Патологическая анатомия : учебник / А. И. Струков, В. В. Серов. - 5-е изд., стер. - М.: Литтерра, 2010. - 848 с. : ил.
|
|
|
|
БОЛЕЗНИ СИСТЕМЫ КРОВИ
Болезни системы крови составляют содержание клинической гематологии, основоположниками которой в нашей стране являются И.И. Мечников, С.П. Боткин, М.И. Аринкин, А.И. Крюков, И.А. Кассирский. Эти болезни развиваются в результате нарушений регуляции кроветворения и кроворазрушения, что отражается на составе периферической крови. Поэтому на основании данных изучения состава периферической крови можно ориентировочно судить о состоянии кроветворной системы в целом. Можно говорить об изменениях красного и белого ростков, а также плазмы крови - как количественных, так и качественных.
Изменения красного ростка системы крови могут быть представлены уменьшением содержания гемоглобина и количества эритроцитов (ане- мии) или их увеличением (истинная полицитемия, или эритремия); нарушением формы эритроцитов - эритроцитопатиями (микросфероцитоз, овалоцитоз) или синтеза гемоглобина - гемоглобинопатиями, или гемоглобинозами (талассемия, серповидно-клеточная анемия).
Изменения белого ростка системы крови могут касаться как лейкоцитов, так и тромбоцитов. Количество лейкоцитов в периферической крови может увеличиваться (лейкоцитоз) или уменьшаться (лейкопения), они могут обретать качества опухолевой клетки (гемобластоз). В равной мере можно говорить об увеличении количества тромбоцитов (тромбоцитоз) или об их уменьшении (тромбоцитопении) в периферической крови, а также об изменении их качества (тромбоцитопатии).
Изменения плазмы крови касаются главным образом ее белков. Количество их может увеличиваться (гиперпротеинемия) или уменьшаться (гипопротеинемия); может изменяться и качество белков плазмы, тогда говорят о диспротеинемиях.
Наиболее полное представление о состоянии кроветворной системы дает изучение пунктата костного мозга (грудины) и трепанобиопсии (гребень подвздошной кости), которыми широко пользуются в гематологической клинике.
Болезни системы крови чрезвычайно разнообразны. Наибольшее значение имеют анемии, гемобластозы (опухолевые заболевания, возникающие из кроветворных клеток), тромбоцитопении и тромбоцитопатии.
Анемии
Анемии (греч. an - отрицательная приставка и haima - кровь), или малокровие, - группа заболеваний и состояний, характеризующихся уменьшением общего количества гемоглобина; обычно оно проявляется в уменьшении его содержания в единице объема крови. В большинстве случаев анемия сопровождается снижением числа эритроцитов в единице объема крови (исключение составляют железодефицитные состояния и талассемия). При анемии в периферической крови нередко появляются эритроциты различной величины (пойкилоцитоз), формы (анизоцитоз), разной степени окраски (гипохромия, гиперхромия); в эритроцитах иногда обнаруживаются включения - базофильные зерна (так называемые тельца Жолли), базофильные кольца (так называемые кольца Кабо) и т.д. При некоторых анемиях в крови выявляются ядерные представители (эритробласты, нормобласты, мегалобласты) и незрелые формы (полихроматофилы) эритроцитов.
На основании изучения пунктата грудины можно судить о состоянии (гипер- или гипорегенерация) и типе эритропоэза (эритробластический, нормобластический, мегалобластический), свойственных той или иной форме анемии.
Этиология и патогенез. Причинами развития анемии могут быть кровопотеря, недостаточная эритропоэтическая функция костного мозга, повышенное кроворазрушение.
При кровопотере анемия возникает в том случае, когда убыль эритроцитов в крови превышает регенераторные возможности костного мозга. То же следует сказать и о кроворазрушении, т.е. гемолизе, который может быть связан с экзогенными и эндогенными факторами. Недостаточность эритропоэтической функции костного мозга зависит от дефицита необходимых для нормального кроветворения веществ: железа, витамина B12, фолиевой кислоты (так называемые дефицитные анемии), или от неусвоения этих веществ костным мозгом (так называемые ахрестические анемии).
Классификация. В зависимости от этиологии и главным образом патогенеза различают три основные группы анемий (Алексеев Г.А., 1970): 1) вследствие кровопотери (постгеморрагические анемии); 2) вследствие нарушенного кровообразования; 3) вследствие повышенного кроворазрушения (гемолитические анемии). В каждой группе выделяются формы анемии. По характеру течения анемии делят на острые и хронические. В соответствии с морфологическим и функциональным состоянием костного мозга, отражающим его регенераторные возможности, анемия может быть регенераторной, гипорегенераторной, гипопластической, апластической, диспластической.
Анемии вследствие кровопотери (постгеморрагические)
Анемии вследствие кровопотери могут иметь острое или хроническое течение.
Острая постгеморрагическая анемия наблюдается после массивных кровотечений из сосудов желудка при язвенной болезни, из язвы тонкой кишки при брюшном тифе, при разрыве маточной трубы в случае внематочной беременности, разъедании ветви легочной артерии при туберкулезе легких, разрыве аневризмы аорты или ранении ее стенки и отходящих от аорты крупных ветвей.
Чем крупнее калибр пораженного сосуда и чем ближе к сердцу он расположен, тем опаснее для жизни кровотечение. Так, при разрыве дуги аорты достаточно потерять менее 1 л крови, чтобы наступила смерть в связи с резким падением артериального давления и дефицитом наполнения полостей сердца. Смерть в таких случаях наступает прежде, чем происходит обескровливание органов, и при вскрытии трупов анемизация органов малозаметна. При кровотечениях из сосудов мелкого калибра смерть обычно наступает при потере более половины общего количества крови. В таких случаях постгеморрагической анемии отмечается бледность кожных покровов и внутренних органов; посмертные гипостазы выражены слабо.
Патологическая анатомия. Если кровотечение оказалось несмертельным, то кровопотеря возмещается благодаря регенераторным процессам в костном мозге. Клетки костного мозга плоских и эпифизов трубчатых костей усиленно пролиферируют, костный мозг становится сочным и ярким. Жировой (желтый) костный мозг трубчатых костей также становится красным, богатым клетками эритропоэтического и миелоидного ряда. Кроме того, появляются очаги внекостномозгового (экстрамедуллярного) кроветворения в селезенке, лимфатических узлах, тимусе, в периваскулярной ткани, клетчатке ворот почек, слизистых и серозных оболочках, коже.
Хроническая постгеморрагическая анемия развивается в тех случаях, когда происходит медленная, но длительная потеря крови. Это наблюдается при небольших кровотечениях из распадающейся опухоли желудочнокишечного тракта, кровоточащей язвы желудка, геморроидальных вен кишечника, из полости матки, при геморрагическом синдроме, гемофилии и т.д.
Патологическая анатомия. Кожные покровы и внутренние органы бледны. Костный мозг плоских костей обычного вида; в костном мозге трубчатых костей наблюдаются выраженные в той или иной степени явления регенерации и превращения жирового костного мозга в красный. Нередко отмечаются множественные очаги внекостномозгового кроветворения. В связи с хронической кровопотерей возникает гипоксия тканей и органов, которая обусловливает развитие жировой дистрофии миокарда, печени, почек, дистрофических изменений в клетках головного мозга. Появляются множественные точечные кровоизлияния в серозных и слизистых оболочках, во внутренних органах.
Анемии вследствие нарушения кровообразования
Анемии вследствие нарушения кровообразования представлены так называемыми дефицитными анемиями, возникающими при недостатке железа, витамина B12, фолиевой кислоты, гипо- и апластическими анемиями.
Анемии вследствие недостатка железа или железодефицитные анемии. Они могут развиваться прежде всего при недостаточном поступлении железа с пищей (алиментарная железодефицитная анемия детского возраста). Они возникают также при экзогенной недостаточности железа в связи с повышенными запросами организма у беременных и кормящих женщин, при некоторых инфекционных заболеваниях, у девушек при «бледной немочи» (ювенильный хлороз). В основе железодефицитной анемии может лежать и резорбционная недостаточность железа, встречающаяся при заболеваниях желудочно-кишечного тракта, а также после резекции желудка (агастрическая анемия) или кишечника (анэнтеральная анемия). Анемии вследствие недостатка железа - гипохромные.
В последнее время выделяют анемии, связанные с нарушением синтеза или утилизации порфиринов. Среди них различают наследственные (Х-сцепленные) и приобретенные (свинцовая интоксикация).
Анемия вследствие недостатка витамина B12 и/или фолиевой кислоты. Их
характеризует извращение эритропоэза. Это мегалобластические гиперхромные анемии.
Витамин B12 и фолиевая кислота являются необходимыми факторами гемопоэза. Витамин B12 поступает в организм через желудочно-кишечный тракт (внешний фактор). Всасывание витамина B12 в желудке возможно только в присутствии внутреннего фактора Касла, или гастромукопротеина, который вырабатывается добавочными клетками фундальных желез желудка. Соединение витамина B12 с гастромукопротеином ведет к образованию белково-витаминного комплекса, который всасывается слизистой оболочкой желудка и тонкой кишки, откладывается в печени и активирует фолиевую кислоту. Поступление витамина B12 и активированной фолиевой кислоты в костный мозг определяет нормальный гормональный эритропоэз, стимулирует созревание клеток красной крови.
Эндогенная недостаточность витамина B12 и/или фолиевой кислоты вследствие выпадения секреции гастромукопротеина и нарушенной ассимиляции пищевого витамина B12 ведет к развитию пернициозной и пернициозоподобных анемий.
Пернициозная анемия впервые описана в 1855 г. Аддисоном, в 1868 г. ее описал Бирмер (анемия Аддисона-Бирмера). Заболевание развивается обычно в зрелом возрасте (после 40 лет). Долгое время, до установления роли витамина B12, фолиевой кислоты и гастромукопротеина в патогенезе пернициозной анемии, она протекала злокачественно (злокачественная анемия) и, как правило, заканчивалась смертью больных.
Этиология и патогенез. Развитие болезни обусловлено выпадением секреции гастромукопротеина в связи с наследственной неполноценностью фундальных желез желудка, завершающейся их преждевременной
инволюцией (описаны случаи семейной пернициозной анемии). Большое значение имеют аутоиммунные процессы - появление трех типов аутоантител: первые блокируют соединение витамина B12 с гастромукопротеином, вторые - гастромукопротеин или комплекс гастромукопротеин - витамин B12, третьи - париетальные клетки. Эти антитела встречаются у 50-90% больных пернициозной анемией. В результате блокады гастромукопротеина и витамина B12 наступает извращение кроветворения, эритропоэз совершается по мегалобластическому типу, причем процессы кроворазрушения преобладают над процессами кроветворения. Распад мегалобластов и мегалоцитов происходит прежде всего в костном мозге и очагах внекостномозгового кроветворения еще до выхода клеток в периферическую кровь. Поэтому эритрофагоцитоз при анемии Аддисона- Бирмера особенно хорошо выражен в костном мозге, значительная часть гемоглобиногенных пигментов (порфирин, гематин) не используется, а только циркулирует в крови и выводится из организма.
С разрушением элементов красной крови связан общий гемосидероз, а с нарастающей гипоксией - жировая дистрофия паренхиматозных органов и нередко общее ожирение. Недостаток витамина B12 ведет к изменениям образования миелина в спинном мозге.
Патологическая анатомия. При наружном осмотре трупа определяются бледность кожных покровов (кожа с лимонно-желтым оттенком), желтушность склер. Подкожный жировой слой развит обычно хорошо. Трупные гипостазы не выражены. Количество крови в сердце и крупных сосудах уменьшено, кровь водянистая. В коже, слизистых и серозных оболочках видны точечные кровоизлияния. Внутренние органы, особенно селезенка, печень, почки, на разрезе ржавого вида (гемосидероз). Наиболее ярко изменения выражены в желудочно-кишечном тракте, костном и спинном мозге.
В желудочно-кишечном тракте имеются атрофические изменения. Язык гладкий, блестящий, как бы полированный, покрыт красными пятнами. При микроскопическом исследовании находят резкую атрофию эпителия и лимфоидных фолликулов, диффузную инфильтрацию подэпителиальной ткани лимфоидными и плазматическими клетками. Эти изменения обозначают как гунтеровский глоссит (по имени впервые описавшего эти изменения Гунтера). Слизистая оболочка желудка (рис. 127), особенно фундальной части, истонченная, гладкая, лишена складок. Железы уменьшены и расположены на значительном расстоянии друг от друга; эпителий их атрофичен, сохранны лишь главные клетки. Лимфоидные фолликулы также атрофичны. Эти изменения слизистой оболочки желудка завершаются склерозом. В слизистой оболочке кишечника развиваются такие же атрофические изменения.
Печень увеличена, плотная, на разрезе имеет буро-ржавый оттенок (гемосидероз). Отложения железа обнаруживают не только в звездчатых ретикулоэндотелиоцитах, но и в гепатоцитах. Поджелудочная железа плотная, склерозирована.
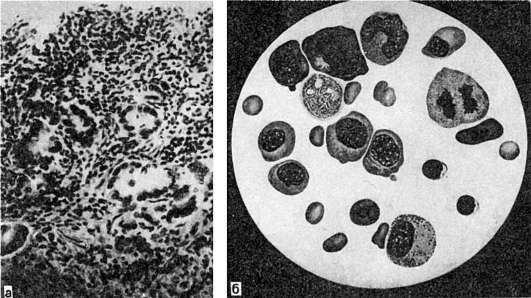 Рис. 127. Пернициозная анемия:
Рис. 127. Пернициозная анемия:
а - атрофия слизистой оболочки желудка; б - костный мозг (трепанобиопсия); среди клеточных элементов много мегалобластов
Костный мозг плоских костей малиново-красный, сочный; в трубчатых костях он имеет вид малинового желе. В гиперплазированном костном мозге преобладают незрелые формы эритропоэза - эритробласты, нормобласты и особенно мегалобласты (см. рис. 127), которые находятся и в периферической крови. Эти элементы крови подвергаются фагоцитозу макрофагами (эритрофагия) не только костного мозга, но и селезенки, печени, лимфатических узлов, что обусловливает развитие общего гемосидероза.
Селезенка увеличена, но незначительно, дряблая, капсула морщинистая, ткань розово-красная, с ржавым оттенком. При гистологическом исследовании обнаруживают атрофичные фолликулы со слабовыраженными зародышевыми центрами, а в красной пульпе - очаги экстрамедуллярного кроветворения и большое число сидерофагов.
Лимфатические узлы не увеличены, мягкие, с очагами экстрамедуллярного кроветворения, иногда на значительном протяжении вытесняющими лимфоидную ткань.
В спинном мозге, особенно в задних и боковых столбах, выражен распад миелина и осевых цилиндров.
Этот процесс называют фуникулярнъм миелозом. Иногда в спинном мозге появляются очаги ишемии и размягчения. Такие же изменения редко наблюдаются в коре головного мозга.
Течение анемии Аддисона-Бирмера обычно прогрессирующее, но периоды обострения болезни чередуются с ремиссиями. За последние годы как клиническая, так и морфологическая картина пернициозной анемии
благодаря лечению препаратами витамина B12 и фолиевой кислоты резко изменилась. Летальные случаи наблюдаются редко.
С дефицитом гастромукопротеина связано развитие пернициозоподобных В12-дефицитных анемий при раке, лимфогранулематозе, сифилисе, полипозе, коррозивном гастрите и других патологических процессах в желудке. При этих патологических процессах в желудке вторично возникают воспалительные, дистрофические и атрофические изменения в железах дна с нарушением секреции гастромукопротеина и эндогенной недостаточностью витамина B12. Такой же генез имеет пернициозоподобная анемия, возникающая спустя несколько лет после удаления желудка (агастрическая B^--дефицитная анемия).
Нарушение всасывания витамина B12 и/или фолиевой кислоты в кишечнике лежит в основе ряда В12(фолиево)дефицитных анемий. Это глистная - дифиллоботриозная - анемия при инвазии широким лентецом, анемия при спру - спру-анемия, а также анемия после резекции тонкой кишки - анэнтеральная В12(фолиево)дефицитная анемия.
Причиной развития B12(фолиево)дефицитных анемий может быть также экзогенная недостаточность витамина B12 и/или фолиевой кислоты алиментарной природы, например у детей при вскармливании козьим молоком (алиментарная анемия) или при лечении некоторыми лекарственными препаратами (медикаментозная анемия).
Гипо- и апластические анемии. Эти анемии являются следствием глубокого угнетения кроветворения, особенно молодых элементов гемопоэза.
Причиной развития таких анемий могут быть как эндогенные, так и экзогенные факторы. Среди эндогенных факторов большое место занимают наследственные, с которыми связано развитие семейной апластической анемии (Фанкони) и гипопластической анемии (Эрлиха).
Семейная апластическая анемия (Фанкони) встречается очень редко, обычно у детей, чаще у нескольких членов семьи. Тяжелая хроническая гиперхромная анемия характеризуется мегалоцитозом, ретикулоцитозом и микроцитозом, лейко- и тромбопенией, геморрагиями, аплазией костного мозга. Она нередко сочетается с пороками развития.
Гипопластическая анемия (Эрлиха) имеет острое и подострое течение, характеризуется прогрессирующей гибелью активного костного мозга, сопровождается кровоточивостью, иногда присоединением сепсиса. В крови наблюдается уменьшение числа всех форменных элементов крови без признаков регенерации.
Для эндогенных гипо- и апластических анемий наиболее характерно поражение эритробластического ростка крови (эритрона) с потерей способности костного мозга к регенерации. Происходит гибель активного костного мозга плоских и трубчатых костей, он замещается желтым, жировым (рис. 128). Среди массы жира в костном мозге встречаются единичные кроветворные клетки. В случаях полного опустошения костного мозга и замещения его жиром говорят о «чахотке» костного мозга - панмиелофтизе.
В качестве экзогенных факторов, ведущих к развитию гипопластических и апластических анемий, могут выступать лучевая энергия (радиа-
ционная анемия), токсические вещества (токсическая, например, бензольная анемия), такие лекарственные препараты, как цитостатические, амидопирин, атофан, барбитураты и др. (медикаментозная анемия).
При экзогенных гипо- и апластических анемиях в отличие от эндогенных анемий полного подавления гемопоэза не происходит, отмечается лишь угнетение регенераторной способности костного мозга. Поэтому в пунктате из грудины можно найти молодые кле-
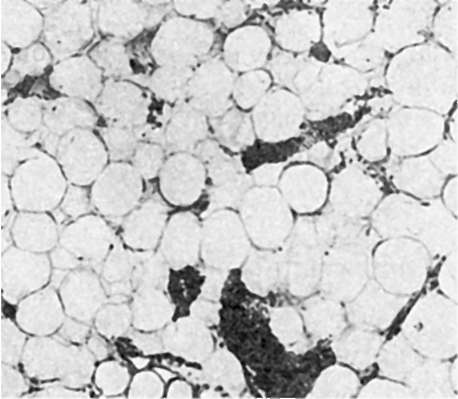 Рис. 128. Апластическая анемия. Активный костный мозг заменен жировым
Рис. 128. Апластическая анемия. Активный костный мозг заменен жировым
точные формы эритро- и миелопо-
этического ряда. Однако при длительном воздействии активный костный мозг опустошается и замещается жировым, развивается панмиелофтиз. Присоединяется гемолиз, возникают множественные кровоизлияния в серозных и слизистых оболочках, явления общего гемосидероза, жировая дистрофия миокарда, печени, почек, язвенно-некротические и гнойные процессы, особенно в желудочно-кишечном тракте.
Гипо- и апластические анемии возникают также при замещении костного мозга лейкозными клетками, метастазами злокачественной опухоли, обычно рака (рак предстательной, молочной, щитовидной желез, желудка), или костной тканью при остеосклерозе (остеосклеротическая анемия). Анемия на почве остеосклероза встречается при остеомиелопоэтической дисплазии, мраморной болезни (остеосклеротическая анемия Альберс-Шенберга) и др. (см. Болезни костно-мышечной системы).
Анемии вследствие повышенного кроворазрушения (гемолитические анемии)
Гемолитические анемии - большая группа заболеваний крови, при которых процессы кроворазрушения преобладают над процессами кровообразования. Разрушение эритроцитов, или гемолиз, может быть как внутрисосудистым, так и внесосудистым (внутриклеточным). В связи с гемолизом при гемолитических анемиях постоянно встречаются общий гемосидероз и надпеченочная (гемолитическая) желтуха, выраженные в той или иной степени в зависимости от интенсивности гемолиза. В ряде случаев развивается «острый нефроз выделения» продуктов гемолиза - гемоглобинурийный нефроз. Костный мозг реагирует на разрушение эритроцитов гиперплазией и поэтому становится розово-красным, сочным в губчатых костях и красным - в трубчатых. В селезенке, лимфатических узлах, рыхлой соединительной ткани возникают очаги экстрамедуллярного кроветворения.
Гемолитические анемии подразделяют на анемии, обусловленные преимущественно внутрисосудистым или преимущественно внесосудистым (внутриклеточным) гемолизом (Кассирский И.А., Алексеев Г.А., 1970).
Гемолитические анемии, обусловленные преимущественно внутрисосудистым гемолизом. Они возникают от разных причин. К ним относятся гемолитические яды, тяжелые ожоги (токсические анемии), малярия, сепсис (инфекционные анемии), переливание несовместимой по группе и резус-фактору крови (посттрансфузионные анемии). Большую роль в развитии гемолитических анемий играют иммунопатологические процессы (иммунные гемолитические анемии). Среди таких анемий выделяют изоиммунные гемолитические анемии (гемолитическая болезнь новорожденных) и аутоиммунные гемолитические анемии (при хроническом лимфолейкозе, карциноматозе костного мозга, системной красной волчанке, вирусных инфекциях, лечении некоторыми лекарственными препаратами; пароксизмальная холодовая гемоглобинурия).
Гемолитические анемии, обусловленные преимущественно внесосудистым (внутриклеточным) гемолизом. Они носят наследственный (семейный) характер. Распад эритроцитов в этих случаях происходит в макрофагах преимущественно селезенки, в меньшей степени костного мозга, печени и лимфатических узлов. Спленомегалия становится ярким клиникоморфологическим признаком анемии. Гемолизом объясняется раннее появление желтухи, гемосидероза. Таким образом, для этой группы анемий характерна триада - анемия, спленомегалия и желтуха.
Гемолитические анемии, обусловленные преимущественно внутриклеточным гемолизом, делят на эритроцитопатии, эритроцитоферментопатии и гемоглобинопатии (гемоглобинозы).
К эритроцитопатиям относят наследственный микросфероцитоз (микросфероцитарная гемолитическая анемия) и наследственный овалоцитоз, или эллиптоцитоз (наследственная овалоцитарная гемолитическая анемия). В основе этих видов анемии лежит дефект структуры мембраны эритроцитов, что обусловливает их нестойкость и гемолиз.
Эритроцитоферментопатии возникают при нарушении активности ферментов эритроцитов. Дефицит в эритроцитах глюкозо-6- фосфатдегидрогеназы - основного фермента пентозофосфатного пути - характеризуется острыми гемолитическими кризами при вирусных инфекциях, приеме лекарств, употреблении в пищу плодов некоторых бобовых растений (фавизм). Аналогичная картина развивается и при дефиците в эритроцитах ферментов гликолиза (пируваткиназы). В ряде случаев при дефиците глюкозо-6-фосфатдегидрогеназы развивается хроническая гемолитическая анемия.
Гемоглобинопатии, или гемоглобинозы, связаны с нарушением синтеза гемоглобина (α- и β-талассемия) и его цепей, что ведет к появлению аномальных гемоглобинов - S (серповидно-клеточная анемия), С, D, Е и др. Нередко сочетание серповидно-клеточной анемии (рис. 129) с другими формами гемоглобинопатии (гемоглобинозы S-группы). Нару-
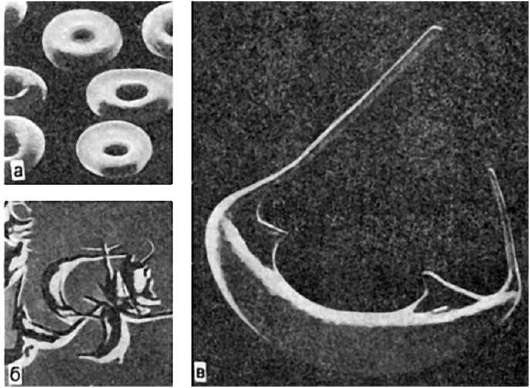 Рис. 129. Серповидно-клеточная анемия (исследование в растровом электронном микроскопе):
Рис. 129. Серповидно-клеточная анемия (исследование в растровом электронном микроскопе):
а - нормальные эритроциты. х5000; б - эритроциты серповидной формы. х1075; в - серповидный эритроцит. х8930 (по Бесси и др.)
шение синтеза гемоглобина, появление аномальных гемоглобинов сопровождаются распадом эритроцитов и развитием гемолитической анемии.
Опухоли системы крови, или гемобластозы
Опухоли системы крови, или гемобластозы, делят на две группы: 1) лейкозы - системные опухолевые заболевания кроветворной ткани; 2) лимфомы - регионарные опухолевые заболевания кроветворной и/или лимфатической ткани.
Классификация опухолей кроветворной и лимфатической ткани I. Лейкозы - системные опухолевые заболевания. A. Острые лейкозы: 1) недифференцированный; 2) миелобластный; 3) лимфобластный; 4) плазмобластный; 5) монобластный (миеломонобластный); 6) эритромиелобластный (ди Гульельмо); 7) мегакариобластный. Б. Хронические лейкозы. Миелоцитарного происхождения: 1) хронический миелоидный; 2) хронический эритромиелоз; 3) эритремия; 4) истинная полицитемия (синдром Вакеза-Ослера). Лимфоцитарного происхождения: 1) хронический лимфолейкоз; 2) лимфоматоз кожи (болезнь Сезари); 3) парапротеинемические лейкозы: а) миеломная болезнь; б) первичная макроглобулинемия (болезнь Вальденстрема); в) болезнь тяжелых цепей (болезнь Франклина).
Моноцитарного происхождения: 1) хронический моноцитарный лейкоз; 2) гистиоцитозы (гистиоцитоз X).
II. Лимфомы - регионарные опухолевые заболевания.
1. Лимфосаркома: лимфоцитарная, пролимфоцитарная, лимфобластная, иммунобластная, лимфоплазмоцитарная, африканская лимфома (опухоль Беркитта).
2. Грибовидный микоз.
3. Болезнь Сезари.
4. Ретикулосаркома.
5. Лимфогранулематоз (болезнь Ходжкина).
Лейкозы - системные опухолевые заболевания кроветворной ткани
Лейкозы (лейкемия) характеризуются системным прогрессирующим разрастанием кроветворных клеток опухолевой природы - лейкозных клеток. Сначала опухолевые клетки разрастаются в органах кроветворения (костный мозг, селезенка, лимфатические узлы), затем гематогенно выселяются в другие органы и ткани, образуя лейкозные (лейкемические) инфильтраты по ходу интерстиция вокруг сосудов, в их стенках; паренхиматозные элементы при этом подвергаются дистрофии, атрофии и погибают. Инфильтрация опухолевыми клетками может быть диффузной (например, лейкозная инфильтрация селезенки, печени, почек, брыжейки), что ведет к резкому увеличению органов и тканей, или очаговой - при образовании опухолевых узлов, прорастающих капсулу органа и окружающие ткани. Обычно опухолевые узлы появляются на фоне диффузной лейкозной инфильтрации, однако они могут возникать первично и быть источником развития диффузной лейкозной инфильтрации.
Для лейкозов весьма характерно появление лейкозных клеток в крови.
Безудержное разрастание лейкозных клеток в органах и тканях, «наводнение» ими крови приводят к анемии и геморрагическому синдрому, тяжелым дистрофическим изменениям паренхиматозных органов. В результате подавления иммунитета при лейкозе развиваются тяжелые язвенно-некротические изменения и осложнения инфекционной природы - сепсис.
Этиология и патогенез. Вопросы этиологии лейкоза и опухолей неразделимы, так как опухолевая природа лейкозов не вызывает сомнений. Лейкозы - полиэтиологические заболевания. В возникновении их могут быть повинны различные факторы, способные вызвать мутацию клеток кроветворной системы.
Среди мутагенов следует назвать вирусы, ионизирующее излучение, ряд химических веществ.
Роль вирусов в развитии лейкоза показана в экспериментах на животных. У человека она доказана для острого эндемического Т-лимфоцитарного лейкоза (ретровирус HTLV-I), волосато-клеточного лейкоза (ретровирус HTLV-II) и для лимфомы Беркитта (ДНК-вирус Эпстайна-Барра).
Известно, что ионизирующее излучение способно вызывать развитие лейкоза (радиационные, или лучевые, лейкозы), причем частота мутаций зависит непосредственно от дозы ионизирующей радиации. После атом-
ного взрыва в Хиросиме и Нагасаки заболеваемость острым лейкозом и хроническим миелозом среди облученных возросла примерно в 7,5 раз.
Среди химических веществ, с помощью которых может быть индуцирован лейкоз, большое значение имеют дибензантрацен, бензпирен, метилхолантрен, т.е. бластомогенные вещества.
Патогенез лейкозов связывают с активацией клеточных онкогенов (протоонкогенов) при воздействии различных этиологических факторов, что ведет к нарушению пролиферации и дифференцировки кроветворных клеток и их злокачественной трансформации. У человека зарегистрировано усиление экспрессии ряда протоонкогенов при лейкозах; ras (1-я хромосома) - при различных лейкозах; sis (22-я хромосома) - при хроническом лейкозе; myc (8-я хромосома) - при лимфоме Беркитта.
Значение наследственных факторов в развитии лейкозов подчеркивается нередко семейным характером заболевания. При изучении кариотипов лейкозных клеток обнаруживаются изменения в наборе их хромосом - хромосомные аберрации. При хроническом миелоидном лейкозе, например, постоянно обнаруживается уменьшение аутосомы 22-й пары хромосом леикозных клеток (Ph'-хромосома, или филадельфийская хромосома). У детей при болезни Дауна, при которой также обнаруживается Ph'-хромосома, лейкоз встречается в 10-15 раз чаще.
Таким образом, мутационная теория патогенеза лейкозов может считаться наиболее вероятной. При этом развитие лейкозов (правда, не всех) подчинено правилам опухолевой прогрессии (Воробьев А.И., 1965). Смена моноклоновости леикозных клеток поликлоновостью лежит в основе появления властных клеток, выселения их из костного мозга и прогрессирования заболевания - бластного криза.
Классификация. Учитывая степень увеличения в крови общего числа лейкоцитов, в том числе и лейкозных клеток, различают лейкемические (десятки и сотни тысяч лейкоцитов в 1 мкл крови), сублейкемические (не более 15 000-25 000 в 1 мкл крови), лейкопенические (число лейкоцитов уменьшено, но лейкозные клетки обнаруживаются) и алейкемические (лейкозные клетки в крови отсутствуют) варианты лейкоза.
В зависимости от степени дифференцировки (зрелости) опухолевых клеток крови и характера течения (злокачественное и доброкачественное) лейкозы делят на острые и хронические.
Для острого лейкоза характерны пролиферация недифференцированных или малодифференцированных, бластных, клеток («бластные» лейкозы) и злокачественность течения, для хронического лейкоза - пролиферация дифференцированных лейкозных клеток («цитарные» лейкозы) и относительная доброкачественность течения.
Руководствуясь гисто(цито)генезом лейкозных клеток, выделяют гисто(цито)генетические формы как острого, так и хронического лейкоза. Гистогенетическая классификация лейкозов в последнее время претерпела значительные изменения в связи с новыми представлениями о кроветворении. Принципиальным отличием новой схемы кроветворения
(Чертков И.Л., Воробьев А.П., 1973) является выделение классов клетокпредшественников разных ростков кроветворения.
Считают, что стволовая лимфоцитоподобная плюрипотентная клетка костного мозга является единственным камбиальным элементом для всех ростков гемопоэза. Ретикулярная клетка потеряла значение «материнской», это не гемопоэтическая, а специализированная стромальная клетка костного мозга. Стволовая кроветворная клетка относится к I классу полипотентных клеток-предшественников. II класс представлен частично детерминированными полипотентными клеткамипредшественниками миело- и лимфопоэза. III класс составляют унипотентные клетки-предшественники В-лимфоцитов, Т-лимфоцитов, лейкопоэза, эритропоэза и тромбоцитопоэза. Клетки-предшественники первых трех классов не имеют морфологических признаков, которые позволили бы отнести их к определенному ростку гемопоэза. IV класс образуют пролиферирующие клетки - прежде всего бласты (миелобласт, лимфобласт, плазмобласт, монобласт, эритробласт, мегакариобласт), которые имеют характерную морфологическую, в том числе и цитохимическую, характеристику (содержание ряда ферментов, гликогена, гликозаминогликанов, липидов). V класс представлен созревающими и VI - зрелыми клетками гемопоэза.
На основании современных представлений о кроветворении среди острых лейкозов выделяют следующие гистогенетические формы: недифференцированный, миелобластный, лимфобластный, монобластный (миеломонобластный), эритромиелобластный и мегакариобластный. Недифференцированный острый лейкоз развивается из клеток-предшественников первых трех классов, лишенных морфологических признаков принадлежности к тому или иному ряду кроветворения. Остальные формы острого лейкоза происходят из клеток-предшественников IV класса, т.е. из клеток-бластов.
Хронические лейкозы в зависимости от ряда созревающих клеток гемопоэза, из которых они возникают, разделяются на: 1) лейкозы миелоцитарного происхождения; 2) лейкозы лимфоцитарного происхождения; 3) лейкозы моноцитарного происхождения. К хроническим лейкозам миелоцитарного происхождения относят: хронический миелоидный лейкоз, хронический эритромиелоз, эритремию, истинную полицитемию. К хроническим лейкозам лимфоцитарного ряда относятся: хронический лимфолейкоз, лимфоматоз кожи (болезнь Сезари) и парапротеинемические лейкозы (миеломная болезнь; первичная макроглобулинемия Вальденстрема; болезнь тяжелых цепей Франклина). К хроническим лейкозам моноцитарного происхождения причисляют моноцитарный (миеломоноцитарный) лейкоз и гистиоцитозы (гистиоцитоз X) (см. классификацию опухолей кроветворной и лимфатической тканей).
Патологическая анатомия имеет определенное своеобразие, касающееся как острых, так и хронических лейкозов, имеется и определенная специфика их многообразных форм.
Острые лейкозы
Диагноз острого лейкоза ставят на основании обнаружения в костном мозге (пунктат из грудины) бластных клеток. Иногда их количество мо-
жет составлять 10-20%, но тогда в трепанате подвздошной кости обнаруживают скопление из многих десятков бластов. При остром лейкозе как в периферической крови, так и в миелограмме находят так называемый лейкемический провал (hiatus leucemicus) - резкое повышение числа бластов и единичные зрелые элементы при отсутствии переходных созревающих форм.
Острые лейкозы характеризуются замещением костного мозга молодыми властными элементами и инфильтрацией ими селезенки, печени, лимфатических узлов, почек, головного мозга, его оболочек, других органов, степень которой различна при разных формах лейкоза. Форма острого лейкоза устанавливается на основании цитохимических особенностей бластных клеток (табл. 11). При лечении острого лейкоза цитостатическими средствами нередко развиваются аплазия костного мозга и панцитопения.
Острые лейкозы у детей имеют некоторые особенности. По сравнению с острыми лейкозами у взрослых они встречаются значительно чаще и характеризуются более широким распространением лейкозной инфильтрации как в кроветворных, так и в некроветворных органах (за исключением половых желез). У детей чаще, чем у взрослых, наблюдаются лейкозы с узловатыми (опухолевидными) инфильтратами, особенно в области вилочковой железы. Чаще встречается острый лимфобластный (Т-зависимый) лейкоз; миелобластный лейкоз, как и другие формы острого лейкоза, обнаруживается реже. Особыми формами острого лейкоза у детей являются врожденный лейкоз и хлоролейкоз.
Острый недифференцированный лейкоз. Он характеризуется инфильтрацией костного мозга (рис. 130), селезенки, лимфатических узлов и лимфоидных образований (миндалины, групповые лимфатические и солитарные фолликулы), слизистых оболочек, стенок сосудов, миокарда, почек, головного мозга, мозговых оболочек и других органов однородного вида недифференцированными клетками гемопоэза. Гистологическая картина этой лейкозной инфильтрации очень однообразна. Селезенка и печень увеличиваются, но незначительно. Костный мозг плоских и трубчатых костей красный, сочный, иногда с сероватым оттенком. В связи с лейкозной инфильтрацией слизистой оболочки полости рта и ткани миндалин появляются некротический гингивит, тонзиллит - некротическая ангина. Иногда присоединяется вторичная инфекция, и недифференцированный острый лейкоз протекает как септическое заболевание.
Лейкемическая инфильтрация органов и тканей сочетается с явлениями геморрагического синдрома, развитие которого объясняется не только разрушением лейкозными клетками стенок сосудов, но и анемией, нарушением тромбоцитообразования в результате замещения костного мозга недифференцированными клетками гемопоэза. Кровоизлияния различного характера возникают в коже, слизистых оболочках, внутренних органах, довольно часто в головном мозге (см. рис. 130). Больные умирают от кровоизлияния в мозг, желудочно-кишечных кровотечений, язвеннонекротических осложнений, сепсиса.
Таблица 11. Цитохимическая характеристика различных форм лейкоза
Форма острого лейкоза | Реакции на питательные вещества | Реакции на ферменты | |||||
гликоген (ШИКреакция) | гликозаминогликаны | липиды (черный Судан) | пероксидаза | кислая фосфатаза | а-нафтилэстераза | хлорацетатэстераза | |
Недифференцированный | Отрицательная | Отрицательная | Отрицательная | Отрицательная | Отрицательная | Отрицательная | Отрицательная |
Миелобластный | Положительная | То же | Положительная | Положительная | Положительная | Слабоположительная | Положительная |
Промиелоцитарный | Резко положительная | Положительная | То же | Резко положительная | Слабоположительная | То же | Резко положительная |
Лимфобластный | Положительная в виде глыбок | Отрицательная | Отрицательная | Отрицательная | Иногда положительная | Отрицательная | Отрицательная |
Монобластный | Слабоположительная | То же | Слабоположительная | Слабоположительная | Высокоположительная | Положительная | То же |
Миеломонобластный | Положительная диффузная | » » | То же | Высокоположительная | Положительная | То же | Слабоположительная |
Эритромиелобластный | Положительная | » » | Реакции зависят от принадлежности бластных элементов к тому или иному ряду (миелобласты, монобласты, недифференцированные бласты) | ||||
Плазмобластный | Выделяется по характерной морфологии клеток и наличию парапротеина в сыворотке крови | ||||||
Мегакариобластный | Выделяется по характерной морфологии клеток | ||||||
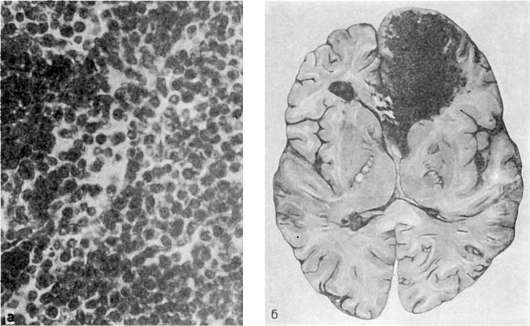 Рис. 130. Острый лейкоз:
Рис. 130. Острый лейкоз:
а - костный мозг, состоящий из однородных недифференцированных клеток; б - кровоизлияние в лобной доле головного мозга
Разновидностью недифференцированного острого лейкоза является хлоролейкоз, который встречается нередко у детей (обычно мальчиков до 2-3 лет). Хлоролейкоз проявляется опухолевыми разрастаниями в костях лицевого черепа, реже - в других костях скелета и совсем редко - во внутренних органах (печень, селезенка, почки). Опухолевые узлы имеют зеленоватый цвет, что послужило основанием для такого названия этого вида лейкоза. Окраска опухоли связана с присутствием в ней продуктов синтеза гемоглобина - протопорфиринов. Узлы опухоли состоят из атипичных недифференцированных клеток миелоидного ростка.
Острый миелобластный лейкоз (острый миелолейкоз). Эта форма острого лейкоза проявляется инфильтрацией костного мозга, селезенки, печени, почек, слизистых оболочек, реже лимфатических узлов и кожи опухолевыми клетками типа миелобластов. Эти клетки имеют ряд цитохимических особенностей (см. табл. 11): содержат гликоген и суданофильные включения, дают положительную реакцию на пероксидазу, α-нафтилэстеразу и хлорацетатэстеразу.
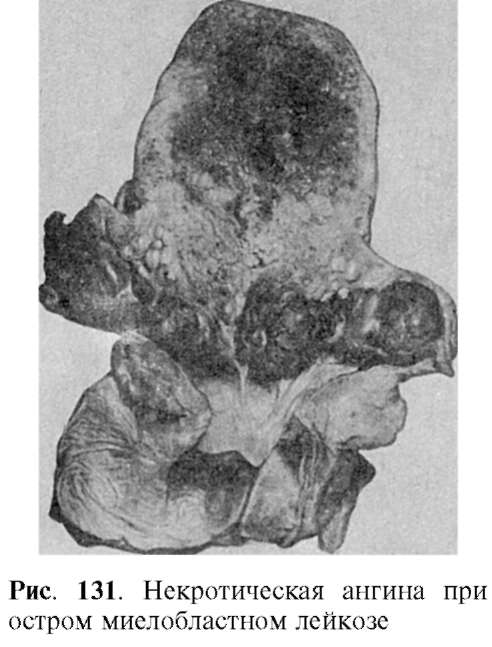
Костный мозг становится красным или сероватым, иногда он приобретает зеленоватый (гноевидный) оттенок (пиоидный костный мозг). Селезенка и печень в результате лейкозной инфильтрации увеличиваются, но больших размеров не достигают. То же можно сказать и о лимфатических узлах. Весьма характерна инфильтрация бластными клетками не только костного мозга, селезенки и печени, но и слизистой оболочки желудочнокишечного тракта, в связи с чем возникают некрозы в полости рта, миндалинах, зеве (рис. 131), желудке. В почках встречаются как диффузные,
так и очаговые (опухолевые) инфильтраты. В 1/3 случаев развивается лейкемическая инфильтрация легких («лейкозный пневмонит»), в 1/4 случаев - лейкозная инфильтрация оболочек мозга («лейкозный менингит»). Резко выражены явления геморрагического диатеза. Кровоизлияния наблюдаются в слизистых и серозных оболочках, в паренхиме внутренних органов, нередко в головном мозге. Умирают больные от кровотечений, язвеннонекротических процессов, присоединившейся инфекции, сепсиса.
В последние годы активная терапия (цитостатические средства, Υ-облучение, антибиотики, антифи-
бринолитические препараты) суще- ственно изменила картину острых
недифференцированного и миелобластного лейкозов. Исчезли обширные некрозы в полости рта и зеве, стали менее выраженными явления геморрагического диатеза. Вместе с тем в результате увеличения продолжительности жизни больных острым лейкозом чаще стали встречаться такие внекостно-мозговые поражения, как «лейкозный пневмонит», «лейкозный менингит» и т.д. В связи с терапией цитостатическими средствами участились случаи язвенно-некротического поражения желудка и кишечника.
Острый промиелоцитарный лейкоз. Его отличают злокачественность, быстрота течения и выраженность геморрагического синдрома (тромбоцитопения и гипофибриногенемия). Для лейкозных клеток, инфильтрирующих органы и ткани, характерны следующие морфологические особенности: ядерный и клеточный полиморфизм, наличие в цитоплазме псевдоподий и гранул гликозаминогликанов (см. табл. 11). Почти все больные этой формой острого лейкоза погибают от кровоизлияния в мозг или желудочно-кишечных кровотечений.
Острый лимфобластный лейкоз. Встречается значительно чаще у детей (в 80% случаев), чем у взрослых. Лейкемическая инфильтрация выражена наиболее резко в костном мозге, селезенке, лимфатических узлах, лимфатическом аппарате желудочно-кишечного тракта, почках и вилочковой железе. Костный мозг губчатых и трубчатых костей малиновокрасный, сочный. Селезенка резко увеличивается, становится сочной и красной, рисунок ее стерт. Значительно увеличиваются и лимфатические узлы (средостения, брыжеечные), на разрезе ткань их бело-розовая, сочная. Такой же вид имеет и вилочковая железа, которая достигает ино-
 гда
гигантских размеров. Нередко лейкозный инфильтрат выходит за пределы
вилочковой железы и прорастает ткани переднего средостения, сдавливая
органы грудной полости (рис. 132).
гда
гигантских размеров. Нередко лейкозный инфильтрат выходит за пределы
вилочковой железы и прорастает ткани переднего средостения, сдавливая
органы грудной полости (рис. 132).
Лейкозные инфильтраты при этой форме лейкоза состоят из лимфобластов, характерной цитохимической особенностью которых является наличие вокруг ядра гликогена (см. табл. 11). Лимфобласты относятся к Т-системе лимфопоэза, чем можно объяснить как быстрое расселение бластов в Т-зависимых зонах лимфатических узлов и селезенки, так и увеличение их размеров одновременно с лейкозной инфильтрацией костного мозга. Выражением прогрессии лейкоза следует считать лимфобластные инфильтраты метастатической природы, появляющиеся за пределами лимфатической ткани. Особенно часто такие инфильтраты встречаются в оболочках и веществе головного и спинного мозга, что называют нейролейкозом.
Острый лимфобластный лейкоз хорошо поддается лечению цитостатическими средствами. У 90% детей удается получить стойкую, нередко длительную (5-10 лет) ремиссию. Без терапии течение этой формы, как и других форм острого лейкоза, прогрессирует: нарастает анемия, развивается геморрагический синдром, появляются осложнения инфекционной природы, и т.д.
Острый плазмобластный лейкоз. Эта форма острого лейкоза возникает из клеток-предшественников В-лимфоцитов, способных к продукции иммуноглобулинов. Эту способность сохраняют и опухолевые плазмобласты. Они секретируют патологические иммуноглобулины - парапротеины, поэтому острый плазмобластный лейкоз относится к группе парапротеинемических гемобластозов. Плазмобластную лейкозную инфильтрацию находят в костном мозге, селезенке, лимфатических узлах, печени, коже и других органах. Большое число плазмобластов обнаруживается и в крови.
Острый монобластный (миеломонобластный) лейкоз. Он мало чем отличается от острого миелобластного лейкоза.
Острый эритромиелобластный лейкоз (острый эритромиелоз ди Гульельмо). Это редкая форма (1-3% всех острых лейкозов), при которой в костном мозге происходит разрастание как эритробластов и других ядросодержащих клеток эритропоэза, так и миелобластов, монобластов
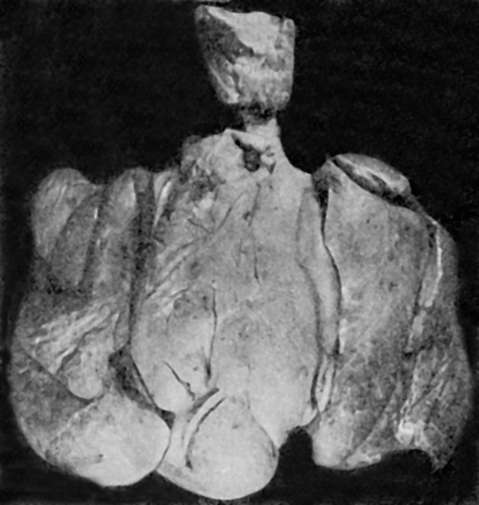 Рис. 132. Опухолевые разрастания в области вилочковой железы при остром лимфобластном лейкозе
Рис. 132. Опухолевые разрастания в области вилочковой железы при остром лимфобластном лейкозе
и недифференцированных бластов. В результате угнетения кроветворения возникают анемия, лейко- и тромбоцитопения. Селезенка и печень увеличиваются.
Острый мегакариобластный лейкоз. Одна из наиболее редких форм острого лейкоза, для которой характерно присутствие в крови и костном мозге наряду с недифференцированными бластами также мегакариобластов, уродливых мегакариоцитов и скоплений тромбоцитов. Число тромбоцитов в крови увеличивается до 1000-1500х109/л.
Врожденный лейкоз, выявляющийся в течение первого месяца после рождения, представляет исключительную редкость. Обычно он встречается в форме миелобластного лейкоза, течет крайне быстро, со сплено- и гепатомегалией, увеличением лимфатических узлов, выраженной диффузной и узловатой лейкозной инфильтрацией многих органов (печень, поджелудочная железа, желудок, почки, кожа, серозные оболочки). Выраженная лейкозная инфильтрация по ходу пупочной вены и портальным трактам печени свидетельствует о гематогенном распространении процесса от матери к плоду, хотя матери больных врожденным лейкозом детей редко страдают лейкозом. Обычно дети умирают от проявлений геморрагического синдрома.
Хронические лейкозы
Хронические лейкозы миелоцитарного происхождения
Эти лейкозы разнообразны, однако основное место среди них занимают хронический миелоидный лейкоз, хронический эритромиелоз, эритремия и истинная полицитемия.
Хронический миелоидный лейкоз (хронический миелоз). Этот лейкоз проходит две стадии: моноклоновую доброкачественную и поликлоновую злокачественную. Первая стадия, которая занимает несколько лет, характеризуется нарастающим нейтрофильным лейкоцитозом со сдвигом до миелоцитов и промиелоцитов, увеличением селезенки. Клетки костного мозга в этой стадии лейкоза морфологически и по способности к фагоцитозу не отличаются от нормальных, однако они содержат так называемую Ph-хромосому (филадельфийскую), возникающую в результате делеции хромосом 22-й пары. Во второй стадии, которая длится от 3 до 6 мес (терминальная стадия), моноклоновость сменяется поликлоновостью. В результате этого появляются бластные формы (миелобласты, реже эритробласты, монобласты и недифференцированные бластные клетки), число которых нарастает как в костном мозге, так и в крови (бластный криз). Отмечаются быстрый рост числа лейкоцитов в крови (до нескольких миллионов в 1 мкл), увеличение селезенки, печени, лимфатических узлов, лейкозная инфильтрация кожи, нервных стволов, мозговых оболочек, появляется тромбоцитопения, развивается геморрагический синдром.
При вскрытии умерших от хронического миелоидного лейкоза в терминальной стадии особенно выраженные изменения находят в костном мозге, крови, селезенке, печени, лимфатических узлах. Костный мозг плоских костей, эпифизов и диафизов трубчатых костей сочный, серокрасный или серо-желтый гноевидный! (пиоидный костный мозг). При
гистологическом исследовании костного мозга обнаруживаются промиелоциты и миелоциты, а также бластные клетки. Встречаются клетки с изменениями ядер (уродливые ядра) и цитоплазмы, явлениями пикноза или кариолиза. В костной ткани иногда отмечаются признаки реактивного остеосклероза. Кровь серо-красная, органы малокровны.
Селезенка резко увеличена (рис. 133), иногда занимает почти всю брюшную полость; масса ее достигает 6-8 кг. На разрезе она темнокрасного цвета, иногда обнаруживаются ишемические инфаркты. Ткань селезенки вытесняет лейкозный инфильтрат в основном из клеток миелоидного ряда, среди которых видны бласты; фолликулы атрофичны. Нередко находят склероз и гемосидероз пульпы. В сосудах встречаются лейкозные тромбы.
Печень значительно увеличена (ее масса достигает 5-6 кг). Поверхность ее гладкая, ткань на разрезе серо-коричневая. Лейкозная инфильтрация обычно наблюдается по ходу синусоидов, значительно реже она видна в портальных трактах и капсуле. Гепатоциты в состоянии жировой дистрофии; иногда отмечается гемосидероз печени.
Лимфатические узлы увеличены значительно, мягкие, серо-красного цвета. В той или иной степени выражена лейкозная инфильтрация их ткани; она наблюдается также в миндалинах, групповых и солитарных лимфа-
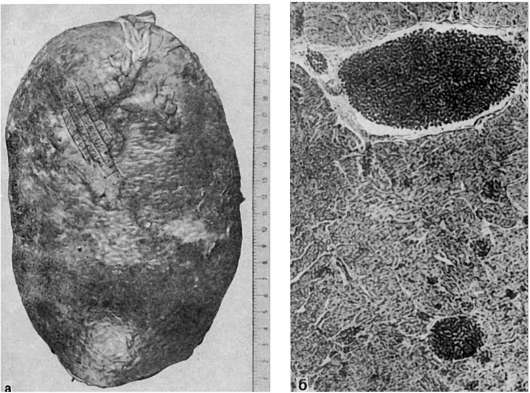 Рис. 133. Хронический миелоидный лейкоз:
Рис. 133. Хронический миелоидный лейкоз:
а - увеличение селезенки (масса 2800 г); б - лейкозные стазы и тромбы в сосудах сердца
тических фолликулах кишечника, почках, коже, иногда головном мозге и его оболочках (нейролейкемия). Большое число лейкозных клеток появляется в просвете сосудов, они образуют лейкозные стазы и тромбы (см. рис. 133) и инфильтрируют сосудистую стенку. В связи с этими изменениями сосудов нередки как инфаркты, так и геморрагии. Довольно часто при хроническом миелоидном лейкозе находят проявления аутоинфекции.
Родственную хроническому миелолейкозу группу составляют остеомиелолейкоз и миелофиброз, при которых наряду с признаками миелоидного лейкоза отмечается замещение костного мозга костной или соединительной тканью. Процесс характеризуется длительным доброкачественным течением.
Терапия цитостатическими средствами ведет к изменениям морфологии хронического миелолейкоза. Наряду с подавлением очагов лейкозной инфильтрации и развитием на их месте фиброза отмечаются омоложение клеточных форм, появление метастатических очагов и опухолевых разрастаний либо аплазия костного мозга и панцитопения.
Хронический эритромиелоз - редкая форма лейкоза. Это опухоль красного и белого ростков кроветворной ткани, при которой в костном мозге, селезенке и печени разрастаются эритрокариоциты, миелоциты, промиелоциты и бласты. Большое число этих клеток обнаруживается и в крови. Отмечается резко выраженная спленомегалия. В ряде случаев присоединяется миелофиброз (форма Вагана хронического эритромиелоза).
Эритремия. Встречается обычно у пожилых и характеризуется увеличением массы эритроцитов в кровяном русле, плеторой. Увеличивается также число тромбоцитов и гранулоцитов, появляются артериальная гипертония, склонность к тромбозам, спленомегалия. В костном мозге происходит разрастание всех ростков, но преимущественно эритроцитарного. Процесс долго течет доброкачественно, но обычно заканчивается трансформацией в хронический миелолейкоз с появлением очагов лейкозной инфильтрации в органах.
Патологоанатомическая картина эритремии достаточно характерна. Все органы резко полнокровны, часто в артериях и венах образуются тромбы. Жировой костный мозг трубчатых костей становится красным. Резко увеличивается селезенка. Возникает гипертрофия миокарда, особенно левого желудочка. В костном мозге, селезенке и печени в ранней стадии эритремии обнаруживаются очаги экстрамедуллярного кроветворения с большим числом мегакариоцитов, а в поздней стадии, при трансформации процесса в миелоидный лейкоз, - фокусы лейкозной инфильтрации.
Истинная полицитемия (болезнь Вакеза-Ослера) близка эритремии. Существует также хронический мегакариоцитарный лейкоз, который встречается исключительно редко.
Хронические лейкозы лимфоцитарного происхождения
Эти формы разделяются на две группы: первую составляют хронический лимфолейкоз и примыкающий к нему лимфоматоз кожи (болезнь Сезари), вторую - парапротеинемические лейкозы.
Хронический лимфолейкоз. Встречается обычно у лиц среднего и пожилого возраста, в ряде случаев у членов одной семьи, развивается из В-лимфоцитов и отличается длительным доброкачественным течением. Содержание лейкоцитов в крови резко увеличивается (до 100х109/л), среди них преобладают лимфоциты. Лейкозные инфильтраты из опухолевых лимфоцитов наиболее выражены в костном мозге, лимфатических узлах, селезенке, печени, что ведет к увеличению этих органов. Опухолевые В-лимфоциты вырабатывают крайне мало иммуноглобулинов. В связи с этим гуморальный иммунитет при хроническом лимфолейкозе резко угнетен, у больных часто возникают осложнения инфекционной природы. Для этой формы лейкоза характерно развитие и аутоиммунных реакций, особенно аутоиммунных гемолитических и тромбоцитопенических состояний.
На фоне доброкачественного течения хронического лимфолейкоза возможны бластный криз и генерализация процесса, что приводит в ряде случаев к летальному исходу. Однако чаще больные умирают от инфекции и осложнений аутоиммунного характера.
На вскрытии основные изменения находят в костном мозге, лимфатических узлах, селезенке, печени и почках.
Костный мозг плоских и трубчатых костей красного цвета, но в отличие от миелоидного лейкоза в диафизах трубчатых костей среди красного костного мозга встречаются участки желтого цвета. При гистологическом исследовании в ткани костного мозга обнаруживаются очаги разрастания опухолевых клеток (рис. 134). В крайних случаях вся миелоидная ткань
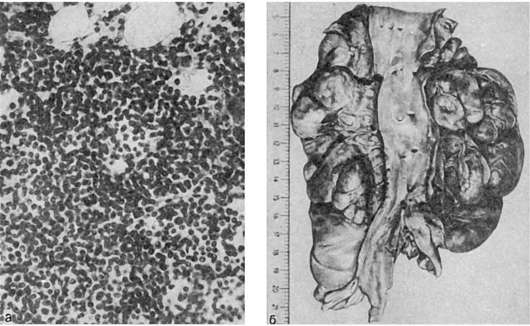 Рис. 134. Хронический лимфолейкоз:
Рис. 134. Хронический лимфолейкоз:
а - костный мозг, опухолевые лимфоциты; б - пакеты увеличенных лимфатических узлов вдоль аорты
костного мозга вытесняется лейкозным лимфоцитарным инфильтратом и остаются сохранными лишь небольшие островки миелоидного кроветворения.
Лимфатические узлы всех областей тела резко увеличены, сливаются в огромные мягкие или плотноватые пакеты (см. рис. 134). На разрезе они сочные, бело-розовые. Увеличиваются размеры миндалин, групповых и солитарных лимфатических фолликулов кишечника, которые также представляют собой сочную бело-розовую ткань. Увеличение лимфатических узлов и лимфатических образований связано с лейкозной их инфильтрацией, которая ведет к резкому нарушению структуры этих органов и тканей; нередко лимфоциты инфильтрируют капсулу лимфатических узлов и окружающие их ткани.
Селезенка достигает значительных размеров, масса ее увеличивается (до 1 кг). Она мясистой консистенции, красного цвета на разрезе; фолликулы сохранены или теряются в пульпе. Лейкозный лимфоцитарный инфильтрат охватывает прежде всего фолликулы, которые становятся крупными и сливаются. Затем лимфоциты разрастаются в красной пульпе, стенках сосудов, трабекулах и капсуле селезенки.
Печень увеличена, плотновата, на разрезе светло-коричневая. Нередко с поверхности и на разрезе видны мелкие серо-белые узелки. Лимфоцитарная инфильтрация происходит главным образом по ходу портальных трактов (рис. 135). Гепатоциты в состоянии белковой или жировой дистрофии.
Почки увеличены, плотноваты, серо-коричневого цвета. Лейкозная инфильтрация их бывает столь резко выражена, что структура почек на разрезе не выявляется.
Лейкемическая инфильтрация отмечается также во многих органах и тканях (средостение, брыжейка, миокард, серозные и слизистые оболочки), причем она бывает не только диффузной, но и очаговой с образованием различных размеров узлов.
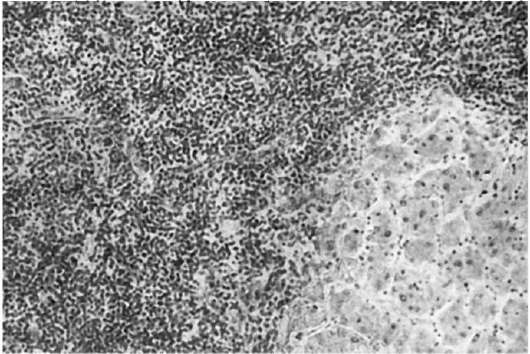 Рис. 135. Лейкозная инфильтрация портальных трактов печени при хроническом лимфолейкозе
Рис. 135. Лейкозная инфильтрация портальных трактов печени при хроническом лимфолейкозе
Описанные изменения, свойственные хроническому лимфолейкозу, дополняются инфекционными осложнениями, например пневмонией, и проявлениями гемолитических состояний - гемолитической желтухой, диапедезными кровоизлияниями, общим гемосидерозом.
Следует иметь в виду, что, помимо генерализованного поражения лимфатических узлов, умеренного увеличения селезенки и печени при хроническом лимфолейкозе, встречаются случаи резкого увеличения лишь определенных групп лимфатических узлов (например, средостения, брыжеечных, шейных, паховых). В таких случаях возникает опасность сдавления соседних органов (например, сдавление сердца, пищевода, трахеи при поражении лимфатических узлов средостения; сдавление воротной вены и ее разветвлений с развитием портальной гипертензии и асцита при поражении лимфатических узлов брыжейки и ворот печени).
Лимфоматоз кожи, или болезнь Сезари. Это своеобразная форма хронического лимфолейкоза, которая характеризуется инфильтрацией опухолевыми Т-лимфоцитами прежде всего кожи. Со временем в процесс вовлекается костный мозг, в крови увеличивается содержание лейкоцитов, появляются характерные клетки (клетки Сезари), увеличиваются периферические лимфатические узлы, селезенка.
Парапротеинемические лейкозы. Эта группа объединяет опухоли, исходящие из клеток В-лимфоцитарной системы (предшественники плазматических клеток), с функцией которых, как известно, связаны реакции гуморального иммунитета. Главной особенностью парапротеинемических лейкозов, которые называют также злокачественными иммунопролиферативными заболеваниями, является способность опухолевых клеток синтезировать однородные иммуноглобулины или их фрагменты - парапротеины (P/g-патологические, или моноклоновые, иммуноглобулины). Патология иммуноглобулинов определяет как клиническое, так и морфологическое своеобразие парапротеинемических лейкозов, к которым относят миеломную болезнь, первичную макроглобулинемию (Вальденстрема) и болезнь тяжелых цепей (Франклина).
Наибольшее значение среди парапротеинемических лейкозов имеет миеломная болезнь.
Миеломная болезнь - довольно распространенное заболевание, описанное впервые О.А. Рустицким (1873) и Калером (1887). В основе заболевания лежит разрастание опухолевых клеток лимфоплазмоцитарного ряда - миеломных клеток (рис. 136) как в костном мозге, так и вне его. Миеломатоз костного мозга ведет к разрушению костей.
В зависимости от характера миеломных клеток различают плазмоцитарную, плазмобластную, полиморфно-клеточную и мелкоклеточную миеломы (Струков А.И., 1959). Полиморфно-клеточную и мелкоклеточную миеломы относят к низкодифференцированным опухолям. Миеломные клетки секретируют парапротеины, которые обнаруживаются в крови и моче больных, а также в самих миеломных клетках. В связи с тем что при миеломной болезни в сыворотке крови и в моче биохимически обнару-
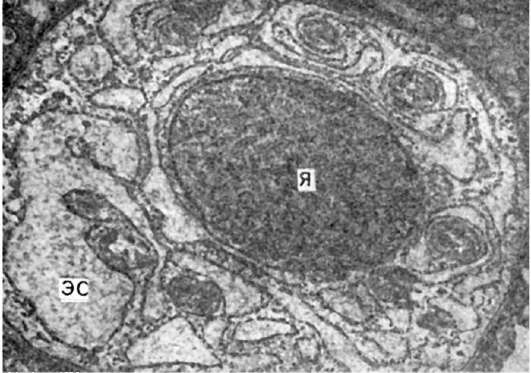 Рис. 136. Миеломная клетка. Резко расширенные канальцы эндоплазматической сети (ЭС) заполнены скоплениями белка - парапротеина.
Рис. 136. Миеломная клетка. Резко расширенные канальцы эндоплазматической сети (ЭС) заполнены скоплениями белка - парапротеина.
Я - ядро. Электронограмма. х23 000.
живают различные виды патологических иммуноглобулинов, различают несколько биохимических вариантов миеломы (А-, D-, Е-миелома, миелома Бенс-Джонса). Обнаруживаемый в моче белок Бенс-Джонса является одним из видов парапротеина, секретируемого миеломной клеткой, он свободно проходит клубочковый фильтр почек, так как обладает крайне малой молекулярной массой.
Обычно миелома протекает по алейкемическому варианту, но возможно и наличие в крови миеломных клеток.
Морфологически в зависимости от характера миеломных инфильтратов, которые обычно локализуются в костном мозге и костях, различают диффузную, диффузно-узловую и множественно-узловую формы миеломной болезни.
О диффузной форме говорят тогда, когда диффузная миеломная инфильтрация костного мозга сочетается с остеопорозом. При диффузноузловой форме на фоне диффузного миеломатоза костного мозга появляются опухолевые узлы; при множественно-узловой форме диффузная миеломная инфильтрация отсутствует.
Разрастание миеломных клеток отмечается чаще в плоских костях (ребра, кости черепа) и позвоночнике, реже - в трубчатых костях (плечевая, бедренная кость). Оно ведет к деструкции костной ткани (рис. 137).
В участках разрастания миеломных клеток в просвете центрального канала остеона или в костной балке под эндостом костное вещество становится мелкозернистым, затем разжижается, в нем появляются остеокласты и эндост отслаивается. Постепенно вся костная балка превращается в так называемую жидкую кость и полностью рассасывается, каналы остеонов становятся широкими. Развивается «пазушное рассасывание» кости, которое объясняет характерный для миеломной болезни остеолизис и остеопороз - образование гладкостенных, как бы штампованных дефектов при отсутствии или очень слабовыраженном костеобразовании. Кости становятся
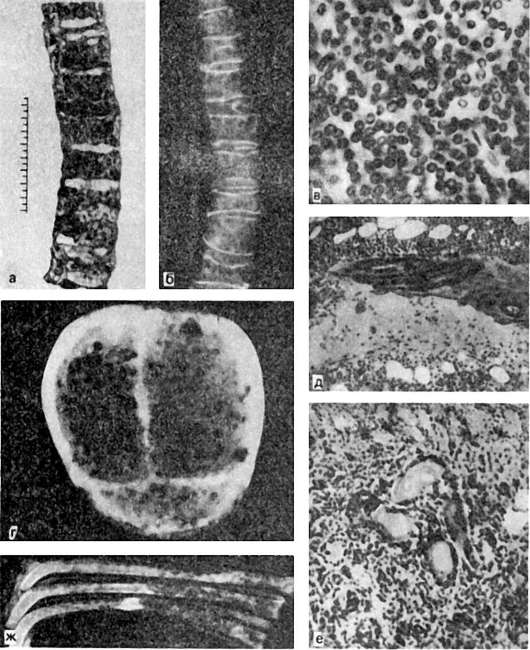 Рис. 137. Миеломная болезнь:
Рис. 137. Миеломная болезнь:
а - позвоночник на распиле - кровоизлияния в межпозвоночные диски; б - рентгенограмма того же позвоночника: остеопороз; в - гистологическая картина: инфильтрация миеломными клетками; г - кости черепа с множественными, как бы штампованными дефектами костного вещества; д - пазушное рассасывание костной балки; е - парапротеинемический нефроз, скопление белковых масс в просвете канальцев почки; ж - миеломатоз ребер
ломкими, чем объясняются частые переломы при миеломной болезни. В связи с разрушением костей при миеломе развивается гиперкальциемия, с которой связано частое развитие известковых метастазов.
Помимо костного мозга и костей, миеломноклеточная инфильтрация почти постоянно отмечается во внутренних органах: селезенке, лимфатических узлах, печени, почках, легких и др.
Ряд изменений при миеломной болезни связан с секрецией опухолевыми клетками парапротеина. К ним относятся: 1) амилоидоз (AL- амилоидоз); 2) отложение в тканях амилоидоподобных и кристаллических веществ; 3) развитие парапротеинемического отека, или парапротеиноза органов (парапротеиноз миокарда, легких, парапротеинемический нефроз), что сопровождается их функциональной недостаточностью. Наибольшее значение среди парапротеинемических изменений имеет парапротеинемический нефроз, или миеломная нефропатия, которая является причиной смерти 1/3 больных миеломой. В основе парапротеинемического нефроза лежит «засорение» почек парапротеином Бенс-Джонса (см. рис. 137), ведущее к склерозу мозгового, а затем коркового вещества и сморщиванию почек (миеломные сморщенные почки). В ряде случаев парапротеинемический нефроз сочетается с амилоидозом почек.
При миеломной болезни в связи с накоплением парапротеинов в крови, белковыми стазами в сосудах развиваются своеобразный синдром повышенной вязкости и парапротеинемическая кома.
В связи с иммунологической беззащитностью при плазмоцитоме нередки воспалительные изменения (пневмония, пиелонефрит), которые возникают на фоне тканевого парапротеиноза и являются выражением аутоинфекции.
Первичная макроглобулинемия - редкое заболевание, которое впервые описано Вальденстремом в 1944 г. Это одна из разновидностей хронических лейкозов лимфоцитарного происхождения, при которой опухолевые клетки секретируют патологический макроглобулин - IgM. Для заболевания характерно увеличение селезенки, печени, лимфатических узлов, что связано с лейозной инфильтрацией их. Деструкция костей встречается редко. Весьма типичен геморрагический синдром, развивающийся в связи с гиперпротеинемией, резким повышением вязкости крови, функциональной неполноценностью тромбоцитов, замедлением кровотока и стазами в мелких сосудах. Наиболее частыми осложнениями являются геморрагии, парапротеинемическая ретинопатия, парапротеинемическая кома; возможен амилоидоз.
Болезнь тяжелых цепей описана Франклином в 1963 г. Опухолевые клетки лимфоплазмоцитарного ряда продуцируют при этом заболевании своеобразный парапротеин, соответствующий Fc-фрагменту тяжелой цепи IgG (отсюда и название болезни). Как правило, наблюдается увеличение лимфатических узлов, печени, селезенки в результате инфильтрации этих органов опухолевыми клетками. Изменения костей отсутствуют, поражение костного мозга не является правилом. Больные умирают
от присоединившейся инфекции (сепсис) в связи с гипогаммаглобулинемией (иммунодефицитное состояние).
Хронические лейкозы моноцитарного происхождения
К этим лейкозам относят хронический моноцитарный лейкоз и гистиоцитозы.
Хронический моноцитарный лейкоз возникает обычно у людей пожилого возраста, протекает длительно и доброкачественно, иногда с увеличением селезенки, но без нарушения костномозгового кроветворения. Однако заканчивается этот лейкоз обычно бластным кризом с разрастанием бластных клеток в костном мозге, появлением их в крови и внутренних органах.
Гистиоцитозы (гистиоцитоз X) объединяют группу так называемых пограничных лимфопролиферативных заболеваний кроветворной ткани. К ней относят эозинофильную гранулему, болезнь Леттерера-Зиве, болезнь Хенда-Шюллера-Крисчена.
Лимфомы - регионарные опухолевые заболевания кроветворной и лимфатической ткани
В эту группу заболеваний входят лимфосаркома, грибовидный микоз, болезнь Сезари, ретикулосаркома, лимфогранулематоз (болезнь Ходжкина).
Лимфомы могут быть В-клеточного и Т-клеточного происхождения. На этом основана классификация лимфом, предложенная Люкез и Коллинз. Согласно этой классификации, В-клеточные лимфомы могут быть: мелкоклеточными (В), центроцитарными, иммунобластными (В), плазмолимфоцитарными, а Т-клеточные лимфомы - мелкоклеточными (Т), из лимфоцитов с перекрученными ядрами, иммунобластными (Т), а также представлены грибовидным микозом и болезнью Сезари. Кроме того, выделяют неклассифицируемые лимфомы. Из этой классификации следует, что мелкоклеточные и иммунобластные лимфомы могут происходить как из В-, так и из Т-клеток. Только из В-клеток развиваются центроцитарная и плазмолимфоцитарная лимфомы и только из Т-клеток - лимфома из лимфоцитов с перекрученными ядрами, грибовидный микоз и болезнь Сезари.
Этиология и патогенез. Лимфомы не имеют каких-либо особенностей по сравнению с лейкозами. Следует подчеркнуть, что в условиях современной терапии цитостатическими средствами некоторые лимфомы (лимфосаркома) нередко «завершают» терминальную стадию лейкоза. Вместе с тем сами они способны «трансформироваться» в лейкоз. Из этого следует, что разграничение опухолей системы крови на «диффузные» и «регионарные», необходимое в интересах нозологии, с позиций онкогенеза весьма условно.
Патологическая анатомия. Каждая из лимфом имеет характерную морфологическую картину.
Лимфосаркома - злокачественная опухоль, возникающая из клеток лимфоцитарного ряда. При этой опухоли поражаются лимфатические
узлы, причем чаще - медиастинальные и забрюшинные, реже - паховые и подмышечные. Возможно развитие опухоли в лимфатической ткани желудочно-кишечного тракта, селезенке и других органах. Вначале опухоль носит локальный, ограниченный характер. Лимфатические узлы резко увеличиваются, спаиваются между собой и образуют пакеты, которые сдавливают окружающие ткани. Узлы плотные, на разрезе серо-розовые, с участками некроза и кровоизлияний. В дальнейшем происходит генерализация процесса, т.е. лимфогенное и гематогенное метастазирование с образованием множественных отсевов в лимфатических узлах, легких, коже, костях и других органах. В лимфатических узлах разрастаются опухолевые клетки типа В- или Т-лимфоцитов, пролимфоцитов, лимфобластов, иммунобластов.
На этом основании различают следующие гисто(цито)логические варианты лимфом: лимфоцитарную, пролимфоцитарную, лимфобластную, иммунобластную, лимфоплазмоцитарную, африканскую лимфому (опухоль Беркитта). Опухоли, состоящие из зрелых лимфоцитов и пролимфоцитов, называют лимфоцитомами, из лимфобластов и иммунобластов - лимфосаркомами (Воробьев А.И., 1985).
Среди лимфосарком особого внимания заслуживает африканская лимфома, или опухоль Беркитта.
Опухоль Беркитта - эндемическое заболевание, встречающееся среди населения Экваториальной Африки (Уганда, Гвинея-Бисау, Нигерия), спорадические случаи наблюдаются в разных странах. Болеют обычно дети в возрасте 4-8 лет. Наиболее часто опухоль локализуется в верхней или нижней челюсти (рис. 138), а также яичниках. Реже в процесс вовлекаются почки, надпочечники, лимфатические узлы. Довольно часто наблюдается генерализация опухоли с поражением многих органов. Опухоль состоит из мелких лимфоцитоподобных клеток, среди которых разбросаны крупные, со светлой цитоплазмой макрофаги, что создает своеобразную картину «звездного неба» (starry sky) (см. рис. 138). Развитие африканской лимфомы связывают с герпесоподобным вирусом, который был выявлен из лимфатических узлов больных с этой опухолью. В лимфобластах лимфомы находят вирусоподобные включения.
Грибовидный микоз - относительно доброкачественная Т-клеточная лимфома кожи, относится к так называемым лимфоматозам кожи. Множественные опухолевые узлы в коже состоят из пролиферирующих крупных клеток с большим числом митозов. В опухолевом инфильтрате находят также плазматические клетки, гистиоциты, эозинофилы, фибробласты. Узлы мягкой консистенции, выступают над поверхностью кожи, напоминая иногда форму гриба, имеют синюшную окраску, легко изъязвляются. Опухолевые узлы находят не только в коже, но и в слизистых оболочках, мышцах, внутренних органах. Ранее развитие опухоли связывали с инвазией мицелия грибов, отсюда и ошибочное название болезни.
Болезнь Сезари - Т-лимфоцитарная лимфома кожи с лейкемизацией; относится к лимфоматозам кожи. Поражение костного мозга, наличие
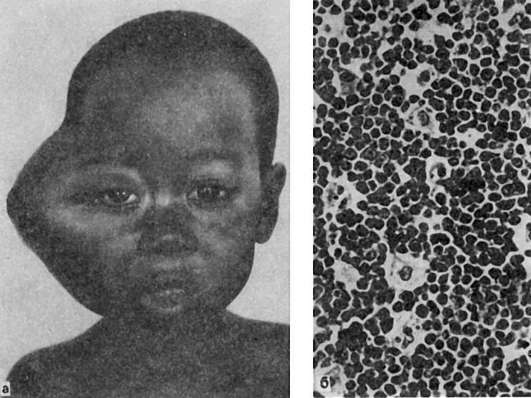 Рис. 138. Африканская лимфома (опухоль Беркитта):
Рис. 138. Африканская лимфома (опухоль Беркитта):
а - локализация опухоли в верхней челюсти; б - гистологическая картина опухоли - «звездное небо» (препарат Г.В. Савельева)
опухолевых клеток в крови, наблюдающиеся при болезни Сезари, послужили основанием для отнесения ее в некоторых случаях к хроническому лимфолейкозу.
Лимфоцитарная инфильтрация кожи завершается формированием опухолевых узлов чаще на лице, спине, голенях. В опухолевом инфильтрате кожи, костном мозге и крови находят атипичные мононуклеарные клетки с серповидными ядрами - клетки Сезари. Возможна опухолевая инфильтрация лимфатических узлов, селезенки, печени, почек, но она никогда не бывает значительной.
Ретикулосаркома - злокачественная опухоль из ретикулярных клеток и гистиоцитов. Следует отметить, что морфологические критерии принадлежности опухолевых клеток к ретикулярным и гистиоцитам весьма ненадежны. Главным гистологическим отличием ретикулосаркомы от лимфосаркомы считают продукцию опухолевыми клетками ретикулярных волокон, которые оплетают клетки ретикулосаркомы.
Лимфогранулематоз (болезнь Ходжкина) - хроническое рецидивирующее, реже остро протекающее заболевание, при котором разрастание опухоли происходит преимущественно в лимфатических узлах.
Морфологически различают изолированный и генерализованный лимфогранулематоз. При изолированном (локальном) лимфогранулематозе поражена одна группа лимфатических узлов. Чаще это шейные, медиа-
стинальные или забрюшинные, реже - подмышечные, паховые лимфатические узлы, которые увеличиваются в размерах и спаиваются между собой. Сначала они мягкие, сочные, серые или серо-розовые, на разрезе со стертым рисунком строения. В дальнейшем узлы становятся плотными, суховатыми, с участками некроза и склероза. Возможна первичная локализация опухоли не в лимфатических узлах, а в селезенке, печени, легких, желудке, коже. При генерализованном лимфогранулематозе разрастание опухолевой ткани обнаруживают не только в очаге первичной локализации, но и далеко за его пределами. При этом, как правило, увеличивается селезенка. Пульпа ее на разрезе красная, с множественными бело-желтыми очагами некроза и склероза, что придает ткани селезенки пестрый, «порфировый», вид («порфировая селезенка»). Развитие генерализованного лимфогранулематоза объясняют метастазированием опухоли из первичного очага.
При микроскопическом исследовании как в очагах первичной локализации опухоли (чаще в лимфатических узлах), так и в метастатических ее отсевах обнаруживают пролиферацию лимфоцитов, гистиоцитов, ретикулярных клеток, среди которых встречаются гигантские клетки, эозинофилы, плазматические клетки, нейтрофильные лейкоциты. Пролиферирующие полиморфные клеточные элементы образуют узелковые образования, подвергающиеся склерозу и некрозу, нередко казеозному (рис. 139). Наиболее характерным признаком для лимфогранулематоза считается пролиферация атипичных клеток, среди которых различают: 1) малые клетки Ходжкина (аналогичны лимфобластам); 2) одноядер-
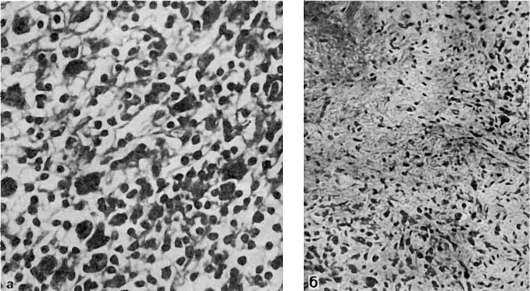 Рис. 139. Лимфогранулематоз:
Рис. 139. Лимфогранулематоз:
а - гранулематозные образования из полиморфных клеток в лимфатическом узле; б - некроз и разрастание грануляционной ткани с атипичными клетками
ные гигантские клетки, или большие клетки Ходжкина; 3) многоядерные клетки Рид-Березовского-Штернберга, которые обычно принимают гигантские размеры. Происхождение этих клеток, вероятно, лимфоцитарное, хотя нельзя исключить и макрофагальную их природу, так как в клетках обнаружены маркерные для макрофагов ферменты - кислая фосфатаза и неспецифическая эстераза.
Лимфогранулематозные очаги претерпевают определенную эволюцию, отражающую прогрессию опухоли, при этом клеточный состав очагов, естественно, меняется. Используя биопсию (чаще лимфатического узла), можно сопоставить гистологические и клинические особенности лимфогранулематоза. Такие сопоставления легли в основу современных клинико-морфологических классификаций лимфогранулематоза.
Клинико-морфологическая классификация. Выделяют 4 варианта (стадии) заболевания: 1) вариант с преобладанием лимфоидной ткани (лимфогистиоцитарный); 2) нодулярный (узловатый) склероз; 3) смешанноклеточный вариант; 4) вариант с подавлением лимфоидной ткани.
Вариант с преобладанием лимфоидной ткани характерен для ранней фазы болезни и локализованных ее форм. Он соответствует I-II стадии болезни. При микроскопическом исследовании находят лишь пролиферацию зрелых лимфоцитов и отчасти гистиоцитов, что ведет к стиранию рисунка лимфатического узла. При прогрессировании заболевания лимфогистиоцитарный вариант переходит в смешанно-клеточный.
Нодулярный (узловатый) склероз характерен для относительно доброкачественного течения болезни, причем первично процесс чаще локализуется в средостении. При микроскопическом исследовании обнаруживают разрастание фиброзной ткани, окружающей очаги клеточных скоплений, среди которых находят клетки Рид-Березовского-Штернберга, а по периферии - лимфоциты и другие клетки.
Смешанно-клеточный вариант отражает генерализацию болезни и соответствует II-III ее стадии. При микроскопическом исследовании выявляются характерные признаки: пролиферация лимфоидных элементов различной степени зрелости, гигантских клеток Ходжкина и Рид-Березовского- Штернберга; скопления лимфоцитов, эозинофилов, плазматических клеток, нейтрофильных лейкоцитов; очаги некроза и фиброза.
Вариант с подавлением (вытеснением) лимфоидной ткани встречается при неблагоприятном течении болезни. Он отражает генерализацию лимфогранулематоза. При этом в одних случаях отмечается диффузное разрастание соединительной ткани, среди волокон которой встречаются немногочисленные атипичные клетки, в других - лимфоидная ткань вытесняется атипичными клетками, среди которых преобладают клетки Ходжкина и гигантские клетки Рид-Березовского-Штернберга; склероз отсутствует. Вариант с вытеснением лимфоидной ткани крайне атипичными клетками получил название саркомы Ходжкина.
Таким образом, прогрессирование лимфогранулематоза морфологически выражается в последовательной смене трех его вариантов: с пре-
обладанием лимфоидной ткани, смешанно-клеточного и с подавлением лимфоидной ткани. Эти клинико-морфологические варианты могут быть рассмотрены как стадии лимфогранулематоза.
Тромбоцитопении и тромбоцитопатии
Тромбоцитопении - группа заболеваний, при которых наблюдается снижение количества тромбоцитов (норма 150х109/л) в связи с повышенным их разрушением или потреблением, а также недостаточным образованием. Повышенное разрушение тромбоцитов - наиболее частый механизм развития тромбоцитопении.
Классификация. Различают наследственные и приобретенные формы тромбоцитопении. При многих наследственных тромбоцитопениях наблюдают изменения различных свойств тромбоцитов, что позволяет рассматривать эти болезни в группе тромбоцитопатий (см. Тромбоцитопатии). Руководствуясь механизмом повреждения мегакариоцитов и тромбоцитов, приобретенные тромбоцитопении делят на иммунные и неиммунные. Среди иммунных тромбоцитопений различают аллоиммунные (несовместимость по одной из систем крови), трансиммунные (проникновение аутоантител матери, страдающей аутоиммунной тромбоцитопенией, через плаценту), гетероиммунные (нарушение антигенной структуры тромбоцитов) и аутоиммунные (выработка антител против собственных неизмененных антигенов тромбоцитов). В тех случаях, когда причину аутоагрессии против тромбоцитов выявить не удается, говорят об идиопатической аутоиммунной тромбоцитопении. Неиммунные тромбоцитопении могут быть обусловлены механической травмой тромбоцитов (при спленомегалии), угнетением пролиферации костномозговых клеток (при радиационном или химическом повреждении костного мозга, апластических анемиях), замещением костного мозга (разрастание опухолевых клеток), соматической мутацией (болезнь Маркиафавы-Микели), повышенным потреблением тромбоцитов (тромбоз - см. ДВС-синдром), недостатком витамина B12 или фолиевой кислоты (см. Анемии). Иммунные формы тромбоцитопении встречаются чаще неиммунных, причем среди первых наиболее часто наблюдается аутоиммуная форма, обычно у взрослых.
Патологическая анатомия. Для тромбоцитопении характерен геморрагический синдром с кровоизлияниями и кровотечениями. Кровоизлияния возникают чаще в коже в виде петехий и экхимозов, реже - в слизистых оболочках, еще реже - в паренхиме внутренних органов (например, кровоизлияние в мозг). Кровотечения возможны как желудочные и кишечные, так и легочные. Нередко отмечается увеличение селезенки в результате гиперплазии ее лимфоидной ткани, увеличение количества мегакариоцитов в костном мозге. Отдельные формы тромбоцитопении имеют свои морфологические особенности. Например, при некоторых аутоиммунных тромбоцитопениях наблюдается увеличение лимфатических узлов (лимфоаденопатия) и размеров тромбоцитов, а уве-
личение селезенки отсутствует. Геморрагии при тромбоцитопении могут приводить к развитию анемии (см. Анемии).
Тромбоцитопатии - большая группа заболеваний и синдромов, в основе которых лежат нарушения гемостаза, обусловленные качественной неполноценностью или дисфункцией тромбоцитов. По сути своей - это группа геморрагических диатезов с геморрагическими проявлениями на уровне сосудов микроциркуляции.
Классификация. Тромбоцитопатии делят на наследственные и приобретенные. Среди наследственных тромбоцитопатии выделяют ряд форм, руководствуясь типом дисфункции, морфологических изменений и биохимических нарушений тромбоцитов. Многие из этих форм рассматриваются как самостоятельные болезни или синдромы (например, тромбастения Гланцмана, связанная с мембранными аномалиями тромбоцитов; синдром Чедиака-Хигаси, развивающийся при недостатке в тромбоцитах плотных телец I типа и их компонентов).
Приобретенные тромбоцитопатии развиваются при разнообразных патогенных воздействиях и встречаются при многих болезнях и синдромах. Выделяют тромбоцитопатии: 1) при гемобластозах; 2) при миелопролиферативных заболеваниях и эссенциальной тромбоцитемии; 3) при В12-дефицитной анемии; 4) при циррозах, опухолях и паразитарных заболеваниях печени; 5) при гормональных нарушениях (гипотиреоз, гипоэстрогении); 6) при цинге; 7) при лучевой болезни; 8) при ДВС-синдроме и активации фибринолиза; 9) при массивных гемотрансфузиях; 10) лекарственные и токсические (при лечении нестероидными противовоспалительными препаратами, ацетилсалициловой кислотой, бруфеном, индометацином, некоторыми антибиотиками, транквилизаторами и др.; при алкоголизме).
Патологическая анатомия. Характеристика тромбоцитопатии сводится к морфологическим проявлениям геморрагического синдрома. При этом следует иметь в виду, что тромбоцитопатии могут протекать с более или менее выраженной тромбоцитопенией.
При решении вопроса о приоритетности тромбоцитопатии или тромбоцитопении в диагнозе следует руководствоваться следующими положениями (Баркаган З.С, 1985): 1) к тромбоцитопатиям относят все формы, при которых выявляются стабильные функциональные, морфологические и биохимические нарушения тромбоцитов, не исчезающие при нормализации их количества в крови; 2) для тромбоцитопатии характерно несоответствие выраженности геморрагического синдрома степени тромбоцитопении; 3) генетически обусловленные формы патологии тромбоцитов в подавляющем большинстве случаев относятся к тромбоцитопатиям, особенно если они сочетаются с другими наследственными дефектами; 4) тромбоцитопатию следует считать вторичной, если качественный дефект тромбоцитов непостоянен, ослабляется или полностью исчезает после ликвидации тромбоцитопении.