Медицинская и клиническая генетика для стоматологов: учебник для вузов / Под ред. О.О. Янушевича., - 2009. - 400 с.
|
|
|
|
ГЛАВА 3. ХРОМОСОМНЫЕ БОЛЕЗНИ
Богомазов Евгений Александрович
3.1. ОБЩИЕ ВОПРОСЫ
Одной из основных фундаментальных задач современной медицинской генетики является изучение закономерностей строения и функционирования хромосом человека в норме и патологии. Материальными носителями наследственной информации являются хромосомы. Наука, занимающаяся изучением функционирования хромосом на всех уровнях их организации (микроскопическом, субмикроскопическом, молекулярном) называется цитогенетикой.
Впервые о хромосомах заговорили в 1880 г., когда В. Флеминг, изучая клетки роговицы глаза человека, обнаружил от 22 до 28 хроматиновых тел. Термин «хромосома» впервые был введен В. Вальдейером в 1888 г. для обозначения окрашенных нитевидных структур, видимых в микроскоп.
Изучение строения и функционирования хромосом человека имеет большое теоретическое и практическое значение для медицинской генетики. Знание того, что представляет собой каждая хромосома человека в химическом, цитологическом, молекулярном и генетическом отношении, важно для правильного понимания происхождения хромосомных нарушений и обусловленных ими аномалий развития, а следовательно, и поиска путей исправления этих отклонений.
Для каждого биологического вида характерно постоянное число хромосом. У большинства высших организмов каждая клетка содержит диплоидный хромосомный набор. Хромосомы отличаются друг от друга определенной формой и своими размерами.
Совокупность количественных и качественных признаков хромосом, определяемая при микроскопировании в единичной клетке, называется кариотипом.
Длительное время (с 1912 по 1956 гг.) из-за несовершенства цитологической техники общее число хромосом человека считалось равным 48. В 1956 г. шведские цитологи Дж. Тио и А. Леван, применив усовершенствованную цитологическую технику, на материале
культуры фибробластов легочной ткани 4 человеческих эмбрионов показали, что модальное число хромосом у человека равно 46. Эти данные были подтверждены в том же году работой английских цитологов - С. Фордом и Дж. Хамертоном. Оба события стали началом бурного развития цитогенетики человека.
Начиная с начала 60-х годов прошлого века благодаря интенсивному развитию цитогенетики встал вопрос об идентификации и систематизации хромосом человека. В США (Денвер) в 1960 г. была проведена первая Международная научная конференция цитогенетиков, которая выработала принципы классификации хромосом человека. В зависимости от морфологической характеристики, учитывающей размеры, форму и положение центромеры, все хромосомы был поделены на 7 групп: А, В, С, D, E, F, G и половые хромосомы Х и У. При световой микроскопии в стадии метафазы и прометафазы хромосомы хорошо различимы. Размеры их колеблются от 1,5 до 2,0 мкм (хромосомы 21, 22 и У) и от 11 до 12 мкм (хромосома 1). Структурно каждая хромосома имеет центромеру (первичная перетяжка), короткое (p) и длинное (q) плечо . Концевые участки каждого плеча хромосомы называются теломерами, которые играют решающую роль в сохранении стабильности хромосом. В настоящее время показано, что в теломере находятся повторяющиеся последовательности ДНК - теломерная ДНК, препятствующие укорочению хромосомы при ее репликации. В зависимости от положения центромеры в хромосоме различают метацентрические, субметацентрические и акроцентрические хромосомы. В некоторых хромосомах присутствуют вторичные перетяжки, отделяющие от плеча хромосомы участок, называемый спутником. Гаплоидный набор человека состоит из 22 аутосом и одной половой хромосомы (Х или У), диплоидный набор представлен 46 хромосомами. Наиболее удобной фазой исследования хромосом является метафаза митоза. При традиционном (рутинном) методе окраски (краситель Романовского-Гимзе) хромосомы окрашиваются равномерно по всей своей длине, поэтому детализация их строения, а тем более индивидуализация каждой хромосомы была затруднена.
С введением в цитогенетическую практику новых методов обработки хромосом и окрашивания удалось обнаружить высокую специфичность метафазных хромосом. Это связано с тем, что в хромосоме наблюдается чередование эухроматиновых и гетерохроматиновых районов, уникальных для каждого конкретного человека. Такая
уникальность обеспечивает широкий хромосомный полиморфизм у человека, для которого характерны наличие определенного варианта строения хромосомы во всех клетках, передачи его от родителей к детям как простого моногенного признака при отсутствии заметного фенотипического эффекта.
Большой вклад в разработку дифференциального способа окраски хромосом наряду с Т. Касперсоном внесли отечественные исследователи А.Ф. Захаров, Н.А. Еголина, Ю.В. Селезнев.
В начале 70-х годов прошлого века Касперсону с сотрудниками удалось идентифицировать Y-хромосому в интерфазных ядрах амниотической жидкости, что позволило определять пол плода внутриутробно. Дистальный участок длинного плеча Y-хромосомы интенсивно светился при обработке флюоресцентными красителями. Это было большим достижением для цитогенетики и, в частности, для клинической цитогенетики, появилась возможность кардинальным способом избавиться от тяжелых заболеваний, наследуемых по Х-сцепленному рецессивному типу. Классическим примером здесь может служить миопатия Дюшена, приводящая к тяжелой инвалидности и ранней смертности у лиц мужского пола.
Вскоре в диагностических целях для упрощения работы с хромосомами в цитогенетическую практику (без применения флюоресцентных красителей) были внедрены три типа дифференциального окрашивания хромосом по длине: G-, C-, Q-методы. Эти методы было проще использовать, применяя краситель Романовского-Гимзе. С помощью этих методов удается получить рисунок строго специфических полос для каждой индивидуальной хромосомы. Число полос на хромосомах зависит от типа окрашивания и специфической стадии разделения клетки, поэтому в настоящее время их широко используют в практической цитогенетике. Самым распространенным методом дифференциального окрашивания в клинической цитогенетике является G-метод (по Гимзе). Число чередующихся темных и светлых полос на кариотип при G-окрашивании варьирует от 350-450 (метафаза) до 800-2500 (прометафаза) на гаплоидный набор. Рисунок и протяженность светлых и темных полос (эу- и гетерохроматических) строго специфичны для каждой пары хромосом. При С-методе выявляются только центромерные и околоцентромерные районы хромосом, в которых содержится структурный гетерохроматин (1, 9, 16 и У-хромосома). Q-метод (окраска флюорохромами) позволяет выявлять ярко светящиеся районы 3, 4, 13-15, 21, 22 и
У-хромосом. Самым информативным при этом методе окрашивания является ярко светящийся сегмент У-хромосомы. Он хорошо различим в интерфазных клетках разных тканей и может служить цитологическим показателем половой принадлежности.
Последние успехи молекулярной цитогенетики человека позволили разработать новые высокоинформативные методы изучения хромосом. Среди них в первую очередь следует назвать метод флю-
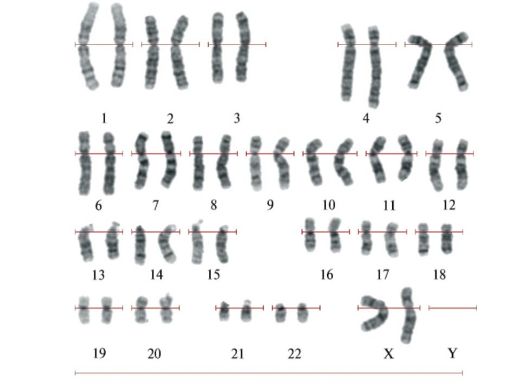 Рис. 3.1. Хромосомы, окрашенные по G-методу
Рис. 3.1. Хромосомы, окрашенные по G-методу
 Рис. 3.2. Хромосомы, окрашенные по С-методу
Рис. 3.2. Хромосомы, окрашенные по С-методу
оресцентной гибридизации in situ, или так называемый FISH-метод (от англ. fluorescent in situ hybridization). с помощью которого можно выявлять специфические последовательности нуклеиновых кислот на метафазных хромосомах, интерфазных ядрах и в других тканях. С помощью этого метода идентифицируются не только индивидуальные хромосомы или отдельные гены, но и расшифровываются сложные межхромосомные перестройки. Этот метод нашел широкое применение в картировании генома, хромосомной локализации генов и последовательностей ДНК и РНК, анализе идентификации хромосомных аномалий, включая микроделеции и микродупликации. Благодаря своей уникальности и специфичности метод гибридизации в последнее время широко применяется при проведении преимплатационной, пренатальной или постнатальной диагностики. Его используют в клинической цитогенетике, онкогенетике, гематологии, при оценке мутагенных воздействий (физических, химических, биологических), диагностике врожденных пороков развития и умственной отсталости.
По мере внедрения в цитогенетику новых метод исследования хромосом человека приходилось регулярно уточнять классификацию хромосом в норме и при хромосомных аномалиях (1963 - Лондон, 1966 - Чикаго, 1971 - Париж); в 1978 г. был разработан важный документ, который получил название «Международная система номенклатуры хромосом человека». В последующие годы (1981, 1985, 1991, 1995, 2004) классификация хромосом человека дорабатывалась и уточнялась и в 2005 г. вышла в новой редакции под названием «Международная система для цитогенетической номенклатуры человека».
Цитогенетический метод
Среди многих методов изучения наследственной патологии человека цитогенетический метод занимает одно из главных. С помощью цитогенетического исследования в генетике человека можно решать такие сложные вопросы, как анализы материальных основ наследственности и кариотипа в норме и патологии, изучать некоторые закономерности мутационного и эволюционного процессов. Все хромосомные болезни у человека были открыты с помощью цитогенетического метода. Для его проведения используют культуру лимфоцитов периферической крови, кожные фибробласты, костный мозг. Классификация хромосом человека, методы индивидуализации
хромосом с помощью различных типов окрашивания, молекулярная организация хромосом, хромосомный пол человека - все эти темы будут освещены в практикуме по медицинской генетике, который выйдет в свет вскоре после настоящей монографии.
Цитогенетика человека занимает особое место в медицинской генетике. Это обусловлено тем, что большая часть множественных пороков и нарушений половой дифференцировки у человека связана с различными структурными и числовыми нарушениями в системе аутосом и гоносом. До недавнего времени с помощью цитогенетического метода можно было судить только о кариотипе - точном числе и структуре хромосом. С введением в практику здравоохранения высокоразрешающих методов молекулярной цитогенетики удалось «подобрать ключи» к патологии, которую не удавалось диагностировать с помощью рутинных методов цитогенетики. Были разработаны и внедрены в клиническую цитогенетику ДНК-диагностика, гибридизация нуклеиновых кислот in situ, которые помогли выяснить природу большого количества микроделеционных синдромов (Ворсанова С.Г. и соавт., 1998, 1999, 2006); появились компьютерные системы для анализа хромосом, которые позволяют проводить автоматический анализ хромосом и внедрять очень эффективную многоцветную детекцию ДНК-зондов. Весьма успешную работу в этом направлении осуществляют лаборатории Научного Центра психического здоровья (руководитель Юров Ю.Б.) и Московского НИИ педиатрии и детской хирургии (руководитель Ворсанова С.Г.), которые создали оригинальную хромосомоспецифическую коллекцию ДНК-зондов на все хромосомы человека и их отдельные участки.
Необходимость цитогенетического исследования диктуется наличием огромного количества хромосомных болезней. Описано уже около 1000 типов хромосомных нарушений, для более 100 из них четко определена клиническая картина. Частота хромосомных аномалий среди новорожденных составляет примерно 1%, среди мертворожденных этот показатель равен 6-7%. У детей, родившихся с задержкой психомоторного развития и имеющих пороки развития внутренних органов, хромосомные болезни встречаются от 1 до 30%. Кроме того, хорошо известно, что по крайней мере около 60% спонтанных абортов в I триместре беременности (в первые дни беременности эти цифры еще выше) связаны с хромосомными аберрациями.
Хромосомные нарушения резко нарушают эмбриогенез. В этот период, период морфогенеза в процессах развития будущего потомства принимают участие до 1000 генов, локализованных во всех хромосомах, поэтому хромосомная или геномная мутация может привести к спонтанному аборту (Бочков Н.П., 2004). Примерно 1/з оплодотворенных яйцеклеток погибает в 1-ю неделю беременности. Во II триместре хромосомные нарушения являются причиной спонтанных абортов в 25-30% случаев. После 20 нед беременности хромосомные аномалии встречаются только в 10% случаев. При отягощенном акушерском анамнезе у супружеских пар с повторными спонтанными абортами, мертворождениями или рождением детей с пороками развития хромосомные аномалии обнаруживаются в 5%.
Среди других контингентов хромосомные аномалии обнаруживаются у детей с олигофренией - в среднем в 15% (в основном из-за структурных перестроек). У больных с нарушением половой дифференцировки частота хромосомных нарушений колеблется от 20 до 50% (в 50% случаев обнаруживается мозаицизм). У больных с первичной и вторичной аменореей частота хромосомных аномалий колеблется от 10 до 50% (более 90% - численные нарушения и мозаицизм). При мужском бесплодии частота аномальных хромосом достигает 10-15% (до 70% - численные нарушения и мозаицизм).
Знания по медицинской генетике, в том числе и по цитогенетике, необходимы акушерам-гинекологам, педиатрам, эндокринологам, психоневрологам, паталогоанатомам, а также другим специалистам. Имеется достаточное количество не только детей, но и взрослых больных, у которых психоневрологические нарушения, нарушения половой сферы или репродуктивной функции связаны с нарушением хромосомного аппарата.
Исторически хромосомные болезни клиницисты начали еще изучать до установления точного числа хромосом человека. Синдромы Дауна, Клайнфельтера и Шерешевского-Тернера клинически были описаны задолго до открытия хромосомной этиологии этих заболеваний.
С открытием «лишней» хромосомы при синдроме Дауна (Лежен Ж. и соавт., 1959) в медицину вошло новое понятие - «хромосомопатии», или «хромосомные болезни».
В настоящее время к хромосомным болезням относят такие формы патологии, при которых наблюдаются, как правило, нарушение психики и множественные врожденные пороки различных
систем организма человека. Генетической основой таких состояний являются численные или структурные изменения хромосом, наблюдаемые в соматических или половых клетках.
Термин «болезнь» по отношению к хромосомным аномалиям употребляется не всегда справедливо. Болезнь - это процессуальность, т.е. закономерная смена симптомов и синдромов во времени. Болезнь имеет продрому, начало, стадию полного развития и исходное состояние. Совокупность же специфических признаков, характеризующих любую хромосомную аномалию, является конституциональной, врожденной, и признаки эти непрогредиентны. Другими словами, врожденные аномалии развития, в основе которых лежат нарушения кариотипа, отличаются от болезней в обычном понимании резким сдвигом процессуальной фазы во времени. Процессуальная фаза в данном случае проходит во время эмбрионального развития. В силу этих соображений употребление термина «хромосомные болезни» необходимо применять при полном осознании его своеобразия.
Одной из важнейших задач медицинской генетики, и в первую очередь клинической цитогенетики человека, является выяснение связи хромосомных аномалий с пороками развития. Положительное решение этой проблемы позволило бы, в свою очередь, установить роль каждой отдельной хромосомы в эмбриональном развитии человека; это, конечно, помогло бы цитогенетикам составить цитологические карты каждого отдельного локуса хромосомы и таким образом определить значение его для развития и жизнедеятельности организма в целом.
3.2. ЭТИОЛОГИЯ И КЛАССИФИКАЦИЯ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
Среди хромосомных нарушений принято выделять геномные и хромосомные нарушения. У человека найдены все формы хромосомных и геномных мутаций. К геномным мутациям относятся аномалии, характеризующиеся увеличением полного набора хромосом (полиплоидии) или изменением количества хромосом по одной из пар (анеуплоидии). К структурным хромосомным мутациям относятся все типы перестроек, которые обнаружены у человека, - делеция (нехватка), дупликация (удвоение), инверсия (перевертывание), инсерция (вставка), транслокация (перемещение).
Можно выделить два основных типа перестроек: внутрихромосомные и межхромосомные. В свою очередь, перестройки могут быть сбалансированными (т.е. в геноме присутствуют все локусы, однако их расположение в хромосомах отличается от исходного - нормального) и несбалансированными. Несбалансированные перестройки характеризуются утратой или удвоением участков хромосомы. Внутрихромосомные перестройки, связанные с перестройками внутри одного плеча хромосомы, называются парацентрическими. Крайние участки без центромеры называются фрагментами, и они обычно утрачиваются в ходе митоза.
Делеция - утрата части хромосомы, происходящая в результате двух разрывов и одного воссоединения, с утратой сегмента, лежащего между разрывами. У человека известна потеря 1/з короткого плеча хромосомы 5, именуемая как синдром «кошачьего крика» и описанная впервые Дж. Леженом в 1963 г.
Дупликация - удвоение сегмента хромосомы, в результате чего клетка организма становится полиплоидной по данному сегменту. Если дупликация находится непосредственно за исходным участком хромосомы, это называется тандем-дупликацией. Кроме того, дупликации могут быть локализованы в других участках хромосомы. Большинство таких перестроек летальны, а те люди, которые с ними выжили, как правило, не способны воспроизвести потомство.
В случае инверсии участок хромосомы разворачивается на 180°, и разорванные концы соединяются в новом порядке. Если в инвертированный участок попадает центромера, такую инверсию называют перицентрической. Если инверсия затрагивает только одно плечо хромосомы, она называется парацентрической. Гены в инвертированном участке хромосомы располагаются в обратном порядке по отношению к исходному в хромосоме.
К межхромосомным перестройкам относят транслокации - обмен сегментами между хромосомами. Различают следующие типы транслокаций:
- реципрокная транслокация, когда две хромосомы взаимно обмениваются сегментами (сбалансированная транслокация); как и инверсия, она не вызывает аномальных эффектов у носителя;
- нереципрокная транслокация - когда сегмент одной хромосомы переносится в другую;
- транслокация типа центрического соединения - когда после разрывов в околоцентромерном районе соединяются два фрагмента с центромерами таким образом, что их центромера соединяется, образуя одну. Взаимное объединение двух акроцентрических хромосом из групп D и G приводит к образованию одной метаили субметацентрической хромосомы. Такую транслокацию называют робертсоновской.
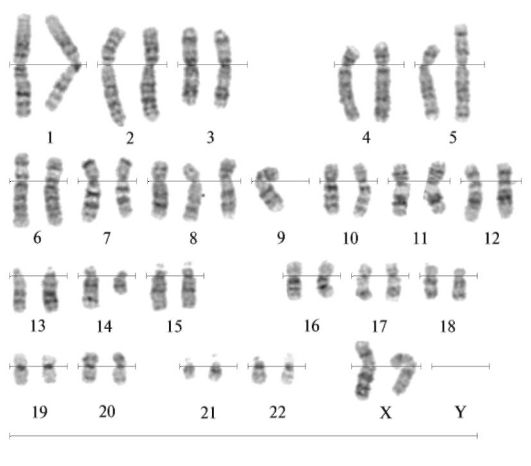 Рис. 3.3. Транслокация t(5;14)
Рис. 3.3. Транслокация t(5;14)
Транслокационный синдром Дауна возникает именно таким образом, при этом больные имеют выраженную симптоматику болезни Дауна, но в их кариотипе всего 46 хромосом, причем хромосом 21 - две, третья транслоцирована обычно на одну из хромосом группы D или G. Исследование кариотипов родителей таких детей показало, что чаще всего фенотипически нормальные родители (как правило, матери) имеют 45 хромосом и точно такую же транслокацию хромосомы 21, как и ребенок.
В основу классификации хромосомных болезней положены тип хромосомной аномалии и характер дисбаланса хромосомного материала соответствующего кариотипа. Исходя из этих принципов хромосомные аномалии делятся на три группы:
- численные нарушения по отдельным хромосомам;
- нарушение кратности полного гаплоидного набора хромосом;
- структурные перестройки хромосом.
Первые две группы относятся к геномным мутациям, а третья группа - к хромосомным мутациям. Кроме этого, необходимо учитывать тип клеток, в которых произошла мутация (в гаметах или зиготе), а также иметь в виду, была ли мутация унаследована или она возникла заново. Таким образом, при постановке диагноза хромосомной болезни необходимо учитывать:
- тип мутации;
- конкретную хромосому;
- форму (полная или мозаичная);
- наследуемый или ненаследуемый случай.
Большая часть хромосомных аномалий, возникающих в хромосомных наборах человека, связана с нарушением числа хромосом. Полиплоидия возникает в результате нарушения нормального митотического цикла: удвоение хромосом не сопровождается делением ядра и клетки. Примерами полиплоидии, которые описаны у человека, являются триплоидии (69,ХХХ; 69,ХХУ) и тетраплоидии (92,ХХХХ; 92,ХХХУ). Эти нарушения несовместимы с жизнью и встречаются в материале спонтанных абортусов или плода и у мертворожденных, а иногда и у новорожденных, продолжительность жизни которых с такими аномалиями составляет, как правило, всего несколько дней.
Анеуплоидия возникает в результате нерасхождения хромосом в мейотических делениях или в митозе. Термин «нерасхождение» означает отсутствие разъединения хромосом (в мейозе) либо хроматид (в митозе) в анафазе. В результате нерасхождения возникают гаметы с аномальным набором хромосом.
Структурные изменения хромосом у человека встречаются намного реже, чем численные аберрации. Структурные перестройки могут быть хромосомными и хроматидными, сопровождаться изменением количества генетического материала (делеции и дупликации) или только сводиться к перемещению его (инверсии, инсерции, транслокации). В перестройку может вовлекаться одна или больше хромосом с несколькими разрывами и соединениями. Иногда в организме могут встречаться клетки с различными кариотипами. Такое сочетание кариотипа обычно обозначают термином «мозаицизм» .
Большинство хромосомных болезней возникает спорадически в результате геномной и хромосомной мутации в гаметах здоровых родителей или на первых делениях зиготы. Хромосомные изменения в гаметах приводят к развитию так называемых полных, или регулярных, форм нарушения кариотипа, а соответствующие изменения хромосом на ранних стадиях развития эмбриона являются причиной возникновения соматического мозаицизма или мозаичных организмов (наличие в организме двух или более клеточных линий с разным числом хромосом). Мозаицизм может касаться как половых хромосом, так и аутосом. У человека чаще всего мозаичные формы обнаруживаются в системе половых хромосом. Мозаики, как правило, имеют более «стертые» формы заболевания, чем люди с измененным числом хромосом в каждой клетке. Так, ребенок с мозаичным вариантом болезни Дауна может иметь фактически нормальный интеллект, но физические признаки этого заболевания все равно остаются.
Число аномальных клеток может быть различным: чем их больше, тем более ярко выражен симптомокомплекс той или иной хромосомной болезни. В некоторых случаях удельный вес аномальных клеток так невелик, что человек кажется фенотипически здоровым.
Установить мозаицизм оказывается не так просто, поскольку клон аномальных клеток имеет в онтогенезе тенденцию к элиминации. Иначе говоря, число таких клеток может быть у взрослого человека относительно мало, в то время как в эмбриональный и ранний постнатальный периоды их удельный вес был достаточно велик, что привело к развитию выраженных клинических симптомов болезни. Однако, несмотря на известные трудности изучения мозаицизма, его открытие и исследование вносят ясность в проблему стертых и рудиментарных форм хромосомных болезней.
Любая из хромосом кариотипа человека может вовлекаться в численные или структурные изменения. Исходя из этого можно наблюдать очень большое разнообразие описанных хромосомных форм. Практическая цитогенетика постоянно сталкивается с обнаружением хромосомных аномалий при исследовании различных клеток и тканей в разные периоды развития человека. Классификация индивидуальных хромосом, которые могут вовлекаться в хромосомные аномалии, а следовательно, и выделение хромосомных синдромов в настоящее время - легко разрешимая проблема в связи с введением в хромосомный анализ методов индивидуализации хромосом: различных типов окрашивания по длине; гибридизации нуклеино-
вых кислот in situ, метода сравнительной геномной гибридизации, спектроскопического метода анализа хромосом. В последнее время при FISH-анализе иногда используют разноцветные ДНК-зонды, позволяющие быстро выявить качественные и количественные перестройки хромосом.
3.3. ПАТОГЕНЕЗ И КЛИНИЧЕСКИЕ ОСОБЕННОСТИ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
Хромосомные аномалии возникают в результате того, что изменения количества или качества генетической информации в сторону ее избытка или недостатка нарушает функционирование нормальной генетической программы онтогенеза (индивидуального развития организма). Характер и тяжесть проявления хромосомных болезней зависит от вида аномалий и вовлеченных хромосом. Хромосомные синдромы обычно характеризуются множественными пороками развития независимо от типа хромосомной аберрации. Многочисленные исследования разнообразных типов повреждения хромосом и вызываемые ими отклонения развития позволяют сделать вывод о том, что в патогенезе хромосомных болезней основное место занимает нарушение физического (соматического) и психического развития.
Общим для всех форм хромосомных аномалий является множественность поражения различных систем и органов. Нарушения развития могут наблюдаться в широких диапазонах - от гибели и элиминации зигот на первых стадиях дробления до нарушений, совместимых с постнатальным существованием. Тщательное клинико-цитогенетическое изучение хромосомных аномалий позволяет выделить ряд признаков, которые в различных сочетаниях и с разной степенью выраженности встречаются у всех пораженных индивидуумов. К таким признакам относят умственную отсталость, пре- и постнатальную задержку развития, аномалии многих органных систем, особенно челюстно-лицевой области, скелета, сердечно-сосудистой и мочеполовой систем. В частности, отмечаются краниофациальная дисплазия, ненормальные форма и расположение ушных раковин, гипертелоризм, эпикант, готическое нёбо, аномалии строения глазных щелей и яблок, специфическое изменение кожного рисунка на ладонях и подошвах, аномалия строения и расположения пальцев нижних и верхних конечностей и др.
Все диагностические признаки, встречающиеся при хромосомных болезнях, можно условно разделить на три группы.
К первой группе можно отнести комплекс признаков, позволяющих лишь заподозрить хромосомную аномалию. Это общие признаки (некоторые из них перечислены выше): физическое недоразвитие, ряд дизморфий мозгового и лицевого черепа, косолапость, клинодактилия мизинцев, некоторые пороки развития внутренних органов (сердца, почек, легких).
Ко второй группе относят признаки, встречающиеся в основном при определенных хромосомных болезнях. Их сочетание позволяет в большинстве случаев диагностировать хромосомную аномалию. Среди характерных, наиболее часто встречающихся признаков при трисомии хромосомы 13 следует назвать глубокую задержку умственного и физического развития (100%), гипертелоризм (90%), низко расположенные уродливые уши (90%). При трисомии хромосомы 18 следует отметить долихоцефалию (90%), тяжелую задержку психомоторного и физического развития (100%), затруднения при глотании, проблемы с кормлением (100%), микрогнатию и короткую грудину (90%).
К третьей группе относят признаки, характерные только для одной хромосомной аномалии, например «кошачий крик» при синдроме 5р-, алопеция при синдроме 18р.
При изучении корреляции фенотипа с кариотипом было сделано важное заключение о том, что чем больше хромосомного материала утрачено или приобретено, тем сильнее отклонения в развитии, тем раньше в онтогенезе они проявляются. Поэтому аномалии по крупным хромосомам встречаются очень редко. Кроме того, нехватка генетического материала сказывается на организме тяжелее, чем его избыток, и поэтому полные моносомии (особенно у живорожденных детей) встречаются гораздо реже, чем полные трисомии. Тяжесть клинической картины зависит не только от размера хромосомы, вовлекаемой в патологический процесс, большое значение имеет и ее качественный состав. Например, полные трисомии у живорожденных чаще всего обнаруживаются по аутосомам 13, 18, 21. Это связано с тем, что данные хромосомы содержат больше гетерохроматина, чем эухроматина. Основу последнего составляют активные районы, содержащие гены, которые контролируют развитие признаков организма. И, естественно, скорее погибнет та клетка, в которой имеется нехватка генов, определяющих продукцию таких белков, которые
участвуют в ключевых биохимических реакциях, обеспечивающих жизнеспособность клетки.
Для хромосомных нарушений характерны увеличение частоты гибели плодов и снижение жизнеспособности живорожденных. Однако при некоторых хромосомных аномалиях возможно выживание до взрослого состояния. В первую очередь это относится к группе синдромов, связанных с патологией в системе половых хромосом. Общее нарушение генного баланса, вызванное аномалиями в системе половых хромосом, гораздо менее фатально для развития организма, чем это имеет место при аутосомных аберрациях, поэтому наличие гоносомных нарушений в кариотипе человека совместимо не только с рождением, но и с нормальной жизнеспособностью и даже иногда с нормальным фенотипом.
Многочисленные исследования, проведенные в больших популяциях новорожденных и здоровых взрослых, а также в различных контингентах умственно отсталых лиц, позволили установить, что аномалии по половым хромосомам среди умственно отсталых людей встречаются в 4-5 раз чаще, чем у новорожденных.
Установлено, что 17-25% мужчин с синдромом Клайнфельтера имеют сниженный интеллект. Лишняя хромосома Х у женщин, вероятно, проявляется в еще большем снижении интеллекта, чем у мужчин.
Отмечена прямая корреляция между числом лишних Х хромосом и степенью умственной отсталости. Если наличие одной лишней хромосомы Х не всегда сопровождается олигофренией (синдромы ХХУ, ХХХ), то наличие лишних двух Х хромосом уже всегда дает картину умственной отсталости (средние значения IQ у больных с кариотипом 48, ХХХУ 52,5, а с кариотипом 49, ХХХХУ - 35,2). Синдром Шерешевского-Тернера более редок среди умственно отсталых женщин.
Причины умственной отсталости при ауто- и гоносомных абберациях, очевидно, заключаются в грубых нарушениях генного баланса и вытекающих отсюда нарушениях множества ферментных функций.
Как уже указывалось выше, клинические проявления одних и тех же форм хромосомных болезней сильно варьируют: от летального эффекта до незначительных отклонений. Почему это происходит, остается неясным: ведущую роль играют то ли генотипические факторы, то ли факторы внешней среды. Например, нет ответа на вопрос,
почему только 2/3 случаев трисомии по хромосоме 21 элиминируется во внутриутробном периоде (примерно такая же картина наблюдается при моносомии ХО).
В формировании клинических (фенотипических) проявлений хромосомных аномалий участвуют многие факторы. Среди них в первую очередь следует отметить:
- генотип организма;
- генный состав индивидуальной хромосомы, вовлекаемой в хромосомную аберрацию;
- тип аберрации и размер недостающего или избыточного хромосомного материала;
- степень мозаичности организма по аберрантным клеткам.
- тяжесть клинических проявлений зависит от соотношения нормальных и аномальных клеточных клонов;
- факторы внешней среды;
- онтогенетическую стадию развития организма.
Исходя из приведенных данных следует сделать вывод о том, что в патогенезе хромосомных аномалий еще много неясного, поскольку пока нет общей четкой схемы развития сложных патологических процессов, каковыми являются хромосомные болезни.
3.4. ЧАСТОТА И РАСПРОСТРАНЕННОСТЬ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
Наиболее полные сведения о частоте и распространенности хромосомных болезней можно получить на основании цитогенетических исследований спонтанных абортов, мертворожденных и новорожденных. Методы учета хромосомных аномалий должны быть строго унифицированы. Цитогенетическое обследование необходимо проводить новорожденным с врожденными пороками развития, недоношенным; больным с олигофренией, нарушением половой дифференцировки, с первичной и вторичной аменореей, спонтанными абортами, лицам с мужским бесплодием. Цитогенетический метод может применяться во многих областях практической и теоретической медицины (акушерство и гинекология, педиатрия, психиатрия, эндокринологи, патологическая анатомия и др.) - вот почему знания хромосомной патологии, ее клинических особенностей, методов диагностики и профилактики играют важную роль в подготовке будущего врача.
Как указывалось ранее, хромосомные аномалии чаще всего наблюдаются при спонтанных абортах - до 60%, у мертворожденных - до 70% и у живорожденных - около 1%.
Клинические и цитогенетические исследования, проводимые у новорожденных с хромосомной патологией, показывают, что жизнеспособность зависит от типа хромосомного нарушения. Большинство новорожденных с аутосомными трисомиями погибают в первые дни жизни. В свою очередь, у больных с аномалиями половых хромосом жизнеспособность снижена незначительно. Это зависит от того, что полная клиническая картина у данного контингента проявляется лишь в период полового созревания, когда начинают функционировать гены, определяющие половое развитие организма и формирование вторичных половых признаков.
Среди эффектов хромосомных аномалий в онтогенезе, кроме спонтанных абортов и врожденных пороков развития, у человека наблюдается явление однородительских дисомий. Однородительская дисомия возникает тогда, когда будущий потомок получает от одного из родителей обе хромосомы одной из пар (кариотип представлен 46 хромосомами). В результате может происходить гомозиготизация по патологическим рецессивным генам, которые могут быть причиной данного заболевания. Примерами однородительской дисомии являются синдромы Прадера-Вилли, Ангельмана, Беквита-Видемана и др.
Хромосомные аномалии возникают не только в ранние периоды онтогенеза. Спонтанный уровень хромосомных перестроек наблюдается у человека на протяжении всей жизни (около 2%). Чаще всего эти перестройки обычно элиминируются, но в какой-то момент могут стать источником злокачественного роста. Известно, что некоторые числовые и структурные хромосомные аномалии либо вызывают злокачественную трансформацию клеток, либо обусловливают предрасположенность к развитию онкологических заболеваний. Опухолевая прогрессия часто возникает в результате появления новых клеточных клонов, несущих различные виды хромосомных перестроек, которые кардинально отличаются от исходного клеточного штамма. В результате анализа огромного количества опухолей (свыше 25 тыс.), который был суммирован и опубликован в пятом издании «Каталога хромосомных аберраций при онкологических заболеваниях», удалось выявить новые гены, изменение которых в некоторых случаях могло привести к злокачественному перерожде-
нию нормальных клеток. По данным ВОЗ, рак это общее обозначение более чем 100 болезней, которые могут поражать любую часть организма, и его считают болезнью генома. Ретинобластома явилась первой опухолью, для которой была выявлена специфическая связь с презиготной хромосомной мутацией в длинном плече хромосомы 13. Классическим примером хромосомной мутации, детерминирующей возникновение хронического миелоидного лейкоза, является так называемая филадельфийская хромосома. Транслокация участков длинных плеч хромосом 9 и 22 приводит к образованию аномальной хромосомы, вызывающей злокачественные изменения белой крови. Известны и другие транслокации хромосом (8;21), (8;14), которые приводят к возникновению соответственно острого миелоидного лейкоза и лимфомы Беркитта.
В середине 60-х годов прошлого века многочисленными исследованиями было доказано, что у больных с врожденными хромосомными аномалиями рак возникает во много раз чаще, чем в популяции, и предрасположенность к новообразованиям при некоторых наследственных синдромах сопровождается (или обусловлена) повышенной частотой спонтанных или индуцированных хромосомных повреждений.
Необходимо помнить, что при старении организма спонтанный уровень хромосомных нарушений увеличивается.
Патологические синдромы, объединяемые термином «хромосомные болезни», являются неоднородными. Описано большое количество многообразных форм хромосомных аномалий у человека. Однако не все из них могут претендовать на «самостоятельность» в виде четко очерченного синдрома или болезни. Это связано с тем, что при некоторых хромосомных нарушениях патологическое состояние не обусловлено непосредственно конкретной хромосомной перестройкой.
Общая частота морфологических пороков развития у детей в возрасте до 1 года составляет примерно 27,2 на 1000 населения. Около 60% из них выявляются в первые 7 дней жизни уже в родовспомогательных учреждениях. Одна из частных причин пороков развития - орофасциальные расщелины, которые входят в «большую пятерку» уродств, занимая по частоте второе место.
По сведениям национального института стоматологии США, 40% населения мира имеют врожденные и наследственные аномалии развития черепно-лицевой области, из которых 15% нуждаются в серьез-
ном хирургическом лечении. По данным ВОЗ, общая встречаемость врожденной расщелины верхней губы и нёба в мире колеблется от 0,8 до 2 случаев на 1000 рождений. Распространение по континентам следующее: в Азии - 1 случай на 500 новорожденных; в Европе - 1 на 700; в Африке - 1 на 1000; в России - 1 на 800. По разным источникам, доля больных с врожденными и наследственными аномалиями черепно-лицевой области в России составляет около 35%, причем ежегодно рождается свыше 50 тыс. детей, которые требуют пристального внимания стоматологической службы.
Одним из самых частых врожденных пороков развития среди всех аномалий челюстно-лицевой области является расщелина губы и нёба, популяционная частота которой по разным источникам колеблется от 1:1000 до 1:460 (ежегодно в Москве этот показатель примерно 1:700). Расщелины губы и/или нёба составляют около 87% от всех врожденных пороков развития лица. Почти каждая пятая типичная расщелина является компонентом тяжелого синдрома.
Из 3 трисомий (синдром Дауна, синдром Патау и синдром Эдвардса), которые встречаются у человека, расщелины губы и/или нёба чаще всего возникают при синдроме Патау (около 70%) и считаются наиболее типичным признаком данного синдрома.
Анализ обращаемости в медико-генетические консультации показывает, что чаще всего к этому специализированному виду медицинской помощи обращаются семьи с хромосомными болезнями, врожденными пороками развития и нервно-психическими заболеваниями. Цитогенетический метод и молекулярно-цитогенетические методы позволяют непосредственно выявить все нарушения кариотипа. Они применяются в тех случаях, когда хромосомная аномалия предполагается как наиболее вероятный этиологический фактор патологии в семье.
Для заболеваний, обусловленных числовыми аберрациями хромосом, вероятность повторного случая в семье крайне мала (не превышает 1%), если известно, что ни у одного из родителей нет хромосомной аномалии, а также отсутствуют другие факторы риска (например, средний возраст матери). Исключение составляют транслокации.
Для семей, в которых уже имеется ребенок с трисомной формой синдрома Дауна, риск рождения еще одного больного ребенка повышен (1 на 50-200 новорожденных в отношении синдрома Дауна и 1 на 100 новорожденных в отношении всех хромосомных аномалий).
При аномалиях половых хромосом повторные случаи любой из них в семье исключительно редки. При синдромах ХХУ и ХХХ обнаружена связь с возрастом матери. В этих случаях риск для сибсов оценивается эмпирически (для каждого типа аномалии) с учетом возраста матери. Наиболее неблагоприятным будет прогноз при транслокациях в том случае, если в гаметах одного из родителей имеется сбалансированная хромосомная мутация.
Показания для проведения цитогенетического обследования:
- возраст женщины более 35 лет;
- наличие у предыдущего ребенка хромосомных аномалий;
- врожденные пороки двух и более систем;
- врожденные пороки в сочетании с олигофренией;
- олигофрения неясной этиологии;
- носительство семейной хромосомной перестройки;
- спонтанные аборты и привычное невынашивание беременности;
- патология плода, выявленная при УЗИ. Правила записи аномальных кариотипов по аутосомам:
Любому врачу, сталкивающемуся в своей практической деятельности с хромосомными аномалиями, необходимо знать правила записи нормальных и абберантных кариотипов. При этом необходимо помнить следующее.
1. В самом начале указывается общее число хромосом.
2. Далее идет состав половых хромосом, например: 46, ХУ - нормальный мужской кариотип; 46, ХХ - нормальный женский кариотип; 45, ХО - синдром Шерешевского-Тернера; 47, ХХУ - синдром Клайнфельтера.
3. Добавочная аутосома обозначается соответствующим номером и знаком «+», который ставится перед хромосомой, например: 47, ХУ, +21 (мужской кариотип с синдромом Дауна). Утрата целой хромосомы обозначается знаком «-», например: 45, ХХ, -13 (женский кариотип с моносомией по 13 хромосоме).
4. Короткое плечо хромосомы, как уже отмечалось, обозначается латинской буквой «р», длинное плечо - «q». Например, 46, ХУ, 5 р- (синдром «кошачьего крика»).
5. Транслокация обозначается буквой «t» с расшифровкой в скобках, например, 45, ХХ, t (14/21) - женщина-носительница сбалансированной транслокации 14/21.
6. Присутствие более чем одной клеточной линии (мозаицизм) обозначается знаком дроби, например: 45, Х/46, ХХ - мозаик по синдрому Шерешевского-Тернера.
Этими символами и терминологией пользуются только при рутинном способе окрашивании хромосом человека. С разработкой и внедрением в цитогенетику человека новых методов окрашивания хромосом, в частности дифференциального окрашивания, появились несколько технических процедур, которые воспроизводят индивидуальную специфическую исчерченность метафазных хромосом. Хромосома стала окрашиваться в темные и светлые полосы (band) . При различных методах обработки хромосомных препаратов одни и те же полосы могут быть либо светлыми, либо темными.
В зависимости от цели исследования в клинической цитогенетике используют два принципиальных типа дифференциального окрашивания. При первом типе применяются методы, окрашивающие хромосому на всем ее протяжении (методы G-, Q-, R-полосы). При втором - целенаправленно окрашиваются специфические хромосомные структуры: конституциональный гетерохроматин (С-полосы), теломерные полосы (Т-полосы) и районы ядрышкового организатора (ЯОР).
Каждая индивидуальная хромосома в кариотипе содержит серию чередующихся полос (светлых и темных), которые располагаются по всей длине плеч хромосом в определенных районах. Нумерация полос и участков идет в направлении от центромеры к теломере каждого плеча. Участками (районами) называются сегменты хромосом, находящиеся между двумя соседними полосами. Для обозначения любой хромосомы придерживаются следующего правила - указывается:
1) номер хромосомы;
2) символ плеча (p and q);
3) номер участка (района);
4) номер полосы (или субполосы) в пределах этого участка. Вышеприведенные обозначения записываются по порядку без
пробелов и пунктуации.
Приведем примеры некоторых записей:
- 46, ХУ, del(5)p12) - эта запись относится к делеции короткого плеча 5 хромосомы, участку 1, полосе 2.
- 45, ХУ, rob(13;21)(q10;q10) - означает, что в данном случае имеется робертсоновская транслокация с утратой коротких
плеч 13 и 21 хромосомы; разрыв и воссоединение произошли в 10-м участке (район центромер) длинных плеч обеих хромосом.
- mos 45, ХО/46, ХХ(r) - в этом случае имеется мозаицизм при синдроме Шерешевского-Тернера с кольцевой Х хромосомой.
Более подробные сведения по номенклатуре и классификации хромосомных аномалий в норме и патологии приведены в авторитетных источниках Прокофьевой-Бельговской А.А. (1969), Ворсановой С.Г. (2006) и в международном документе «Международная система для номенклатуры в цитогенетике человека» (2005).
3.5. ЛЕЧЕНИЕ ХРОМОСОМНЫХ БОЛЕЗНЕЙ
Лечение хромосомной патологии - в основном симптоматическое. Цель такой терапии заключается в том, чтобы скорректировать такие фенотипические проявления, как умственная отсталость, замедленный рост, недостаточная феминизация или маскулинизация, недоразвитие гонад, устранение или исправление различных костных дефектов и т.д. Для этого широко используют различные виды терапии, в том числе анаболические гормоны, андрогены и эстрогены, гормоны гипофиза и щитовидной железы, различные витамины и общеукрепляющие средства. Очень широко применяется хирургическое, симптоматическое лечение: удаление катаракты, лишнего (шестого) пальца на ноге или руке, пластические операции при незаращении верхней губы и/или нёба, устранение стеноза привратника и врожденных пороков сердца, удаление различных опухолей и т.д. Перечисленные дефекты часто сопровождают трисомии по хромосомам 13, 18 и 21, триплоидию, синдромы 4р- и 5р- и иные хромосомные аномалии. Из других видов симптоматической терапии следует отметить климатотерапию, бальнеолечение, разные виды электротерапии, теплолечение, рентгенорадиологическое облучение.
Несмотря на широкое разнообразие симптоматической терапии, применяемой для лечения хромосомных болезней, они до сих пор неизлечимы. Учитывая этот фактор, в настоящее время основное внимание уделяется предупреждению рождения детей с хромосомными аномалиями.
3.6. КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА ХРОМОСОМНЫХ БОЛЕЗНЕЙ
К хромосомным болезням относят группу врожденных патологий, которые возникают в результате нарушения числа и структуры хромосом в соматических и половых клетках человека. Общая популяционная частота таких аномалий - около 1%. Как правило, это спорадические случаи; большинство хромосомных заболеваний (90%) возникает за счет новых мутаций. Исключение составляют транслокационные варианты, которые являются результатом сбалансированных транслокаций родителей.
3.6.1. Аутосомные синдромы
Переходя к общей характеристике аутосомных синдромов, следует помнить, что все моносомии по любой из аутосом обычно приводят к внутриутробной гибели плода. Чаще всего в материалах спонтанных абортусов встречаются моносомии. При трисомиях аутосом летальность гораздо меньше, однако родившиеся дети имеют тяжелейшие врожденные пороки развития. Наиболее благоприятное положение наблюдается при наличии в организме мозаицизма. Дети с мозаичным кариотипом обладают повышенной жизнеспособностью, а клиническая картина у них менее выражена. Кроме численных хромосомных нарушений, у человека описано большое количество структурных перестроек.
Известно, что среди живорожденных с аутосомными синдромами чаще всего встречаются полные трисомии по 13, 18 и 21 хромосомам, среди которых 75% приходится на долю синдрома Дауна. Из других полных трисомий по аутосомам зарегистрированы единичные случаи родов по хромосомам 8, 9, 14 и 22.
Синдром Дауна
Первое клиническое описание этой аномалии относится к 1866 г. и принадлежит английскому врачу Ленгтону Дауну. Спустя почти 100 лет цитогенетическую природу синдрома Дауна установил французский исследователь Ж. Лежен в 1959 г., обнаружив у больных лишнюю 21 хромосому. Еще до открытия Ж. Лежена в 1932 г. Варденбург предположил, что причина болезни Дауна, возможно, связана с аномалиями хромосом. К настоящему времени болезнь Дауна изучена достаточно полно, она представляет собой одну из самых частых
хромосомных болезней (встречается с частотой 1:700-1:800). Среди всех умственно отсталых детей больные с синдромом Дауна составляют 10-12%. Соотношение полов при этом заболевании 1:1. На частоту рождения больных с синдромом Дауна не влияют расовые, географические и популяционные различия при сравнении одинакового возраста родителей. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца.
Причиной возникновения болезни Дауна является простое нерасхождение хромосом в мейозе. Вклад материнского нерасхождения составляет от 80 до 90%, а отцовского - от 10 до 20%. Чем старше мать, тем больше риск появления ребенка с синдромом Дауна. Если возраст матери достигает 35-46 лет, вероятность рождения больного ребенка может вырасти до 4%. Вероятность повторного возникновения синдрома Дауна в семье, где родители имеют нормальные кариотипы, не превышает 1-2%.
 Рис. 3.4. Синдром Дауна (трисомия по 21-й хромосоме)
Рис. 3.4. Синдром Дауна (трисомия по 21-й хромосоме)
Цитогенетически болезнь Дауна представлена 3 формами:
- простая (регулярная) трисомия по 21-й хромосоме (94-95% случаев);
- транслокация хромосомы 21 обычно на хромосомы группы D и G (3-4%);
- мозаицизм (1-2%).
Большая часть транслокаций при данном заболевании возникают за счет мутаций de novo. Одна четверть всех случаев транслокаций носит семейный характер, при этом повторный риск достигает 15% и во многом зависит от типа транслокации и от того, кто из родителей несет симметричную перестройку. Если же наследуемая транслокация представлена сочетанием двух хромосом 21q21, то повторный риск рождения больного ребенка 100%.
При молекулярно-генетических исследованиях удалось обнаружить критический район хромосомы 21, который, по мнению многих исследователей, несет ответственность за фенотипические проявления болезни Дауна. Полагают, что основную роль в возникновении умственной отсталости при этом заболевании играет увеличенная доза гена фермента супероксиддисмутазы, находящегося в районе длинного плеча хромосомы 21 (21q22).
Больные с синдромом Дауна обычно невысокого роста, отличаются слабоумием и многочисленными физическими пороками. Они имеют характерную внешность и во многом очень похожи друг на друга. Диагностика этой болезни для акушера и педиатра не представляет особых затруднений даже у больных различных этнических групп.
Характерные признаки: мышечная гипотония, уплощенное лицо, монголоидный разрез глаз, эпикант, брахицефалия, короткий нос с широкой плоской переносицей, маленькие деформированные уши, полуоткрытый рот с высунутым утолщенным бороздчатым языком. Отмечаются катаракты, пятна Брушвильда (очаги белого цвета на границе наружной и средней трети радужки), косоглазие, разболтанность суставов.
При дерматоглифическом исследовании часто обнаруживается длинная поперечная складка на ладони (так называемая обезьянья борозда). В общей популяции этот признак встречается приблизительно у 1%, в то время как при синдроме Дауна его частота достигает 40%. Кроме того, у больных на мизинце имеется всего одна единственная складка (20-25%), которая довольно часто бывает симметричной на обеих руках.
Особенно часто у детей с болезнью Дауна наблюдаются пороки сердечно-сосудистой системы: дефект межжелудочковой перегородки, тетрада Фалло или незаращение артериального протока, иногда отмечаются пороки желудочно-кишечного тракта, гораздо реже встречаются пороки развития почек и мочевыводящих путей.
У больных с синдромом Дауна чаще возникают инфекционные и злокачественные заболевания, что, по-видимому, связано с нестабильностью и слабостью иммунной системы при этом заболевании.
При паталогоанатомическом исследовании размер и масса мозга больных из-за недоразвития уменьшены, ствол мозга и мозжечок маленькие, борозды и извилины развиты не полностью.
Одним из самых важных симптомов при синдроме Дауна является общее психическое недоразвитие. Олигофрения наблюдается от легких до тяжелых форм. Чаще всего у этих больных встречается имбецильность (65-90%), дебильность и идиотия диагностируются примерно в равном соотношении. На первом году жизни дети с таким заболеванием заметно отстают в моторном и психическом развитии. Они позже начинают сидеть и ходить, их мышцы резко гипотоничны, объем движений в суставах увеличен. Обучение во вспомогательных школах возможно, но не всегда; мыслительные процессы заторможены, читают и пишут они с трудом, пересказывают только по вопросам, самостоятельный пересказ вызывает у них большие затруднения. Проведенные в последнее время исследования при болезни Дауна показали, что у них наблюдается более раннее развитие болезни Альцгеймера.
За последнее время продолжительность жизни больных с синдромом Дауна значительно увеличилась. Если раньше такие больные умирали в раннем детстве от различных инфекционных болезней, то теперь они доживают до 30 лет и более. Снижение продолжительности жизни в основном связано со снижением клеточного и гуморального иммунитета; у больных с синдромом Дауна нарушаются процессы репарации ДНК, они подвержены преждевременному старению, смерть часто наступает от сердечно-сосудистой недостаточности, инфекций, ряда онкологических заболеваний.
Лечение болезни Дауна малоэффективно, в основном оно симптоматическое. Широко применяется стимулирующая терапия. Медикопсихологические, медико-педагогические и лечебные мероприятия позволяют адаптировать некоторых больных к посильной трудовой деятельности.
Диагноз болезни Дауна проводится на основании тщательного клинического обследования и обязательного цитогенетического анализа.
Если один из родителей является носителем сбалансированной транслокации с вовлечением 21-й хромосомы, то при планировании
деторождения в такой семье необходимо проводить дородовую диагностику, основанную на цитогенетическом и ультразвуковом обследовании плода. Кроме того, дородовую диагностику плода целесообразно проводить и у женщин старше 35 лет из-за повышенного риска рождения детей с синдромом Дауна.
Синдром Патау
Синдром Патау (синдром трисомии 13-й хромосомы) впервые был описан в 1960 г. Частота встречаемости этого синдрома в популяции - 1:6000-1:13 000 рождений; соотношение полов 1:1. Как и при болезни Дауна, дети с синдромом Патау чаще рождаются у матерей старшего возраста; средний возраст матерей, родивших детей с трисомией 13, около 33 лет, отцов - 34 года. Цитогенетически этот синдром представлен двумя вариантами: простой трисомией и транслокационной формой. В основе синдрома Патау лежит нерасхождение хромосом в мейозе у одного из родителей (в основном у матери) по 13-й паре хромосом. В кариотипе больного наблюдается 47 хромосом с лишней хромосомой 13. Этот вариант встречается у больных с частотой от 80 до 85%; остальные 15-20% представлены транслокационными вариантами. При транслокационной форме в кариотипе больного имеется 46 хромосом. Уменьшение числа хромосом происходит чаще всего в результате слияния двух хромосом группы D или хромосом групп D и G. Реже обнаруживаются и другие цитогенетические варианты (изохромосома, мозаицизм и другие транслокации). Следует заметить, что средний возраст матерей, родивших детей с транслокацией хромосом D/D, не превышает 25 лет.
При рождении у детей с синдромом Патау отмечается пренатальная гипоплазия (масса тела не превышает 2,5 кг); беременность осложняется многоводием (встречается до 50%).
Внешний вид больных с синдромом Патау весьма специфичен. Клинически отмечается резкая умственная отсталость, выраженная микроцефалия, тригоноцефалия, неправильно сформированные и низко расположенные уши, аномалии глазного яблока (микрофтальмия и анофтальм), гипертелоризм, колобома радужки, помутнение хрусталика, одноили двустороннее незаращение губы и нёба, полидактилия, повышенная гибкость суставов, врожденные пороки внутренних органов (кардиоваскулярной и мочевой систем, желудочно-кишечного тракта), часто наблюдаются судороги. Из других клинических симптомов следует отметить гемангиомы на коже
лица и рук, флексорную деформацию пальцев кисти, деформацию стопы, пупочные и пахово-мошоночные грыжи, крипторхизм, глухоту. Глухота у больных с трисомией 13 встречается в 80-85% случаев. Чаще всего изменения ограничены средним и нижней частью внутреннего уха.
При паталогоанатомическом исследовании выявляются множественные внешние и внутренние уродства практически всех органов и систем. Масса мозга уменьшена, часто отсутствует передний мозг; наблюдаются дефекты межжелудочковой и межпредсердной перегородок, камеры сердца расширены. Отмечается аномалия почек, мочеточников.
Из всех перечисленных аномалий ведущими, основными признаками синдрома Патау являются расщелина верхней губы и нёба, полидактилия (часто двусторонняя) и глубокие поражения центральной нервной системы; в ряде случаев отмечаются достаточно грубые пороки - циклопия, этмоцефалия, цебоцефалия и др.
На основании клинических, дерматоглифических и паталогоанатомических данных диагноз поставить несложно. Окончательно он подтверждается цитогенетически. Дифференциальную диагностику следует проводить с врожденными пороками развития (синдромы Меккеля, Мора, тригоноцефалия Опица). Следует отметить крайне важное для практического врача обстоятельство - трисомные и транслокационные формы синдрома Патау по клиническим признакам неотличимы друг от друга, поэтому цитогенетическое исследование у больных для дифференциальной диагностики этих форм обязательно. При транслокационном варианте трисомии 13 вероятность повторного рождения аномального потомства высока, а при трисомном варианте она, вероятно, не превышает аналогичных показателей при болезни Дауна (1-2%).
Прогноз при синдроме Патау неблагоприятен, продолжительность жизни редко превышает 1 год, дети умирают от тяжелых пороков развития, несовместимых с жизнью.
Успешных методов лечения нет.
Синдром Эдвардса
Синдром Эдвардса описан в 1960 г. Частота его среди новорожденных колеблется от 1 на 7000 до 1 на 10 000 детей; девочки поражаются в 3 раза чаще, чем мальчики. Так же как и при синдромах Дауна и Патау, имеется четкая зависимость частоты рождаемости
детей с этим синдромом от возраста матери, но эта зависимость менее выражена. Риск родить больного ребенка не превышает 0,8%. Цитогенетически синдром Эдвардса представлен простой трисомией хромосомы 18 (90%), в 10% случаев наблюдается мозаицизм, который встречается значительно чаще у девочек, чем у мальчиков: вероятнее всего, это связано с большей жизнестойкостью женского организма.
Больные дети часто рождаются недоношенными или переношенными, отмечаются слабая активность плода, многоводие. Дети часто рождаются в асфиксии, с низкой массой тела (2200-2400) и резкой гипотрофией. Череп маленький, сбоку сдавлен, затылочная часть вытянута, лоб маленький, уши расположены низко и их форма почти всегда аномальная, глазные щели узкие, наблюдаются гипертелоризм, эпикант, птоз, часты колобомы, микрофтальмия, катаракта, рот маленький, высокое нёбо, иногда с расщелиной. Шея короткая, иногда с крыловидной складкой, короткая грудная клетка, сердечный горб. Характерно расположение пальцев кистей - они согнуты. Второй палец перекрывает третий, остальные искривлены. Типична форма стопы в виде «качалки» (80%), часто наблюдается косолапость. Постоянны пороки сердца, почек, пищеварительного тракта. У 100% больных отмечается сниженный интеллект, часто идиотия и имбецильность, реже дебильность. Во всех случаях наблюдается нарушение развития головного мозга.
Дерматоглифическая картина типична: на кончиках пальцев рук преобладают дуги или плоские петли (реже), в результате чего общий гребневой счет чрезвычайно низкий. Часто наблюдается поперечная складка ладони.
Цитогенетически у 80% больных обнаруживаются трисомия по хромосоме 18, у 10% - мозаицизм; в остальных случаях имеются другие хромосомные нарушения.
Дифференциальная диагностика очень сложна.
Цитогенетическое исследование должно проводиться во всех случаях для подтверждения диагноза и определения риска рождения будущего потомства.
Клинические проявления при синдроме Эдвардса гораздо тяжелее, чем при синдроме Дауна.
Продолжительность жизни чаще не более 6 мес, лишь 50% детей доживают до 2-месячного возраста, около 10% живут 1 год; некоторые дети доживают до 10 лет. Причина смерти: сердечная недостаточность или инфекционные заболевания.
3.6.2. Синдромы частичных анеуплоидий
При всех хромосомных мутациях (делециях, инверсиях, дупликациях, инсерциях, транслокациях) возникают различные хромосомные аномалии. Большая часть частичных анеуплоидий (частота их составляет 4% обследованных беременностей) не повторяют фенотип полных трисомий и моносомий. Их можно отнести к самостоятельным нозологическим формам. Частичная трисомия, частичная моносомия или их сочетание обычно вызывают нарушение интеллекта, скелетные аномалии и пороки внутренних и наружных органов и систем. Но не все хромосомные мутации приводят к хромосомным заболеваниям. Имеется часть людей, которые являются носителями сбалансированных транслокаций и которые внешне практически здоровы. Тем не менее среди них отмечается пониженная фертильность, спонтанные аборты, рождение потомства с различными аномалиями.
Синдром Вольфа-Хиршхорн
Синдром впервые был описан в 1965 г. Цитогенетически он обусловлен частичной утратой короткого плеча хромосомы 4 (около 80% всех аномалий), причем критическим районом является 4р16 (теряется половина короткого плеча).
Частота встречаемости в популяции: 1 случай на 100 000; соотношение полов 1:1. Средняя масса тела при рождении низкая - не более 2000 гр. Постнатальное развитие очень медленное. Все больные имеют глубокую умственную отсталость. У больных детей наблюдаются микроцефалия, асимметричный череп, гипертелоризм, эпикант, косо расположенные глазные щели, птоз, нистагм, колобома радужки. Отмечается небольшой рот с опущенными углами, расщелины верхней губы и/или нёба, гемангиомы кожи небольших размеров в области лица. Ушные раковины крупные, низко расположенные, нередко оттопыренные, шея короткая и тонкая, туловище вытянутое, конечности тонкие, с ямками на локтях и коленях, пальцы длинные, тонкие с заостренными концами и узкими выпуклыми ногтями. Из внутренних органов чаще всего поражаются сердце и почки, у мальчиков наблюдаются гипоспадия и крипторхизм.
Продолжительность жизни у детей с синдромом 4р- резко снижена; большинство из них не доживают до 1 года.
Для уточнения диагноза больного и определения риска будущего потомства в обязательном порядке показано цитогенетическое обследование.
3.6.3. Синдром «кошачьего крика»
Синдром «кошачьего крика» впервые описал Дж. Лежен с соавторами в 1963 г. у 3 детей с множественными аномалиями, глубокой умственной отсталостью и характерным плачем, который напоминал кошачий крик. Цитогенетически у всех больных обнаруживается укорочение приблизительно на треть и более короткого плеча одного из гомологов 5-й хромосомы. В коротком плече находится участок (15,1-15,2), который непосредственно вызывает развитие этого синдрома. Кроме обычной делеции, хромосомная перестройка может быть представлена другими вариантами (кольцевая хромосома, транслокация, мозаицизм по делеции). Около 85% всех случаев синдрома «кошачьего крика» являются спорадическими, 15% наследуются от фенотипически нормальных родителей - носителей сбалансированных перестроек. Этот синдром встречается гораздо чаще других синдромов, связанных с делециями аутосом; частота его примерно - 1 на 45 тыс.
Клинически синдром «кошачьего крика» очень полиморфен. Корреляцию между величиной делеции хромосомного материала и клиническими симптомами установить весьма трудно. Без своеобразного крика у больного надежный диагноз до цитогенетического исследования установить невозможно, так как большинство клинических симптомов этой болезни встречается и при других хромосомных аномалиях. В типичных случаях у детей с синдромом «кошачьего крика» клинически отмечают круглое лицо с гипертелоризмом, антимонголоидные глазные щели, косоглазие, эпикант, уменьшенный подбородок, плоскую спинку носа, деформированные и низко расположенные уши, короткую шею, нижнюю синдактилию, укороченные пальцы, клинодактилию, врожденные пороки сердца и половых органов, аномалии почек, атрофию зрительного нерва. С возрастом некоторые клинические признаки постепенно исчезают и среди них «кошачий крик», лунообразное лицо, мышечная гипотония. В то же время нарастают отставание умственного и физического развития, косоглазие, микроцефалия.
При паталогоанатомическом исследовании находят микрогирию и гипоплазию мозжечка, уменьшенный мозг, расширенные желудочки мозга, гипоплазию лобных долей, аринэнцефалию, различные пороки сердца, аномалии почек, крипторхизм, экзофтальм, гемангиомы.
Продолжительность жизни больных с данным синдромом зависит от тяжести врожденных пороков развития; большинство из них
умирает рано, некоторые доживают до 10-летнего возраста и более (около 14%).
Лечения нет (паллиативная терапия).
Во всех случаях для уточнения диагноза у больного и расчета риска прогноза потомства в семье показано цитогенетическое обследование, так как среди некоторых семей наблюдаются носители сбалансированных транслокаций.
В клинической практике встречаются и другие частичные анеуплоидии (трисомии, моносомии): 9р+;1q+;18р-; 18q-; 21q-; 22q-; делеции коротких плеч акроцентрических хромосом (13-15; 21-22) практически не имеют каких-либо клинических проявлений. Более подробную информацию об этих и других хромосомных аномалиях можно узнать из монографии С.Г. Ворсановой и др. (2006).
3.6.4. Аномалии половых хромосом
Аномалии половых хромосом у человека представлены различными типами трисомий и моносомий. Оба типа аномалий возникают при слиянии двух видов гамет - нормальной и патологической (с лишней половой хромосомой или без нее). Причиной таких аномалий является нерасхождение хромосом в мейозе или митозе во время первых делений зиготы. Суммарная частота хромосомных аномалий по половым хромосомам составляет от 1,5 до 2,5 на 1000 рождений, большая часть которых составляют полисомии ХХХ, ХХУ и ХУУ.
Характерной особенностью гоносомных аномалий является мозаицизм, т.е. существование в организме клеток с различным числом половых хромосом. Всевозможные сочетания различных клонов клеток (нормальных и аномальных) обусловливают разную клиническую симптоматику у больных с одним и тем же синдромом. Мозаичные формы составляют примерно 25%. Мозаицизм может возникать не только за счет увеличения или уменьшения количества половых хромосом в кариотипе, но и за счет характера комбинации нормальных и патологических клонов хромосомных аномалов. Помимо числовых нарушений, в системе половых хромосом встречаются и структурные перестройки в виде кольцевых хромосом и делеций, которые в большинстве своем обусловливают неправильное формирование наружных и внутренних половых органов. Как правило, численные нарушения в системе половых хромосом (трисомии и моносомии) не вызывают таких тяжелых последствий, как аутосомные аномалии.
У женщин наиболее часто встречаются аномалии половых хромосом, проявляющиеся синдромами Шерешевского-Тернера (ХО) и трипло-Х (ХХХ), а у мужчин - синдромами Клайнфельтера (ХХУ) и двойной У-хромосомы (ХУУ).
Синдром Шерешевского-Тернера
Впервые клиническую картину данного синдрома описал Н.А. Шерешевский в 1925 г. Классическое описание принадлежит Х.Х. Тернеру (1938). Цитогенетическую природу заболевания открыл С.Е. Форд в 1959 г., обнаружив кариотип 45, ХО.
Это единственная форма моносомии, обнаруженная у человека. Частота встречаемости синдрома ХО по разным источникам колеблется от 1 на 1000 до 1 на 7000 и более. Такое разночтение частоты встречаемости данного синдрома может быть объяснено не только присутствием в кариотипе мозаичных вариантов, но и тем, что при различных структурных перестройках Х-хромосомы (изохромосомы, делеции короткого и длинного плеча, кольцевые хромосомы, Х-транслокации) наблюдается одна и та же клиническая картина. По последним уточненным данным, моносомия по Х-хромосоме встречается с частотой от 0,1 до 0,4 на 1000. Синдром ШерешевскогоТернера обнаруживается приблизительно при 1% всех зачатий, среди спонтанных абортусов его находят в 19% случаев; 95% зигот с хромосомным набором погибает внутриутробно.
Кариотип 45, ХО характеризуется большой цитогенетической и клинической вариабельностью. Приблизительно у 60% больных в кариотипе содержится только одна Х-хромосома, в остальных случаях наблюдаются различные типы структурных и числовых нарушений Х-хромосомы. В 80-85% случаев единственная Х-хромосома имеет материнское происхождение и лишь в 15-20% - отцовское.
Клинические симптомы заболевания проявляются с первых дней жизни. Масса тела детей при рождении снижена, отмечается лимфатический отек верхних и нижних конечностей, низкий рост волос на шее. Отек стоп и голеней может держаться от 2 до 3 лет. В течение 1-го года жизни ребенок постепенно отстает в росте, особенно заметно замедление роста в 9-10 лет. В дальнейшем для таких больных низкий рост является одним из самых характерных признаков; у взрослых он не превышает 140-145 см.
Для больных с синдромом Шерешевского-Тернера характерны кожные крыловидные складки на короткой шее (до 60%), широкая
грудная клетка (60%), Х-образное искривление голеней (56%). При полной форме синдрома Шерешевского-Тернера наблюдаются половой инфантилизм, первичная аменорея и бесплодие (90%), внешние и внутренние половы органы недоразвиты, отсутствуют матка и фаллопиевые трубы; наблюдается недоразвитие вторичных половых признаков, связанное с недостатком эстрогенов, которые приводят к недоразвитию молочных желез и скудному оволосению на лобке и в подмышечных впадинах. Поражаются сердечно-сосудистая, мочеполовая, скелетная и кожные системы. При дерматоглифическом обследовании отмечаются дистально расположенный осевой трирадиус, поперечная ладонная складка, увеличение частоты узоров в области гипотенара и высокий гребневой счет. Интеллектуальное развитие нормальное или близкое к норме.
При патологоанатомическом исследовании вместо гонад у таких больных находят недифференцированный тяж, не содержащий фолликулов и секреторных клеток. В 60% случаев встречаются аномалии мочевой системы, чаще подковообразная почка, удвоение почек и мочевыводящих путей; реже описывают врожденные аномалии
сердца (20%).
Предварительный диагноз синдрома Шерешевского-Тернера основан на характерной клинической картине и исследовании полового хроматина, окончательный - на результатах цитогенетического анализа и применении высокоразрешающих молекулярно-цитогенетических методов. Последние методы необходимо применять в случаях определения происхождения маркерных хромосом (минихромосомы) и низкого содержания мозаичных клеток в кариотипе
(до 20%).
Дифференциальную диагностику проводят с синдромом БоневиУльриха - аутосомно-доминантной болезнью, при которой у некоторых больных сохраняется генеративная функция, наблюдается передача патологического гена или генов из поколения в поколение и отсутствует характерная цитогенетическая картина (ХО). Кроме того, синдром ХО необходимо отличать от синдрома Нунан, смешанной дисгенезии гонад, чистой дисгенезии гонад 46, ХХ и чистой дисгенезии гонад 46, ХУ
Лечение в основном симптоматическое и обычно направлено на коррекцию вторичных половых признаков. Лечебные мероприятия проводят обычно эндокринологи (эстрогены, гормон роста), пластические хирурги (удаление крыловидных складок), психотерапевты;
при стертых мозаичных формах синдрома показана гормональная заместительная терапия.
Синдром трипло-Х
Впервые синдром трисомии по Х-хромосоме был описан П. Джекобс и соавторами в 1959 г.
Они обнаружили в ядрах эпителия слизистой оболочки щеки больной два тельца полового хроматина. В среднем женщины с кариотипом ХХХ встречаются с частотой 1-1,4 на 1000 родившихся девочек. Кроме обычного трисомного варианта 47, ХХХ у женщин описаны полисомии по Х-хромосоме с кариотипами 48, ХХХХ и 49,
ХХХХХ.
Клиническая картина этого заболевания чрезвычайно разнообразна. Психиатр, эндокринолог и гинеколог могут встретиться как с отчетливыми клиническими проявлениями этого синдрома, так и со стертыми формами. Всем больным свойственно только присутствие в кариотипе трех хромосом Х. Около 30% таких больных сохраняют генеративную функцию и имеют нормальных детей.
Клинически больные с трипло-Х имеют недоразвитые яичники, гипоплазию матки, нерегулярный менструальный цикл; у них рано наступает вторичная аменорея или бывает преждевременный климакс. У многих больных обнаруживаются неспецифические соматические дизморфии различной выраженности; грубых аномалий развития наружных половых органов не обнаружено. Довольно часто у женщин с ХХХ-хромосомным комплексом отмечается незначительное снижение интеллекта в стадии дебильности. Доказано, что среди них в несколько раз чаще можно встретить лиц с психопатическими чертами и наклонностью к расстройствам шизофреноподобного круга. По данным Ю.И. Филлипова (1971), у взрослых больных с синдромом трипло-Х шизофрения протекает неблагоприятно с выраженными изменениями личности; они склонны к проявлению эпилепсии, особенно в детском возрасте. У таких больных частота трипло-Х в несколько раз выше популяционных показателей. На цитогенетическое исследование больные чаще всего попадают из психиатрических лечебниц и домов инвалидов для детей, которые страдают умственной отсталостью.
Многие исследователи отмечают своеобразную особенность: с увеличением числа Х- хромосом в кариотипе до 4, 5 и более клинические проявления синдрома усиливаются. Больные, имеющие 4,
5 или более Х-хромосом, умственно более отсталые и, как правило, из-за эндокринного дисбаланса у них резко нарушается генеративная функция.
Предварительный диагноз синдрома трипло-Х основан на исследовании полового хроматина. Этот метод позволяет различать больных с аномальным комплексом Х-хромосом и первичной эндокринной патологией. Окончательный диагноз устанавливается по результатам кариологического исследования.
Больные с трипло-Х могут иметь потомство.
Лечение в основном симптоматическое и направлено на коррекцию эндокринного дисбаланса, в первую очередь на устранение нарушений функции яичников.
Синдром Клайнфельтера
Клинически синдром Клайнфельтера описан в 1942 г., а цитогенетически - в 1959 г. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетания (мозаицизма). Обнаружено несколько типов полисомии по хромосомам Х и У у лиц мужского пола: 47, ХХУ; 48, ХХХУ; 49, ХХХХУ; 47, ХУУ; 48, ХУУУ; 48, ХХУУ; 49, ХХХУУ. Наиболее распространен полисомный по хромосоме Х синдром Клайнфельтера (ХХУ). Общая частота его в популяции 1,2 случая на 1000 новорожденных. Примерно у 10% больных с синдромом Клайфельтера наблюдается мозаицизм 46, ХУ/47, ХХУ. Считают, что добавочная Х-хромосома в 60% случаев наследуется от матери.
Общеизвестно, что с увеличением числа половых хромосом в кариотипе больных в большей степени проявляется задержка умственного развития (встречается примерно у 25% больных) и возникает ряд нетяжелых пороков и микроаномалий (пороки сердца, сколиоз, катаракта, изменение дерматоглифических рисунков и т.д.). По сравнению с аутосомными аномалиями нарушения в системе половых хромосом слабо влияют на фенотип. Это связано, вероятнее всего, с тем, что в организме больного активна одна лишь Х-хромосома (остальные инактивированы); псевдоаутосомный участок Х-хромосомы значительно короче любой из аутосом и к тому же число генов в У-хромосоме невелико.
Основные клинические проявления, как правило, у таких больных проявляются в пре- и пубертатном периоде. В период новорожденности они не особенно отличаются от своих сверстников: иногда
выявляется незначительная задержка психомоторного развития и гипоплазия яичек.
Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности, евнухоидизм и гинекомастия (50%), нарушенный сперматогенез и в результате этого бесплодие, уменьшенные яички, повышенное выделение женских половых гормонов, склонность к ожирению, скудное оволосение в подмышечных впадинах и на лобке.
Как уже указывалось, лишняя Х-хромосома обусловливает разнообразные нарушения психики. Больные с этим синдромом очень внушаемы, вялы, апатичны, у них часто отмечается умственная отсталость (обычно дебильность). В период полового созревания повышается титр гонадотропинов в моче; при электроэнцефалографическом исследовании у некоторых больных отмечают эпиактивность и различные аномалии биоэлектрической активности мозга. При патологоанатомическом и гистологическом исследовании в яичках обнаруживают гипоплазию, более или менее выраженный гиалиноз и склерозирующую дегенерацию семенных канальцев; в гипофизе находят недостаток хромофобных и избыток ацидофильных клеток.
Диагностировать синдром Клайнфельтера, особенно у взрослых лиц, нетрудно. Своеобразное сочетание высокого роста, строения скелета по женскому типу, гинекомастии, ожирения и снижения интеллекта позволяет даже без исследования полового хроматина предполагать синдром Клайфельтера. При определении в соскобе слизистой оболочки щеки тельца полового хроматина и тем более при кариотипировании лишней Х-хромосомы диагноз этой болезни не вызывает никаких сомнений.
Из сопутствующих заболеваний у больных с синдромом Клайнфельтера могут быть рак молочной железы, сахарный диабет, болезни щитовидной железы, хронические обструктивные заболевания легких.
Повторный риск рождения для синдрома Клайфельтера не превышает общепопуляционные показатели и составляет 1 случай на 2000 новорожденных.
Специфического лечения нет; при симптоматической терапии применяют гормональные препараты (прогестерон, эстрадиола пропионат, тестостерона пропионат и др.), которые направлены на коррекцию вторичных половых признаков. Однако пациенты даже
после терапии остаются бесплодными. Психотерапия направлена на социальную адаптацию таких больных в обществе.
Синдром ХУУ
Своеобразной разновидностью синдрома Клайфельтера является полисомный по У-хромосоме синдром 47, ХУУ, который впервые был описан в 1962 г. у фенотипически нормального мужчины. Данная хромосомная аномалия встречается у мужчин с частотой 1 случай на 1000 новорожденных.
Клинически синдром ХУУ в общих чертах напоминает синдром Клайфельтера. Однако у мужчин с хромосомным комплексом ХУУ рост гораздо выше - в среднем более 180-185 см; пубертатное ускорение роста наступает раньше и продолжается дольше, чем обычно. Как правило, у большинства индивидов с полисомией по У-хромосоме интеллект сохранен, но умственное развитие соответствует низкой или средней норме; некоторые из этих лиц отличаются агрессивностью и антисоциальным поведением. Большинство больных с ХУУ синдромом выявляются в специализированных учреждениях (психиатрических лечебницах, лечебно-профилактических учреждениях для содержания особо опасных лиц и в тюрьмах). Какихлибо специфических соматических нарушений у большинства таких людей нет, поэтому они часто не попадают в поле зрения врачей. Как и при синдроме Клайнфельтера, у больных с ХУУ синдромом наблюдаются бесплодие, эндокринный дисбаланс, гипогенитализм и азоспермия. При гистологическом исследовании выявляются уменьшение герминативных клеток семенных канальцев, гиалинизация и утолщение базальных мембран.
Радикального лечения нет.
3.6.5. Микроцитогенетические синдромы
В связи с развитием и внедрением в клиническую цитогенетику высокоразрешающих молекулярно-генетических и молекулярноцитогенетических методов диагностики хромосомных болезней за последнее время удалось выделить особую группу синдромов, обусловленных микроперестройками некоторых хромосом. Разработка и внедрение высокоразрешающих методов в цитогенетику человека не в последнюю очередь были связаны с тем, что возникли затруднения при диагностике хромосомных аномалий, вызванных микро-
перестройками, которые невозможно было обнаружить с помощью классических рутинных методов окраски хромосом. Достаточно часто такие микроделеции и микродупликации хромосом, вызывающие умственную отсталость и врожденные пороки развития, относились к менделирующим точечным мутациям, наследуемым по аутосомно-доминантному типу. По материалам пре- и постнатальной цитогенетической диагностики, частота встречаемости аномальных минихромосом в кариотипе человека колеблется от 1 до 2 случаев на 1000. С помощью молекулярно-цитогенетических методов наконец-то удалось установить истинные причины группы заболеваний с нетрадиционным типом наследования. Частота их встречаемости колеблется от 1:50 000 до 1:100 000 новорожденных.
В основе болезней с нетрадиционным типом наследования могут лежать феномены однородительской дисомии и геномного импринтинга. Следует напомнить, что под явлением однородительской дисомии понимается наличие двух гомологичных хромосом (или хромосомных сегментов) одного из родителей (матери или отца); под геномным импринтингом подразумевают различную экспрессию аллелей в зависимости от их родительского происхождения. Различная активность отцовских или материнских генных локусов может оказывать свое влияние на степень развития плаценты, вес плода, развитие и функционирование других органов и систем. В настоящее время для некоторых хромосом (7, 11, 15) геномный импринтинг твердо установлен для других (2, 3, 6, 14 и 20) предполагается. Геномный импринтинг и однородительская диссомия обусловливают ряд заболеваний человека. К таким заболеваниям относятся синдромы Прадера-Вилли, Ангельмана, Лангера-Гидеона, Видемана-Беквита и др.
Клиническая картина этих заболеваний очень вариабельна. Это может зависеть от протяженности хромосомной микроаномалии, качественного состава вовлеченных генов, от материнского или отцовского происхождения перестройки и т.д. Анализ возникновения микроперестройки и причастность ее к той или иной хромосоме играет очень важную роль при планировании деторождения в семье. Возникать такие аномалии могут на любом этапе гаметогенеза, в том числе и после завершения половой клеткой обоих мейотических делений. Другой причиной возникновения частичных анеуплоидий могут стать родительские сбалансированные перестройки (инверсии, транслокации).
Синдром Прадера-Вилли
Впервые синдром Прадера-Вилли был описан в 1956 г. Причиной возникновения этого синдрома является потеря функции хромосомных участков, расположенных в проксимальной части длинного плеча хромосомы 15 (15q11-13). Делеция имеет отцовское происхождение и наблюдается у 70% больных, у 5% заболевание связано с перестройкой хромосомы 15. В большинство случаев заболевание возникает de novo, в 25% случаев синдром возникает в результате однородительской дисомии. У некоторых больных хромосомную аномалию не удается идентифицировать, но у них наблюдается характерная клиническая картина синдрома Прадера-Вилли.
Основными клиническими признаками являются отставание умственного развития, неадекватное поведение, задержка физического развития, низкорослость, гипотония. Одни клинические признаки при этом заболевании можно наблюдать до 3-летнего возраста (мышечная гипотония, малый вес и трудности вскармливания), другие начинают преобладать после 6-месячного возраста (ожирение, усиление аппетита, нарастание умственной отсталости, отставание в росте). Наряду с диспластическими признаками (опущенные углы рта, высокое нёбо, гипертелоризм, эпикант, маленькие стопы и кисти, миндалевидный разрез глаз, аномалии дерматоглифика), выявляются гипогонадизм, обусловленный низким уровнем половых гормонов, гипопигментация (у 75% больных). Следует отметить, что синдром Прадера-Вилли характеризуется широким клиническим полиморфизмом, поэтому необходимо проводить дифференциальную диагностику с синдромами Коэна, Опица-Фриаса, Барде-Бидля.
Продолжительность жизни составляет 25-30 лет.
Диагностика заболевания осуществляется с помощью ДНК-анализа или методом FISH. Риск для сибсов пробанда - около 1%.
Синдром Ангельмана
Если для возникновения синдрома Прадера-Вилли основной причиной являлась делеция проксимальной части длинного плеча хромосомы 15 отцовского происхождения, то аналогичная потеря той же части длинного плеча хромосомы 15, но только материнского происхождения обусловливает развитие другой патологии - синдрома Ангельмана. При этом заболевании развивается совсем другая клиническая картина. Для синдрома Ангельмана характерно: выраженная олигофрения, задержка речи, гиперактивное поведение,
судороги, большая нижняя челюсть, макростомия, гипопигментация (у 40% больных). Они поздно начинают ходить, для них характерна походка с широко расставленными ногами, локтевые суставы согнуты; отмечается насильственный немотивированный смех, имеются выраженные расстройства координации движений.
Дифференциальную диагностику следует проводить с синдромами Петерса-Пласа, Ретта и с тригоноцефалией Опица.
Частота синдрома в популяции составляет 1:20 000.
Примерно 20-30% больных не имеют делеции проксимальной части длинного плеча хромосомы 15; у незначительного числа больных причиной является однородительская дисомия. Диагностика синдрома осуществляется теми же методами, что и при синдроме Прадера-Вилли, т.е. проводится ДНК-анализ и метод FISH. С помощью них можно установить этиологию около 90% случаев заболевания. Риск для сибсов пробанда не известен.
Синдром Видемана-Беквита
Не менее интересным и познавательным в плане феномена геномного импринтинга является терминальный район другой хромосомы - район короткого плеча хромосомы 11, структурные и функциональные аномалии которого обусловливают широко распространенный синдром Видемана-Беквита, нефробластому, некоторые опухоли детского возраста.
Синдром Видемана-Беквита достаточно распространенное заболевание, встречающееся с частотой 1:10 000 - 1:12 000 новорожденных.
Характерными клиническими признаками этого синдрома являются: гигантизм, пупочная грыжа, макроглоссия, гипоплазия верхней челюсти, прогнатизм, долихоцефалия и другие диспластические стигмы; при рождении отмечается гипогликемия; у больных отмечается повышенная предрасположенность к возникновению опухолей.
Первоначально предпологалось, что синдром Видемана-Беквита наследуется по аутосомно-доминантному и аутосомно-рецессивному типу наследования. Не исключалась возможность и мультифакториального наследования. Но в настоящее время принято считать, что синдром наследуется аутосомно-доминантно с неполной пенетратностью и вариабельной экспрессивностью, причем при наследовании большую роль играет феномен геномного импринтинга (в некоторых семьях клинические проявления отмечаются в 15%). Интерес же
цитогенетиков был связан с тем, что при синдроме Видемана-Беквита с помощью высокоразрешающих молекулярных методов удалось установить частичную трисомию дистального участка короткого плеча 11-й хромосомы отцовского происхождения. Кроме того, в 20% случаев причиной синдрома Видемана-Беквита является однородительская дисомия, а также в некоторых семьях описаны сбалансированные транслокации между 11-й и 22-й хромосомами, которые вызывают аналогичный синдром. В общей сложности структурные аномалии короткого плеча 11-й хромосомы встречаются в 2% и могут быть унаследованы как от матери, так и от отца.
При определении риска у потомства в семьях с синдромом Видемана-Беквита необходимо проводить тщательное молекулярно-генетическое и молекулярно-цитогенетическое обследование.