значение придается их конденсации и набуханию, а также появлению митохондриальных включений. Конденсация и набухание митохондрий (см. рис. 10) могут отражать функциональное напряжение клетки, но чаще нарастающее кислородное голодание. Эти изменения нередко обратимы, однако, прогрессируя, ведут к тяжелой деструкции митохондрий и гибели клетки. Тогда к набуханию митохондрий присоединяются уплотнение их внутреннего пространства, деформация крист и потеря митохондриальных гранул, гомогенизация матрикса и появление в нем хлопьевидного материала, очагов обызвествления; в финале возникают разрывы наружной мембраны митохондрий.
Митохондриальные включения представлены хлопьевидным электронноплотным материалом (липидные вещества), очагами обызвествления (гидрооксиапатитоподобные кристаллы - рис. 16), миелиновыми фигурами, филаментоподобными и пластинчатыми структурами, белковыми кристаллами. Включения в митохондрии, как правило, встречаются при патологических состояниях, отражая неспецифическую реакцию митохондрий на повреждение клетки.
Размеры митохондрий колеблются в широких пределах - от гигантских до резко редуцированных форм. Гигантские митохондрии, которые образуются за счет гипертрофии или слияния митохондрий, встречаются только в патологических условиях (рис. 17). Такие митохондрии, нередко с кристаллическими включениями, как правило, обнаруживают, напри-
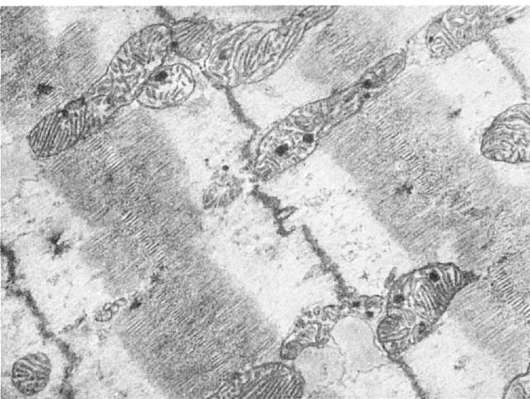 Рис. 16. Включения солей кальция в матриксе митохондрий мышечного волокна при ишемии. х18 500
Рис. 16. Включения солей кальция в матриксе митохондрий мышечного волокна при ишемии. х18 500
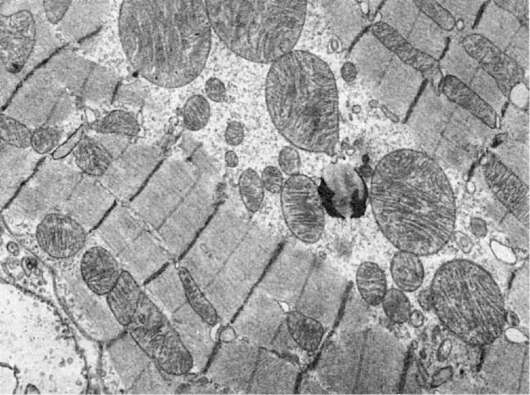 Рис. 17. Гигантские митохондрии кардиомиоцитов. Миокард собаки при синдроме длительного раздавливания. х16 000
Рис. 17. Гигантские митохондрии кардиомиоцитов. Миокард собаки при синдроме длительного раздавливания. х16 000
мер, в гепатоцитах при алкоголизме. Митохондрии, в том числе и гигантские, могут быть различной формы: сигарообразные, каплеобразные, извитые и т.д.
Число митохондрий крайне вариабельно. Увеличение числа митохондрий (т.е. гиперплазия), отражающее усиление протекающего в них окислительного фосфорилирования, характерно для клеток с активацией специализированной функции, что имеет место при гипертрофии, пролиферации и трансформации клеток, особенно после повреждения ткани. Большое число митохондрий крайне характерно для онкоцитов, в том числе и онкоцитарньгх опухолей. Уменьшение числа митохондрий типично для так называемых регрессивных процессов - старения клеток, их атрофии.
Изменения крист митохондрий
Изменения крист митохондрий, как и самих митохондрий, могут касаться также их структуры, размеров, формы и числа.
Структурные изменения разнообразны: пластинчатые кристы появляются при усилении активности митохондрий. Деформация и агрегация крист встречаются при понижении этой активности. Форма крист также отражает повышенную или пониженную активность митохондрий. Размеры крист, как правило, соответствуют размерам митохондрий: гигантские кристы в гигантских митохондриях, редукция крист при редукции митохондрий. В такой же мере и число крист отражает активность митохондрий: увеличение числа крист митохондрий - свидетельство возрастающих функциональных потребностей клетки; уменьшение числа крист (редукция) митохондрий - свидетельство снижения этих потребностей.
Митохондриальный транспорт кальция и повреждение клетки
Одной из важных функций митохондрий является транспорт кальция. Кальций может накапливаться митохондриями в весьма значительных количествах, особенно параллельно с неорганическим фосфатом. Высвобождение кальция из митохондрий происходит двумя путями. Один из путей накопления кальция (митохондрии клеток сердца, мозга, скелетных мышц, экзокринных и эндокринных желез) стимулируется натрием и, видимо, представляет собой обмен Са2+ на Na+; другой путь (митохондрии клеток почек, печени, легких) нечувствителен к натрию, механизм его неясен.
Морфологическим подтверждением транспорта кальция митохондриями является обнаружение в митохондриальном матриксе электронноплотных гранул диаметром 20-50 нм, которые, возможно, служат местом аккумуляции двухвалентных ионов. Увеличение размера, плотности и числа этих гранул обнаружено не только при обработке тканей высокими концентрациями Са2+, но и в интактных клетках тех тканей, которые вовлечены в активный транспорт кальция - остеокластах, остеобластах и др. Та же ситуация обнаружена и при гормонально-обусловленных гиперкальциемиях - кальцинозах. При некоторых болезнях (коронарная болезнь сердца), синдромах (хроническая почечная недостаточность) и патологических состояниях (отравления тиоацетатамидом, папаином, йодоформом и т.д.) клетки отвечают на повреждение появлением в митохондриальном матриксе многочисленных крупных плотных гранул кальция (см. рис. 16). При этом кальцификация митохондрий предшествует некрозу клетки и часто бывает обратимой.
Внутримитохондриальная кальцификация может быть связана как с избыточным притоком кальция в клетку вследствие первичного повреждения плазматической мембраны, так и с первичными нарушениями транспорта кальция митохондриями. При первичном повреждении плазматической мембраны избыточный приток кальция в клетку приводит к накоплению его в митохондриях, что «отнимает» энергию АТФ и повреждает саму систему генерации энергии - митохондрии. Первичные нарушения митохондриального транспорта кальция встречаются при заболеваниях скелетных мышц - миопатиях (болезнь Люфта, синдром Кернса-Сайра). При этих болезнях митохондрии, несмотря на высокий уровень эндогенного кальция, могут дополнительно накапливать значительные его количества. В таких случаях можно говорить о «болезнях» нарушенного митохондриального транспорта.
Лизосомы
Лизосомы не только «органы» внутриклеточного пищеварения, о чем говорит их название, но и «убийцы» клетки; они причастны как к фагоцитозу, так и аутофагии. Физиологическая и патологическая активность лизосом зависит в основном от двух факторов: состояния (стабилизации) мембран лизосом и активности их ферментов. Поэтому повреждения клетки, к которым могут быть причастны лизосомы, возникают либо при дестабилизации лизосомных мембран, позволяющей проявиться гидролазной активности
ферментов, либо при лизосомной ферментопатии, ведущей к накоплению в клетке ряда исходных или промежуточных продуктов обмена.
Дестабилизация мембран лизосом и патология клетки
К дестабилизации (лабилизации) мембран лизосом могут привести воздействия различных веществ и агентов - лабилизаторов мембран лизосом (например, так называемые провоспалительные гормоны, витамины A, D, К и др.). Выраженным повреждающим мембраны лизосом действием отличаются некоторые микотоксины, различные канцерогенные вещества, фосфолипазы, активаторы и продукты перекисного окисления, двуокись кремния. Дестабилизирующе на мембраны лизосом действуют гипоксия, нарушения кислотноосновного состояния, голодание и белковая недостаточность, изменения гормонального статуса, шок, травмы, обширные оперативные вмешательства. Антагонистами лабилизаторов мембран лизосом являются их стабилизаторы (например, так называемые противовоспалительные гормоны, хлороксин, фенерган, холестерол и др.).
В патологических условиях возникают конкурентные взаимоотношения между лабилизаторами и стабилизаторами лизосомных мембран, и, если они в пользу первых, проницаемость мембран становится достаточной для выхода гидролаз в цитоплазму и взаимодействия с субстратом, которым могут стать и субклеточные структуры. Часть клетки или вся клетка погибают. Тот же механизм дестабилизации мембран лизосом имеется при фагоцитозе, когда после контакта первичных лизосом с фагосомами образуются фаголизосомы (рис. 18) и цитолизосомы. Подобный механизм лежит и в основе клеточной аутофагии. Как видно, патология мембран лизосом может определять и патологию фагоцитоза.
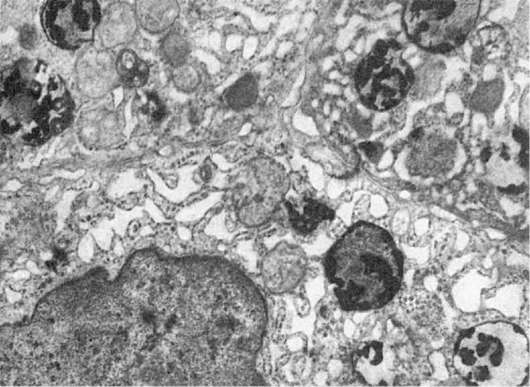 Рис. 18. Фаголизосомы в гепатоцитах. х18 500
Рис. 18. Фаголизосомы в гепатоцитах. х18 500
Нарушения функций лизосом и наследственные болезни
Среди наследственных болезней, связанных с нарушениями функций лизосом и называющихся лизосомными болезнями, прежде всего следует назвать наследственные лизосомные энзимопатии. Они являются следствием первичной генной мутации и проявляются либо полным блоком синтеза ферментного белка, либо синтезом белковых молекул со сниженной биокаталитической активностью. Дефект (отсутствие) одного или нескольких лизосомных ферментов ведет к накоплению в клетке веществ, которые в норме метаболизируют эти ферменты. Поэтому наследственные лизосомные энзимопатии включены в группу болезней накопления, или тезаурисмозов. Группа наследственных лизосомных энзимопатии достаточно велика. Особенно отчетливо она представлена среди гликогенозов (болезнь Помпе), ганглиозидозов (болезни Тея-Сакса, Сандхофа, ювенильный ганглиозидоз), гепатозов (болезнь Дабина-Джонсона), ожирения (недостаточность липаз адипозоцитов).
Другую группу наследственных болезней, обусловленных нарушением функции лизосом, можно связать с нарушением мембранных взаимодействий органелл клетки, что приводит к образованию гигантских органелл, в том числе гигантских лизосом (рис. 19). Эта группа невелика: синдром Чедиака-Хигаси, так называемая циклическая нейтропения.
Лизосомы и липопигменты
Содержимое телолизосом представлено липопигментами, т.е. продуктами, которые энзимы лизосом расщепляют с трудом или вообще не рас-
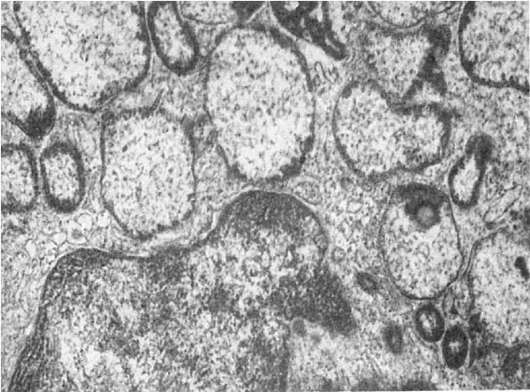 Рис. 19. Гигантские светлые лизосомы звездчатого ретикулоэндотелиоцита при врожденной недостаточности α-1-антитрипсина. х21 000
Рис. 19. Гигантские светлые лизосомы звездчатого ретикулоэндотелиоцита при врожденной недостаточности α-1-антитрипсина. х21 000
щепляют. После растворения лизосомальной мембраны они долгое время находятся в цитоплазме, лишь изредка покидают клетки.
Липопигментами обозначают группу цитоплазматических гранул и включений от желтого до темно-коричневого цвета, содержащих белки и труднорастворимые липиды. Их цвет обусловлен продуктами окисления и полимеризации ненасыщенных жирных кислот. Лизосомное происхождение липопигментов подтверждено биохимически, гистохимически и электронно-микроскопически. Липопигменты делят на липофусцин, встречающийся в паренхиматозных и нервных клетках, и цероид, образующийся в макрофагах (см. Дистрофия).
Микротельца (пероксисомы)
Изменения микротелец, касающиеся их числа и структурных компонентов, встречаются при многих болезнях человека. Будучи вторичными, они отражают нарушения оксидазно-каталазной активности клетки. Но изменения микротелец могут быть и первичными, что позволяет говорить о «пероксисомных болезнях», имеющих характерные клинические проявления первичной каталазной недостаточности.
Изменения числа и структуры микротелец, их нуклеоидов и матрикса Увеличение числа пероксисом и повышение каталазной активности в гепатоцитах (рис. 20) и нефроцитах можно вызвать в эксперименте с помощью ряда лекарственных препаратов, обладающих гиполипопротеинемическим действием, а в кардиомиоцитах - при длительной даче этанола. У человека повышение числа пероксисом отмечено в гепатоцитах при вирусном гепатите, лептоспирозе.
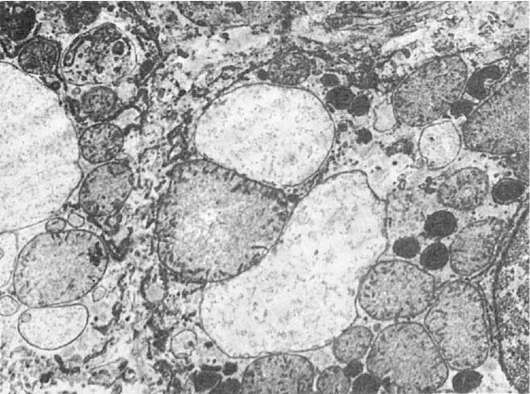 Рис. 20. Увеличение количества пероксисом в гепатоцитах. х22 000
Рис. 20. Увеличение количества пероксисом в гепатоцитах. х22 000
Уменьшение числа пероксисом, особенно в гепатоцитах, вызывают в эксперименте с помощью веществ, тормозящих синтез каталаз, или отмены стимуляторов этого синтеза. У человека уменьшение числа пероксисом и снижение синтеза их ферментов наблюдаются в печени при воспалении, а также при опухолевом росте. Значительные дефекты пероксисомной системы, разрушение пероксисом находят при гиперлипидемии и гиперхолестеринемии, причем разрушение пероксисом происходит путем аутолиза или аутофагии.
Нуклеоиды пероксисом разрушаются в эксперименте на животных при введении веществ, уменьшающих липидемию, или после облучения. У человека при одних заболеваниях (гепатоцеребральная дистрофия) происходит деградация нуклеоидов пероксисом, при других (идиопатический холестаз) - новообразование нуклеол в пероксисомах.
Пероксисомный матрикс разрушается у животных при введении им ингибиторов синтеза каталаз. У человека разрушение матрикса пероксисом находят при ишемическом некрозе, вирусном гепатите.
Пероксисомные болезни
Известны три наследственных метаболических расстройства, которые могут рассматриваться как пероксисомные болезни: акаталаземия, цереброгепаторенальный синдром Целлвегера и системная недостаточность карнитина.
При акаталаземии активность каталазы в печени и других органах крайне низка вследствие сниженной ее термостабильности. Единственный клинический синдром этого заболевания - гангренозные изъязвления полости рта.
Цереброгепаторенальный синдром Целлвегера характеризуется отсутствием пероксисом в гепатоцитах; эндоплазматическая сеть их редуцирована, митохондрий мало; цитоплазма заполнена гликогеном и липидами. Каталазная активность печени у этих больных составляет примерно 20% нормы. Результатом недостаточности пероксисом при этом синдроме является нарушение синтеза желчных кислот.
Системная недостаточность карнитина клинически характеризуется миопатией с периодическими нарушениями функций печени и головного мозга. Выраженный дефицит карнитина обнаруживается в скелетных мышцах, печени, плазме крови; в мышцах не происходит окисления жирных кислот.
Цитоскелет и патология клетки
«Скелет» клетки выполняет опорную, транспортную, контрактильную и двигательную функции. Он представлен 3 видами филаментов (фибрилл) - микрофиламентами, промежуточными филаментами и микротрубочками - макрофиламентами. Каждый из филаментов, выполняя ряд общих функций клетки, специализирован в отношении преимущественно одной из них - контракции (микрофиламенты), статики (промежуточные филаменты) или движения органелл и транспорта (микротрубочки). Цитоскелет претерпевает различные изменения при многих
болезнях и патологических состояниях, что, естественно, влияет на специализированные функции клетки.
Микрофиламенты
Микрофиламенты имеют прямое отношение к актину и миозину. Актиновые филаменты, как и миозин, обнаружены почти во всех клетках. Для миозина, независимо от того, принадлежит он мышечным или немышечным клеткам, характерна одна способность - обратимо связываться с актиновыми филаментами и катализировать гидролиз АТФ, что требует присутствия самого актина. Количество миозина в мышечных клетках в 50 раз больше по сравнению с немышечными, кроме того, миозиновые филаменты мышечных клеток длиннее и толще, чем филаменты немышечных клеток.
Патология микрофиламентов довольно разнообразна. С их дисфункцией связывают, например, определенные виды холестаза и даже первичный билиарный цирроз. Считают, что циркуляция желчи в печени регулируется микрофиламентозной системой (рис. 21), так как микрофиламенты в большом количестве окружают желчные канальцы и, прикрепляясь к плазматической мембране гепатоцитов, могут влиять на размер просвета желчных канальцев. Показано, что воздействия на микрофиламенты, угнетающие их сократительную способность, ведут к застою желчи. Возможно, что подобный механизм лежит в основе некоторых видов холестаза. Резкое увеличение микрофиламентов находят в эпителии желчных протоков при первичном билиарном циррозе, что может быть причиной нарушения кинетики билиарной системы, холестаза и после-
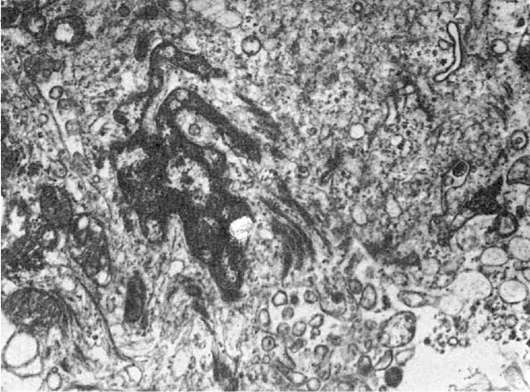 Рис. 21. Увеличение количества микрофиламентов в эпителиальной клетке желчного протока при холестазе. х20 000
Рис. 21. Увеличение количества микрофиламентов в эпителиальной клетке желчного протока при холестазе. х20 000
дующего гранулематоза холангиол, характерного для этого заболевания. Однако вопрос о том, первична или вторична аккумуляция микрофиламентов в эпителии билиарной системы при первичном билиарном циррозе, еще не решен. Увеличение количества микрофиламентов описано в клетках злокачественных опухолей, особенно в зонах инвазии опухоли. Микрофиламентозная активность характерна и для ряда репаративных процессов, например для заживления ран.
Микрофиламентозная система служит также секреторным процессам, фагоцитозу и митозу.
Промежуточные филаменты
Промежуточные филаменты достаточно специализированы в зависимости от типа клеток, в которых встречаются: цитокератины находят в эпителиях, скелетин (десмин) - в мышечных клетках, виментин - в мезенхимальных клетках, нейрофиламенты - в клетках центральной и периферической нервной системы, глиальные филаменты - в клетках глии. Однако в клетках одного и того же происхождения могут встречаться промежуточные филаменты разного типа. Так, в гладких мышцах пищеварительной, дыхательной и мочеполовой систем промежуточные филаменты представлены главным образом скелетином, а в гладких мышечных клетках сосудов, как и во многих мезенхимальных клетках, - виментином. В связи с этим понятными становятся функциональные возможности гладких мышечных клеток сосудов (фагоцитоз, фибробластическая трансформация и др.).
С патологией промежуточных филаментов, преимущественно их аккумуляцией, пытаются связать многие патологические процессы: образование алкогольного гиалина (телец Мэллори), нейрофибриллярных сплетений в нервных клетках и сенильных бляшек при старческом слабоумии и болезни Альцгеймера. С аккумуляцией промежуточных филаментов связывают и развитие некоторых форм кардиомиопатии.
Алкогольный гиалин, формирующий тельца Мэллори, обнаруживают обычно в гепатоцитах, реже в эпителии желез поджелудочной железы и нервных клетках головного мозга, при хроническом алкоголизме, индийском детском циррозе, гепатоцеребральной дистрофии (болезни Вильсона-Коновалова), первичном билиарном циррозе. Он имеет характерную ультраструктуру (рис. 22). Однако образование алкогольного гиалина из промежуточных филаментов признается далеко не всеми исследователями. Многие считают, что при алкоголизме алкогольный гиалин является продуктом извращенного синтеза при воздействии на клетку (гепатоцит) этанола с участием в этом процессе цитоскелета.
Патологические изменения нейрофиламентов представлены образованием нейрофибриллярных сплетений, которые описаны при многочисленных патологических состояниях. Нейрофибриллярные сплетения вдоль аксонов периферических нервов и в нервных сплетениях характерны для своеобразного заболевания - наследственной нейропатии гигантских аксонов. Нейрофибриллярные сплетения лежат в основе так называемых
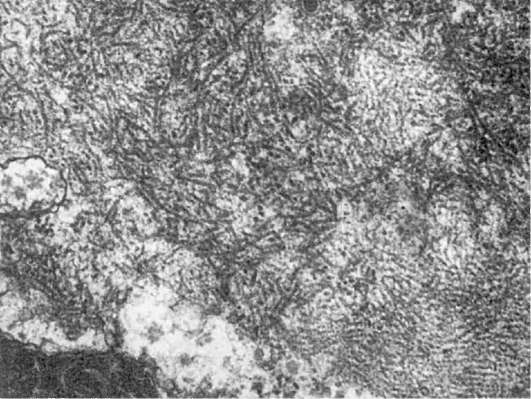 Рис. 22. Фибриллярный алкогольный гиалин в цитоплазме гепатоцита при остром алкогольном гепатите. х20 000
Рис. 22. Фибриллярный алкогольный гиалин в цитоплазме гепатоцита при остром алкогольном гепатите. х20 000
сенильных бляшек головного мозга, патогномоничных для старческого слабоумия и болезни Альцгеймера. Однако в случаях появления амилоида в сенильнъгх бляшках, т.е. при локальной церебральной форме старческого амилоидоза, нет оснований для заключения о том, что амилоид строят нейрофиламенты и их сплетения.
Некоторые формы кардиомиопатий рассматриваются в настоящее время как вторичные по отношению к нарушениям метаболизма промежуточных филаментов (десмина). Описана необычная форма кардиомиопатий с прогрессирующей недостаточностью миокарда, характеризующаяся массивными отложениями в кардиомиоцитах AS-негативного материала, состоящего из промежуточных филаментов. Аккумуляция промежуточных филаментов является морфологическим маркером хронического алкоголизма, при котором скопления их находят в клетках эпителиального и мезенхимального происхождения (рис. 23).
Микротрубочки
Как известно, микротрубочки выполняют множество разнообразных функций: определяют движение и ориентацию хромосом, митохондрий, рибосом, цитоплазматических гранул; принимают участие в секреции, митотическом делении клетки, осуществляют цитоплазматический транспорт. Не менее разнообразна и патология микротрубочек. При воздействии на микротрубочки рядом веществ, активирующих их функции (винбластин, изофлуран и др.), размеры микротрубочек увеличиваются в 2-3 раза. Они образуют скопления, связанные с рибосомами, к ним прилежат паракристаллические включения из гексогонально упакованных
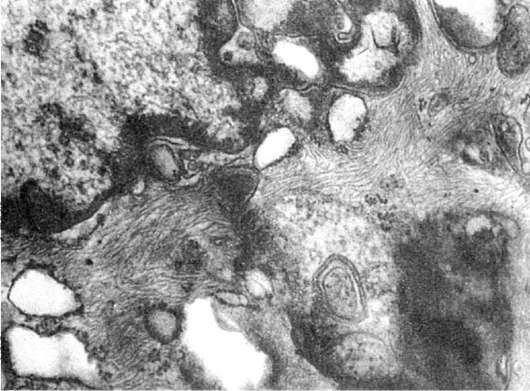 Рис. 23. Аккумуляция промежуточных филаментов в цитоплазме эндотелиоцитов сосудов кожи при хроническом алкоголизме. х20 000
Рис. 23. Аккумуляция промежуточных филаментов в цитоплазме эндотелиоцитов сосудов кожи при хроническом алкоголизме. х20 000
субъединиц. К тяжелому повреждению микротрубочек ведет ионизирующее излучение, при этом страдает генетический аппарат клетки, возникают патологические митозы. Резко уменьшается число микротрубочек (особенно в гепатоцитах) при воздействии этанолом, они округляются, вытесняются промежуточными филаментами.
Патология микротрубочек может быть основой некоторых клиникоморфологических синдромов. Таков, например, синдром неподвижныхресничек, ранее известный как синдром Картагенера. При этом врожденном синдроме реснички покровного эпителия дыхательных путей и слизистой оболочки среднего уха, основой строения которого являются дефектные микротрубочки, малоподвижны. Поэтому мукоцеллюлярный транспорт резко ослаблен или отсутствует, что ведет к хроническому воспалению дыхательных путей и среднего уха. У таких больных неподвижны также и сперматозоиды, так как их хвост эквивалентен ресничкам.
Плазматическая мембрана
Плазматической мембране свойственны различные функции, из которых основные - информационная, транспортно-обменная, защитная и контактная. Информационная функция обеспечивается рецепторами мембраны, транспортно-обменная и защитная - самой мембраной, контактная - клеточными стыками.
Клеточная рецепция и патология клетки
Плазмолемма (ее гликокаликс) содержит сложные структуры - рецепторы, воспринимающие различные раздражения («сигналы») внеш-
ней среды. Они специализированы для восприятия «сигналов» гормонов, многих биологически активных веществ, антигенов, иммуноглобулинов и их фрагментов, компонентов комплемента и т.д. Рецепторы представлены обычно гликопротеидами, они способны свободно перемещаться как по поверхности клеточной мембраны, так и внутри ее - так называемая латеральная диффузия рецепторов. Поэтому рецепторы можно рассматривать как своеобразные многокомпонентные мембранные комплексы.
Механизм реализации рецепторного сигнала довольно универсален, так как рецепторы связаны с аденилатциклазой. Эта связь представлена трехкомпонентной системой (Авцын А.П., Шахламов В.А., 1979): рецептор на внешней поверхности мембраны, трансдуктор (фосфолипиды) и катализатор на внутренней поверхности мембраны (аденилатциклаза). Аденилатциклаза катализирует внутриклеточное превращение АТФ в АМФ, который в отношении стимуляции клеточных ферментов универсален. Считают, что изменения в любом компоненте рецептора (надмембранном, внутримембранном или подмембранном) должны привести к молекулярным изменениям клеток. Таким образом, основное значение в нарушении рецепторной информации придается разобщению звеньев рецепторного комплекса.
Ряд болезней связан с отсутствием или блокадой рецепторов клетки. Так, отсутствие апо- и В, Е-рецепторов у паренхиматозных и мезенхимальных клеток ведет к развитию гомозиготной гиперлипопротеинемии 11а типа, известной также как семейная эссенциальная гиперхолестеринемия. Пересадка печени с сохранными апо-В, Е-рецепторами при гомозиготной гиперлипопротеинемии снижает уровень холестерина крови до нормы, ведет к исчезновению проявлений атеросклероза и коронарной болезни. С врожденным дефектом рецепторов к Fc-фрагментам иммуноглобулинов у мезангиоцитов связывают идиопатическую мембранозную нефропатию.
Блокаду рецепторов клетки нередко вызывают аутоантитела. Возникает одна из разновидностей цитотоксических реакций (реакции инактивации и нейтрализации), проявляющаяся антительными болезнями рецепторов. Среди них миастения, в развитии которой участвуют антитела к ацетилхолиновым рецепторам нервно-мышечной пластинки, а также инсулинрезистентный сахарный диабет, при котором антитела против клеточных рецепторов к инсулину блокируют эти рецепторы и не позволяют клетке отвечать на инсулиновый сигнал.
Нарушение проницаемости плазматической мембраны и состояние клетки
Существует два принципиально различных механизма проникновения взвешенных частиц в клетку через плазмолемму: микропиноцитоз (образование микропиноцитозных везикул) и диффузия. При воздействии на клетку факторов, нарушающих проницаемость плазмолеммы, может преобладать один из этих механизмов.
Изменения плазмолеммы при нарушении ее проницаемости. Характерными ультраструктурными проявлениями нарушенной проницаемости плазматической мембраны являются (Авцын А.П., Шахламов В.А., 1979):
усиленное везикулообразование; увеличение поверхности плазмолеммы за счет мембран микропиноцитозных везикул; образование цитоплазматических отростков и инвагинаций плазмолеммы; микроклазматоз и клазматоз; утолщение плазмолеммы; образование «крупных» микропор; «бреши» в плазмолемме; «штопка» локально разрушенной плазмолеммы; образование миелиноподобных структур.
Усиленное везикулообразование (усиленный эндоцитоз), как правило, отражает повышение проницаемости цитолеммы и приводит к дефициту ее поверхности («минус-мембрана»).
Увеличение поверхности плазмолеммы за счет мембран микропиноцитозных пузырьков является признаком резкого набухания клетки. Общая площадь плазмолеммы, испытывающей предельное натяжение, при этом увеличивается («плюс-мембрана»). В результате срыва такой адаптации цитолеммы к нарастающему отеку клетки возникает ее гибель.
Образование цитоплазматических отростков и инвагинаций плазмолеммы встречается при воздействии на клетку самых различных патогенных факторов и свидетельствует об активности цитоплазматической мембраны.
Микроклазмацитоз и клазмацитоз - отделение части цитоплазмы наружу, которая затем распадается и нередко реутилизируется в межклеточной среде. Механизм его сводится к образованию цитоплазматических ограниченных мембраной выростов, что ведет к отрыву части цитоплазмы от клетки. К усилению микроклазмацитоза и клазмацитоза ведут различные воздействия на клетку (антигены, иммунные комплексы, гипоксия).
Утолщение плазмолеммы возникает по ряду причин и может влиять на мембранную проницаемость. Одной из причин является уменьшение ионов кальция во внеклеточной жидкости, при этом изменяется проницаемость мембраны для ионов натрия и калия, в клетке накапливается жидкость. Другой причиной может быть удаление фосфолипидов из мембраны воздействием фосфолипаз.
Образование «крупных» микропор в цитоплазматической мембране связано с нарушением обменной диффузии в клетке. В нормально функционирующей клетке, т.е. при нормально протекающей обменной диффузии (ионы калия и натрия, анионы хлора и др.), микропоры не превышают 0,4-0,6 нм; при нарушении обменной диффузии они могут достигать 9 нм. Появление «крупных» микропор ведет к изоосмотическому набуханию клетки, перерастяжению, а в дальнейшем и к разрыву клеточных мембран.
«Бреши» в плазмолемме (локальные разрушения мембраны), размеры которых могут достигать 1 мкм, связаны с лизисом мембраны, который может быть вызван самыми разными агентами. «Бреши» в мембране, независимо от того, «сквозные» они или «поверхностные», ведут к осмотическому набуханию клетки и ее гибели.
«Штопка» локально разрушенной плазмолеммы осуществляется с помощью мембран мелких везикул, которые сосредоточиваются в месте повреждения.
Своеобразным изменением плазмолеммы, встречающимся не только при нарушении ее проницаемости, является образование миелиноподобных структур (рис. 24). Эти структуры появляются в связи с перекисным окислением липидов мембран, усиливающимся под воздействием разных агентов. Высвобождающиеся из разрушающихся при перекисном окислении мембран фосфолипиды (дезагрегация и реагрегация мембраны) образуют сложные миелиноподобные структуры. Подобные структуры появляются и при скручивании удлиненных цитоплазматических отростков.
Изменения клетки при повреждении плазмолеммы. Повреждение плазмолеммы ведет к утрате так называемого активного мембранного транспорта: концентрации интра- и экстрацеллюлярного натрия и калия выравниваются, внутрь клетки проникают низкомолекулярные анионы, а затем и катионы, повышается внутриклеточное осмотическое давление. Таким образом, резко нарушается мембранный водно-электролитный транспорт, следствием чего становятся набухание и отек клетки. Нарушение активного мембранного транспорта может приводить также к избирательному поступлению в клетку определенных продуктов обмена (белки, липиды, углеводы, пигменты) и накоплению их после истощения ферментных систем, метаболизирующих эти продукты. Так развиваются клеточные дистрофии инфильтрационного генеза (жировая дистрофия гепатоцитов при гиперлипидемиях; гиалиново-капельная дистрофия нефроцитов при нефротическом синдроме). При резком повреждении плазмолеммы и поступлении в клетку ряда токсических или биологически активных веществ возможна деструкция структурных комплексов клетки
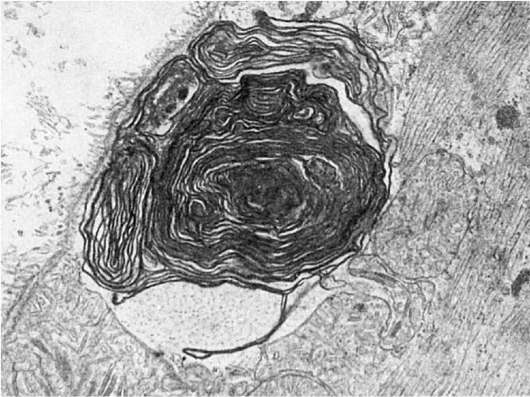 Рис. 24. Миелиноподобные структуры под плазматической мембраной мышечного волокна при ишемии. х22 500
Рис. 24. Миелиноподобные структуры под плазматической мембраной мышечного волокна при ишемии. х22 500
с высвобождением составляющих их химических веществ (белки, липиды и т.д.), что ведет к их накоплению. Возникают клеточные дистрофии декомпозиционного генеза (жировая дистрофия миокарда при дифтерии, гидропическая дистрофия гепатоцитов при вирусном гепатите). Следует заметить, что инфильтрационный механизм развития дистрофии может сменяться декомпозиционным и наоборот. В ряде случаев повреждения плазмолеммы позволяют проникнуть в клетку веществам, способным извратить синтез того или иного продукта. Тогда возникают клеточные дистрофии извращенного синтеза (синтез алкогольного гиалина гепатоцитом под воздействием этанола). Финалом тяжелого повреждения плазмолеммы является гибель клетки - ее некроз (см. Дистрофия, Некроз).
Патология клеточных стыков
В тканях человека клеточные стыки ответственны за три главные функции: межклеточную адгезию, «тесное общение» клеток и герметизацию слоя эпителиальных клеток.
Межклеточную адгезию как чисто механическую функцию ранее связывали в первую очередь с десмосомами. В настоящее время установлено, что в межклеточной адгезии участвуют все типы клеточных стыков.
Медиаторами «тесного общения» (или сопряжения) клеток считают щелевидные стыки, которые обеспечивают прямое сообщение между клетками, перенос ионов и малых молекул без потери их во внеклеточное пространство. Это способствует регуляции метаболических процессов в клетках и их дифференцировке.
Герметизация клеток эпителиального пласта обеспечивается плотными стыками, степень ее коррелирует с количеством стыков и внутримембранных тяжей. Плотные стыки отвечают за поддержание осмотических и электрохимических градиентов эпителиального пласта и отчасти за состояние внеклеточных структур, окружающих этот пласт.
Изменение межклеточной адгезии. Показано, что степень межклеточной адгезии ослабевает при опухолевом росте, причем уже на ранних стадиях онкогенеза. Количество и распределение клеточных стыков на поверхности опухолевых клеток могут быть одним из критериев характеристики роста опухоли.
Изменение «тесного общения» клеток. Как уже говорилось, «тесное общение» клеток предопределяет их непосредственный контакт для обмена информационными молекулами и обычно осуществляется с помощью щелевидных стыков, гидрофильные каналы которых пропускают ионы и молекулы с молекулярной массой до 1000. Считают, что дефекты «тесного общения» клеток могут играть важную роль в развитии и поведении опухолей.
Нарушения межмембранных связей клеток тканевых барьеров. Плотные стыки являются структурной основой таких тканевых барьеров, как кровь - мозг, кровь - легкие, кровь - желчь, кровь - почки. Поэтому эти стыки находятся, как правило, в эпителии. Они предотвращают «про-
извольный обмен» белками и другими макромолекулами между клеточными «партнерами» барьеров. Наиболее частым следствием повреждения тканевых барьеров является увеличение проницаемости плотных стыков клеток (рис. 25), что ведет к «трансэпителиальной протечке» (например, при повышении внутрисосудистого гидростатического давления, мозговой коме, холестазе, шоке, нефротическом синдроме).
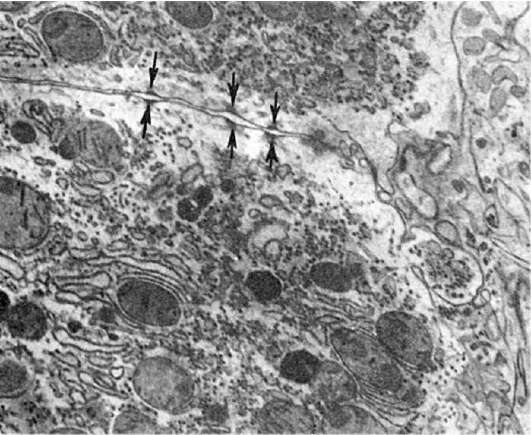 Рис. 25. Расхождение
десмосомальных контактов между гепатоцитами (показано стрелками) вблизи
желчного канальца при первичном билиарном циррозе. х23 500
Рис. 25. Расхождение
десмосомальных контактов между гепатоцитами (показано стрелками) вблизи
желчного канальца при первичном билиарном циррозе. х23 500
Структурные изменения клеточных стыков. Эти изменения касаются прежде всего десмосом. Псевдодесмосомы («несовершенные» десмосомы) с хорошо развитой пластинкой лишь у одной клетки могут возникать в результате разрыва дефектных стыков, неполной сборки стыка, диссоциации клеток. В основе асимметричных десмосом с недоразвитой пластинкой у одной из клеток лежат, вероятно, те же механизмы. К структурным изменениям клеточных стыков следует отнести и нарушения их топографии, т.е. появление их на поверхности клеток, где они в обычных условиях жизнедеятельности клеток не встречаются.
Изменения структуры десмосом, как и других типов клеточных стыков, находят при метаплазии, дисплазии, опухолевом росте, в эмбриональных тканях (асимметричные десмосомы); они найдены при таких заболеваниях, как ревматоидный артрит, псориаз.
В заключение следует сказать, что патология клетки как интегративное понятие является необходимой базой общей патологии человека.