Клиническая генетика : учебник / Н. П. Бочков, В. П. Пузырев, С. А. Смирнихина ; под ред. Н. П. Бочкова. - 4-е изд., доп. и перераб. - 2011. - 592 с. : ил.
|
|
|
|
Глава 4. ГЕННЫЕ БОЛЕЗНИ
ЭТИОЛОГИЯ
Генные болезни - разнородная по клиническим проявлениям группа заболеваний, обусловленных генными мутациями. Основой для объединения их в одну группу служат этиологическая генетическая характеристика и соответственно закономерности наследования в семьях и популяциях. Коль скоро мутации в индивидуальных генах являются этиологическим фактором генных болезней, то закономерности их наследования соответствуют менделевским правилам расщепления в потомстве, т.е. формальная генетика генных наследственных болезней ничем не отличается от «поведения» в семьях любых менделирующих признаков. «Поведение» некоторых патологических генов может отклоняться от менделевско-моргановских правил в связи с фенотипическими эффектами (летальность, стерильность). Необходимо, однако, сразу сделать пояснения в отношении содержания понятий «генные мутации» и «менделирующая наследственность» у человека.
Во-первых, согласно многочисленным исследованиям разных наследственных болезней и генома человека в целом, можно говорить о многообразии видов мутаций в одном и том же гене, которое является причиной наследственных болезней. У человека описаны все типы генных мутаций, обусловливающие наследственные болезни: миссенс, нонсенс, сдвиг рамки считывания, делеции, вставки (инсерции), нарушения сплайсинга, увеличение числа (экспансия) тринуклеотидных повторов. Любой из этих видов мутаций может вести к наследственным болезням. Даже одна и та же генная болезнь может быть обусловлена разными мутациями одного и того же гена. Например, в гене муковисцидоза описано около 300 вызывающих болезнь мутаций (всего их более 1500) следующих типов: делеции, миссенс, нонсенс, сдвиг рамки считывания, нарушения сплайсинга. Для гена фенилкетонурии известно более 30 патологических мутаций (миссенс, нонсенс, делеции, нарушения сплайсинга).
Во-вторых, современная генетика, принимая в полной мере менделизм, делает поправки в определенных случаях. К ним относятся
условность понятий о доминантности и рецессивности, неоднородность проявления аллеля, унаследованного от отца или матери (импринтинг), сложное взаимодействие генов, гонадный мозаицизм и т.д. Более того, установлено, что мутации в разных частях одного гена ведут к различным болезням. Например, мутации в разных частях RET-онкогена ведут к 4 клинически разным наследственным болезням: двум формам полиэндокринного аденоматоза (ZA) и (ZB), семейной медуллярной тиреоидной карциноме, семейной болезни Гиршпрунга.
Мутации, вызывающие наследственные болезни, могут затрагивать структурные, транспортные и эмбриональные белки, ферменты.
Белковые классы, ассоциированные с моногенными болезнями, имеются фактически во всех составных элементах клетки (табл. 4.1).
Таблица 4.1. Примеры классов белков, ассоциированных с моногенными болезнями
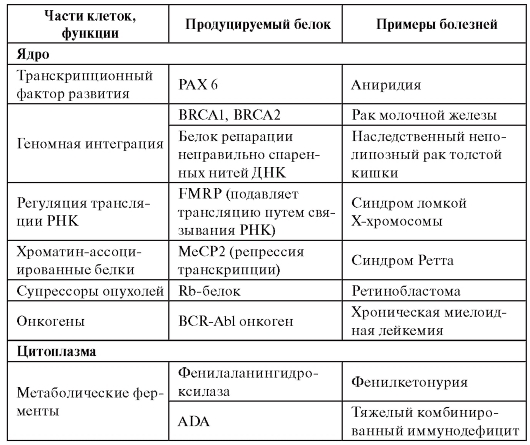
Продолжение таблицы 4.1
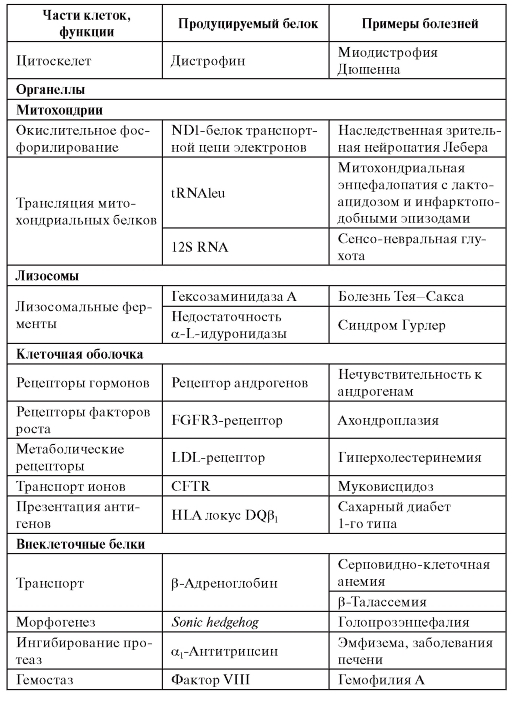
Окончание таблицы 4.1
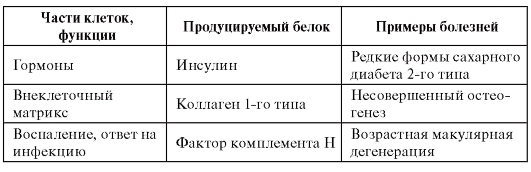
Примечание. CFTR - кистофиброзный трансмембранный регулятор. HLA - человеческий лейкоцитарный антиген (Human Leucocyte Antigen). RNA - рибонуклеиновая кислота.
Существует несколько уровней регуляции синтеза белков: претранскрипционный, транскрипционный, трансляционный. Можно предположить, что на всех этих уровнях, обусловленных соответствующими ферментативными реакциями, могут проявляться наследственные аномалии. Если принять, что у человека примерно 30 000 генов и каждый ген может мутировать и контролировать синтез белка с другим строением, а многим генам свойственно еще явление альтернативного сплайсинга, то, казалось бы, должно быть не меньшее число наследственных болезней. Более того, по современным данным, в каждом гене может возникать до нескольких сотен вариантов мутаций (разные типы в различных участках гена). На самом деле более чем для 50% белков изменения генетической природы (первичная структура) приводят к гибели клетки, и мутация не реализуется в наследственную болезнь. Такие белки называются мономорфными. Они обеспечивают основные функции клетки, консервативно сохраняя стабильность видовой организации этой клетки.
Так или иначе, число генных болезней действительно велико. Менделирующих болезней, согласно справочнику OMIM, на 2008 г. зарегистрировано около 4000, из них 84% (более 3300) обусловлены мутациями в 1990 генах. Оставшиеся 16% болезней (OMIM) отчетливо менделирующие, но мутантные гены их еще неизвестны. Количество болезней с известными генетическими причинами и количество генов, мутации в которых могут вызывать болезни, не совпадают. Обусловлено это тем, что разные мутации в одном и
том же гене могут вызывать различные болезни, а мутации в разных генах могут вызывать неразличимые болезни. Непосредственно с развитием генетических болезней связано около 8% генов. Это, безусловно, пока существенная недооценка. Впереди еще большая работа по «инвентаризации» наследственных болезней, если их рассматривать не только с клинической (фенотипической), но и с генетической точек зрения.
При рассмотрении генных болезней как менделирующих признаков организма речь идет о так называемых полных формах, т.е. формах, обусловленных гаметическими (в зародышевых клетках) мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. Следовательно, в этих случаях патологические гены присутствуют во всех клетках организма. Однако теоретически можно представить возможность появления и мозаичных, а не только полных форм, подобно хромосомным болезням. Любые мутации, в том числе и генные, могут возникать на ранних стадиях дробления зиготы в одной из клеток, и тогда индивид будет мозаичен по данному гену. В одних клетках у него будет функционировать нормальный аллель, в других - мутантный или патологический. Если эта мутация доминантная, то она проявится в соответствующих клетках и, очевидно, приведет к развитию менее тяжелой формы болезни. Если возникшая мутация в одной из клеток на ранних стадиях развития зародыша рецессивная, то ее эффект проявится только у гетерозиготы. Вероятность появления двух рецессивных мутаций в одном и том же локусе гомологичных хромосом в одной соматической клетке чрезвычайно мала.
Проблема мозаичных форм генных болезней и в генетическом, и в клиническом плане исследована недостаточно. Частота возникновения мозаичных форм не может быть высокой, поэтому выявлять их трудно. Современные молекулярно-генетические методы позволяют диагностировать мозаицизм на клеточном или тканевом уровне. В одной и той же ткани обнаруживают клетки, несущие разные генотипы по изучаемой патологической мутации. Соматические мутации, появляющиеся на ранних стадиях развития организма, дают больший эффект, чем мутации на поздних. В последние годы соматический мозаицизм был доказан при 30 генных болезнях, среди которых нейрофиброматоз I типа, миотоническая дистрофия, миодистрофия Дюшенна, гемофилия А и В, синдром Альпорта, синдром Марфана, синдром андрогенной нечувствительности, тубероз-
ный склероз и др. Соматический мозаицизм был обнаружен также при злокачественных новообразованиях (колоректальный рак и рак предстательной железы).
С мозаичными формами генных болезней не следует путать мозаицизм гонад. Мозаицизм гонад - частный случай органного мозаицизма, возникающего на более поздних стадиях эмбрионального развития в процессе органогенеза. Мозаицизм гонад у клинически здорового индивида может обусловить несколько случаев рождения детей с полной формой доминантной наследственной болезни.
На рис. 4.1 приведена родословная здоровых родителей (французская семья), у которых трое из четверых детей больны ахондроплазией.
Ахондроплазия - аутосомно-доминантное заболевание с полной пенетрантностью гена. Клиническая и рентгенологическая диагностика этой болезни (в частности, в упомянутой выше семье) не вызывает сомнений. Объяснить семейные случаи можно гонадным мозаицизмом у отца, поскольку больные дети родились в двух его браках. Возможно еще одно объяснение подобных случаев: болезнь возникла в результате премутации в одном из аллелей этого гена у
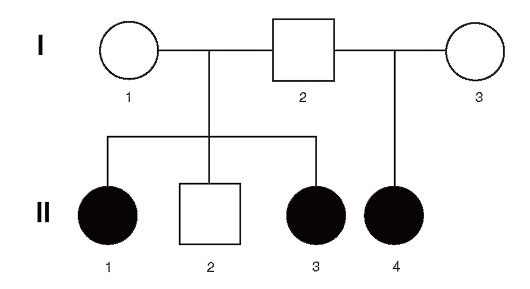
Рис. 4.1. Родословная семьи с 3-мя случаями ахондроплазии в одном поколении от двух браков: I: 1 - рост 163 см; 2 - рост 166 см; 3 - рост 164 см; клинических и радиологических признаков ахондроплазии не имели; II: 1 - родилась от неродственного брака (мать 17 лет, отец 30 лет). Ахондроплазия заподозрена при рождении, позже подтверждена; 2 - здоровый мальчик; 3 - больная ахондроплазией. Диагноз установлен после рождения; 4 - во втором браке отца диагноз ахондроплазии у его ребенка был установлен внутриутробно на 7-м месяце [сначала с помощью ультразвукового исследования (УЗИ), затем рентгенографически]. При рождении диагноз подтвержден
родителя, которая реализуется в мутацию при прохождении через мейоз. Однако в гене ахондроплазии премутантных состояний пока не обнаружено.
Современные молекулярно-генетические исследования показали, что родительский мозаицизм (в том числе гонадный) ответствен не менее чем за 5-15% случаев доминантных и Х-сцепленных рецессивных болезней. Мозаицизм гонад у здоровых родителей убедительно доказан в случаях рождения детей (по соответствующим генам) с несовершенным остеогенезом, синдромом Элерса-Данло, гемофилией (факторы VIII и IX).
В связи с многообразием мутаций в одном и том же гене возникает вопрос об этиологической зависимости клинической картины болезни от характера мутаций. Ответ на этот вопрос неоднозначен и пока не всегда ясен. Определенно можно сказать, что в большинстве случаев такой зависимости нет, хотя в некоторых случаях она присутствует. Объяснение этому можно найти в первичных механизмах развития генных болезней, т.е. в первичных эффектах мутантных аллелей. С клинико-генетической точки зрения эти аллели называют патологическими для отличия от других мутантных состояний этого же гена, которые ведут к биологическим межиндивидуальным вариациям без патологических проявлений признака.
Первичные эффекты мутантных аллелей могут проявляться в 4-х вариантах: отсутствии синтеза полипептидной цепи (белка); синтезе аномальной по первичной структуре полипептидной цепи (белка); количественно недостаточном синтезе полипептидной цепи (белка); количественно избыточном синтезе полипептидной цепи (белка).
Независимо от характера изменений первичного продукта гена эффект мутаций может выражаться в разных вариантах нарушения функций:
- Потеря функции белка в результате либо ингибирования процессов транскрипции/трансляции, либо изменения его структуры и функциональных свойств.
- Появление новой функции. При мутациях такого типа у мутантного белка наряду с нормальной функцией появляются новые цитотоксические свойства, приводящие к гибели клеток.
- Доминантный негативный эффект проявляется тогда, когда первичный продукт мутантного аллеля ингибирует функцию нормальных белков.
- Изменение дозы гена (делеции или дупликации) может приводить к нарушению пространственной структуры молекулярного продукта.
На основе первичного эффекта мутантного аллеля развертывается весь сложнейший патогенез генной болезни, проявляющийся в разнообразных фенотипических эффектах или клинической картине.
Результатом действия патологической мутации (фенотипический эффект) может быть, прежде всего, летальность на ранних стадиях развития зародыша, до имплантации. Механизмы такой летальности еще не выяснены, но ее существование у человека не вызывает сомнений. Это проявляется в виде несостоявшегося зачатия (имплантации) у фертильных женщин при нормальной половой жизни. У молодых женщин зачатие наступает в среднем через 3 мес регулярной половой жизни (без контрацепции). Примерно 50% несостоявшихся зачатий обусловлены гибелью зиготы по генетическим причинам (генные, хромосомные и геномные мутации). Если развитие эмбриона с патологической генной мутацией не остановилось на ранних стадиях, то фенотипические эффекты в зависимости от вовлеченного гена и характера мутации формируются в виде 3 вариантов: дисморфогенеза (врожденных пороков развития), нарушенного обмена веществ, смешанных эффектов (дисморфогенеза и аномального обмена веществ).
Влияние патологических мутаций начинает реализовываться в разные периоды онтогенеза - от внутриутробного до пожилого возраста. Большая часть патологических мутаций проявляется внутриутробно (до 25% всей наследственной патологии) и в допубертатном возрасте (45%). Еще 20% проявляется в пубертатном и юношеском возрасте, и лишь 10% моногенных болезней развивается в возрасте старше 20 лет.
Для понимания природы генных болезней очень важно иметь представление о том, что клиническая картина заболевания может сформироваться вследствие включения разных патогенетических звеньев, которые могут быть обусловлены фенотипическими эффектами мутаций разных генов. Следовательно, в одну группу будут включены разные с генетической точки зрения заболевания (мутации в разных локусах). Такие случаи называются генокопиями. Наряду с этим, хотя и редко, могут встречаться фенокопии генных болезней. Это те случаи, при которых повреждающие внешние факторы, действующие, как правило, внутриутробно, вызывают болезнь, по
клинической картине в общих чертах сходную с наследственной. Противоположное состояние, когда при мутантном генотипе индивида в результате средовых воздействий (лекарства, питание и т.п.) болезнь не развивается, называют нормокопированием.
Понятия о гено- и фенокопиях помогают установить правильный диагноз, а также более точно определить прогноз здоровья пациента или вероятность рождения больного ребенка. Понимание принципов нормокопирования дает врачу возможность предупредить развитие болезни у ребенка, унаследовавшего патологический ген.
КЛАССИФИКАЦИЯ
Как и для любой группы заболеваний, классификация генных болезней условна и многокомпонентна. В основу классификации генных болезней можно положить генетический, клинический или патогенетический принцип.
В соответствии с генетическим принципом классификации генные болезни можно подразделить на группы согласно типам наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные доминантные, Х-сцепленные рецессивные, Y-сцепленные (голандрические) и митохондриальные. Отнесение болезни к той или иной группе помогает врачу сориентироваться относительно ситуации в семье и определить вид медико-генетической помощи. Выше (см. главу 3) были рассмотрены характеристики наследования каждой из этих групп.
Клинический принцип классификации генных болезней учитывает систему или орган, наиболее вовлеченный в патологический процесс. Так, различают наследственные болезни нервные, нервномышечные, кожные, глазные, опорно-двигательного аппарата, эндокринные, крови, сердечно-сосудистой системы, психические, мочеполовой системы, желудочно-кишечного тракта (ЖКТ), легких. Для некоторых групп болезней установились даже специальные термины: нейрогенетика, дерматогенетика, офтальмогенетика. Условность клинического принципа классификации очевидна. Некоторые болезни у одних больных больше проявляются в одной системе, у других - в другой. Например, муковисцидоз может преимущественно поражать или ЖКТ, или легкие. Нейрофиброматоз I типа может выражаться либо кожными изменениями (пигментные пятна, нейрофибромы),
либо опухолями нервных стволов и мозга. Несмотря на некоторую условность, клиническая классификация помогает врачу соответствующего профиля концентрировать внимание на наследственных болезнях, встречающихся в его практике.
Патогенетическая классификация наследственных болезней подразделяет их на 3 группы в зависимости от того, в чем проявляется основное патогенетическое звено. Патогенез болезни может привести к нарушенному обмену веществ, аномалиям морфогенеза или комбинации того и другого. В соответствии с этим различают наследственные болезни обмена веществ, врожденные пороки развития (моногенной природы) и комбинированные состояния. Наследственные болезни обмена веществ, в свою очередь, подразделяют по типам обмена (углеводный, аминокислотный, обмен витаминов, липидов, металлов и др.).
ОБЩИЕ ЗАКОНОМЕРНОСТИ ПАТОГЕНЕЗА
Начало патогенеза любой генной болезни и его ключевая точка связаны с первичным эффектом мутантного аллеля, поэтому принципиальные звенья патогенеза генных болезней можно представить следующим образом: мутантный аллель - патологический первичный продукт (качественно или количественно) - цепь последующих биохимических процессов - клетки - органы - организм. Это главная и общая закономерность развития генных болезней при всем их многообразии.
Мутации могут вызывать болезни через множество различных механизмов. Они могут затрагивать посттрансляционный процессинг, формирование клеточных компартментов, функцию белка и взаимодействие первичных продуктов. Молекулярные основы патогенеза большинства болезней еще не полностью понятны. Предстоит проследить последствия мутации от эффектов на молекулярном уровне до физиологии и клиники болезни, что является задачей молекулярной медицины.
Функция большинства генов определяется трехмерной структурой их белковых продуктов. Хотя многие патологические мутации в генах локализованы в некодирующих областях, большинство охарактеризованных мутаций поражает структуру и функцию белков. В целом, речь при этом идет о болезнях, ассоциированных с одним геном с
высокой пенетрантностью. Большие делеции, вставки или инверсии в протеинкодирующих областях гена почти неизбежно нарушают функцию белка. Наследуемые болезни обусловлены патологическими мутациями, не ведущими к смерти гетерозиготного носителя до пострепродуктивного возраста.
Патогенез болезни на молекулярном уровне
В зависимости от контролируемого конкретным геном продукта и от характера его нарушения при мутации соответствующим образом развертывается патогенез болезни на молекулярном уровне.
Если в результате мутации будет вырабатываться избыточное количество продукта, то патогенез болезни в целом будет обусловлен усиленной генной активностью. Существование таких вариантов можно предполагать, но обнаружен он лишь в единичных формах наследственных болезней. Пример такой генной мутации - мутация в гене FII, приводящая к усиленному синтезу протромбина.
При другом варианте патологического эффекта мутантного гена синтезируется аномальный белок. За этим следуют нарушения той системы (клетки, органа), функции которой обеспечиваются нормальным белком. Эти нарушения первоначально развертываются на молекулярном уровне. Примером такого варианта патогенеза болезни может быть серповидно-клеточная анемия. В результате замены урацила на аденин в кодоне GUA синтезируется цепь молекулы глобина с глютамином, заменившим валин. Замены одной аминокислоты оказывается достаточно, чтобы изменить функциональные свойства гемоглобина (пониженную растворимость, повышенную полимеризацию). Такой гемоглобин уже не может выполнять кислородакцепторную функцию и кристаллизуется при недостатке кислорода, а эритроциты приобретают серповидную форму (отсюда и название болезни), склеиваются, тромбируют капилляры и т.д. (рис. 4.2).
Третий вариант патологического эффекта мутантного аллеля - отсутствие выработки первичного продукта. Этот вариант, очевидно, встречается наиболее часто. Естественно, что в этих случаях нарушается тот или другой процесс нормального биохимического гомеостаза. Это выражается в накоплении токсичных продуктов-предшественников (рис. 4.3). На схеме представлены
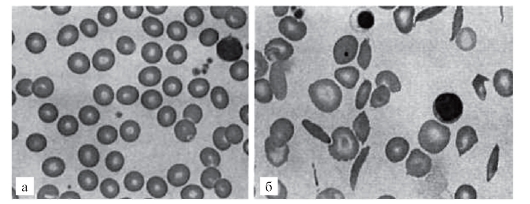
Рис. 4.2. Мазок крови больного серповидно-клеточной анемией (б) по сравнению с нормой (а). Патология: серповидные эритроциты, пойкилоцитоз, анизоцитоз, склеенные эритроциты. Пояснения в тексте
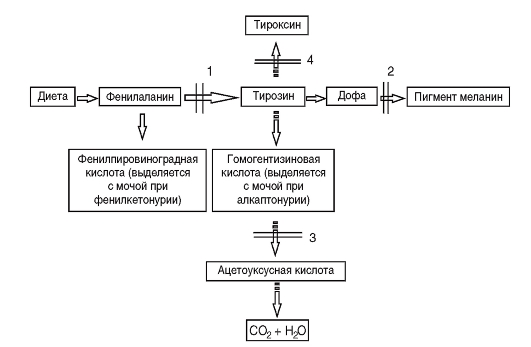
Рис. 4.3. Биохимические «блоки» при наследственных нарушениях обмена аминокислот: 1 - фенилкетонурия; 2 - альбинизм; 3 - алкаптонурия; 4 - врожденная недостаточность тироксина (дисгормоногенез)
результаты наследственных нарушений аминокислот фенилаланина и тирозина. При фенилкетонурии в крови накапливаются фенилаланин и продукты его патологического метаболизма (1), поскольку он из-за отсутствия фенилаланингидроксилазы не превращается в тирозин. Нарушение обмена тирозина приводит к патологии образования меланина (2) или тироксина (4). Недостаточность синтеза оксидазы гомогентизиновой кислоты (сущность мутации в этом гене) ведет к накоплению гомогентизиновой кислоты в крови (3), которая из-за высокой концентрации откладывается в хрящах и клапанах сердца. В конечном счете с возрастом это приводит к артритам и порокам сердца. Возможны и другие (обходные) пути обмена, часто также с патологическим исходом. В результате отсутствия первичного продукта гена может задерживаться какой-либо важный процесс, постоянно осуществляющийся в организме. Так, мутации генов, детерминирующих синтез ферментов репарации ДНК, приводят к невозможности восстановления постоянно возникающих нарушений в структуре ДНК, что обусловливает развитие злокачественных новообразований (пигментная ксеродерма, атаксиятелеангиэктазия).
Известен и 4-й вариант первичного патологического эффекта мутантного аллеля - выработка уменьшенного количества нормального первичного продукта (β-талассемия, акаталазия). Патогенез таких заболеваний различен, поскольку наряду с нормальным путем обмена веществ будут протекать и патологические процессы.
Выше были описаны общие закономерности патогенеза генных болезней на молекулярном уровне на примерах нарушения обмена веществ. Тот же самый принцип патогенеза (мутантный аллель - патологический первичный продукт) действует и для генов морфогенетического контроля, мутации в которых приводят к врожденным порокам развития (полидактилия, синдромы Холта- Орама, Крузона, Нунан, Лоренса-Муна, Меккеля, Робертс, Эллиса-Ван-Кревельда, Гольтца: рис. 4.4, 4.5). Начальное звено врожденного порока развития связано с нарушением дифференцировки клеток. Запрограммированные в геноме дифференцировка клеток, а затем и органогенез осуществляются путем смены процессов активации и выключения определенных генов в строго
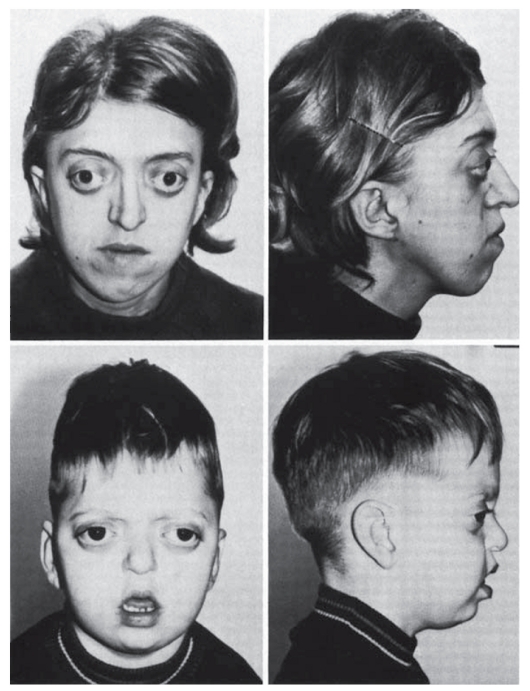
Рис. 4.4. Синдром Крузона. Мать и сын
ограниченных временных (по отношению к онтогенезу) промежутках (транскрипционные факторы). Если первичный продукт морфогенетического гена аномальный, то необходимая для дальнейшего правильного развития органа дифференцировка клеток не последует. Естественно, что морфогенетических генов много, они действуют в разные периоды онтогенеза. Соответственно мутации в них будут приводить к специфическим врожденным порокам развития.
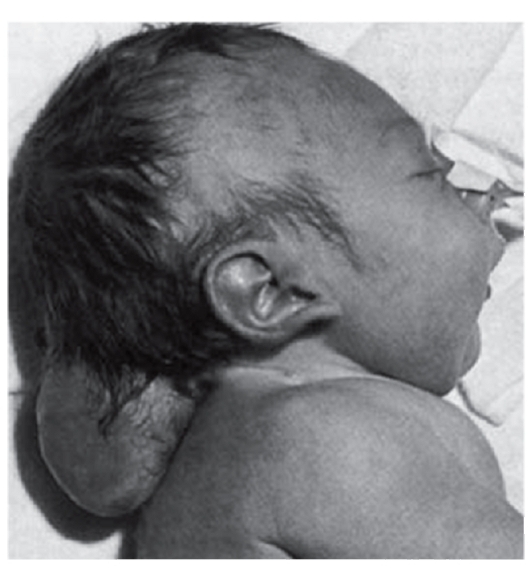
Рис. 4.5. Синдром Меккеля (энцефалоцеле)
Клеточный уровень патогенеза генных болезней
Патогенез генных болезней не заканчивается на молекулярном уровне даже в первичных звеньях. Для многих болезней главное звено патогенеза - клетка. Во всех генетических процессах клетка - дискретная самостоятельно регулируемая единица, и в ней осуществляются все процессы реализации генетической информации (транскрипция, трансляция, синтез белка). Это - общебиологическая аксиома. Клеточный уровень патогенеза генных болезней означает, что в определенных типах клеток разыгрываются основные патологические процессы, присущие конкретной нозологической форме. Клетка как бы не выпускает из себя патологические явления, а принимает на себя удар первичного патогенного эффекта гена. Точкой приложения первичного действия мутантного гена являются отдельные структуры клетки, разные при различных болезнях (лизосомы, пероксисомы, мембраны, митохондрии).
Патогенетические процессы на клеточном уровне развертываются при болезнях накопления (или лизосомных) в связи с нарушением активности лизосомных ферментов. Так, накопление в клетках, а затем и в основном межклеточном веществе, гликозаминогликанов (мукополисахаридов) приводит к развитию тяжелых заболеваний - мукополисахаридозов. Причина избыточного содержания полимеров - гликозаминогликанов - заключена в отсутствии их деградации в лизосомах. Нарушение деградации гликозаминогликанов связано с дефектами в группе специфических
ферментов, катализирующих весь цикл деградации. Более подробную информацию о клинической картине, диагностике и лечении мукополисахаридозов см. в статье С.В. Михайловой с соавт. «Мукополисахарозы: дифференциальня диагностика и лечение» на компакт-диске.
Другим примером болезней накопления могут служить гликогенозы. В клетках печени и мышц накапливаются полимеры гликогена, которые не подвергаются деградации даже тогда, когда организму необходима глюкоза в крови. Патогенез гликогенозов принципиально такой же, как и мукополисахаридозов. В клетках печени и мышц отсутствует определенный фермент (их уже известно много), который участвует в цикле расщепления гликогена до глюкозы.
Другие внутриклеточные структуры - пероксисомы - также могут быть точкой приложения первичного действия мутантного гена. В этих случаях развиваются так называемые пероксисомные болезни. Описано уже 18 нозологических форм. Основное патологическое звено при всех пероксисомных болезнях локализовано в пероксисомах в виде биохимических нарушений, обусловленных генными мутациями. Биохимическая сущность многих пероксисомных болезней уже раскрыта на уровне мутантных ферментов. Клинически болезни проявляются в виде множественных врожденных пороков развития, в целом сходных при разных нозологических формах (множественных черепно-лицевых дисморфиях, катаракте, кожных складках на шее, почечных кистах и др.). Пероксисомные болезни - пример наследственных болезней обмена, при которых множественные пороки развития объясняются молекулярным дефектом.
Различают 3 группы пероксисомных болезней: генерализованные с измененным числом пероксисом (пример - цереброгепаторенальный синдром, или синдром Целлвегера); с неизмененным числом пероксисом и нарушением нескольких биохимических функций (пример - целлвегерподобный синдром); с неизмененным числом пероксисом и нарушением единственной биохимической функции (пример - болезнь Рефсума).
Мембраны, так же, как и структуры клеток, могут быть ключевыми элементами патогенеза генных болезней. Так, отсутствие специфических белковых молекул-рецепторов на клеточной поверхности, связывающих ЛПНП, приводит к семейной гиперхолестеринемии.
Синдром полной нечувствительности к андрогенам (синоним: синдром тестикулярной феминизации) вызывается мутациями в Х-сцепленном гене, который кодирует синтез внутриклеточного
рецептора андрогенов. Отсутствие чувствительности клеток к андрогенам приводит к развитию женского фенотипа при хромосомном наборе XY. У таких больных, несмотря на женское строение наружных половых органов, имеются семенники в брюшной полости и нормальный уровень андрогенов в крови.
Клиника витамин-D-резистентного рахита (аутосомно-доминантное заболевание) обусловлена дефектом рецепторов 1,25-ди- гидроксихолекальциферола.
При муковисцидозе нарушается регуляция транспорта хлоридов через мембраны эпителиальных клеток. Такая регуляция в норме осуществляется белком-продуктом гена, названным кистофиброзным трансмембранным регулятором (CFTR). Одни мутации в гене CFTR ведут к снижению синтеза данного белка из-за незавершенности процессинга РНК, другие - к качественным изменениям мембранных хлорных каналов. Одна первичная биохимическая аномалия (нарушение транспорта хлоридов) обусловливает возникновение мультиорганного патологического процесса (прогрессирующее поражение дыхательных путей, хронические синуситы, недостаточность экзокринной секреторной функции поджелудочной железы, стерильность у мужчин).
Клеточный уровень патогенеза генных болезней может проявляться не только в конкретных органеллах, но и в виде нарушения скоординированности функций клетки. Так, мутации, затрагивающие области онкогенов, ведут к снятию контроля размножения клеток (репрессия антионкогенов) и соответственно к злокачественному росту (наследственный рак толстой кишки, ретинобластома).
Клетка может быть главным звеном при реализации патогенеза на молекулярном уровне. Так, прекращение синтеза мышечного белка дистрофина при мутациях в соответствующем гене приводит к постепенной деградации мышечных клеток. Это спусковой крючок патогенеза тяжелой наследственной болезни - миопатии Дюшенна.
Органный уровень патогенеза
Органный уровень патогенеза наследственных болезней, безусловно, производный от молекулярного и клеточного. В результате первичных или вторичных процессов при разных болезнях мишенью патологического процесса служат различные органы. Например, отложение меди в печени и экстрапирамидной системе мозга при гепатолентикулярной дегенерации (болезнь Вильсона-Коновалова) - первичный процесс, а гемосидероз паренхиматозных органов при пер-
вичном гемохроматозе или талассемии развивается вторично вследствие усиленного распада эритроцитов. При алкаптонурии отложение гомогентизиновой кислоты в хрящах суставных поверхностей и клапанах сердца - вторичный процесс, обусловленный высокой концентрацией гомогентизиновой кислоты в крови (она не превращается в малеилацетоуксусную кислоту в результате мутационно обусловленного отсутствия оксидазы гомогентизиновой кислоты). Это ведет к медленному развитию пороков сердца и тугоподвижности суставов (примерно к 40 годам).
Организменный уровень
В целом организме взаимосвязь патогенетических процессов проявляется сочетанно на молекулярном, клеточном и органном уровнях. Патологический процесс, запущенный первичным эффектом мутантного аллеля, приобретает целостность с закономерными межиндивидуальными вариациями. Тяжесть и скорость развития болезни при прочих равных условиях (пол ребенка, одинаковый характер мутации) зависят от генотипа организма (соматический мозаицизм, гены-модификаторы) и условий среды.
Патогенез любой наследственной болезни у разных индивидов хотя и сходен по первичным механизмам и этапам, но формируется строго индивидуально.
ГЛАВНЫЕ ЧЕРТЫ КЛИНИЧЕСКОЙ КАРТИНЫ
Общие характеристики клинической картины обусловлены генетической природой болезней этой группы, т.е. принципами экспрессии, репрессии и взаимодействия генов. В то же время очевидно, что в полном объеме все общие черты клинической картины при одном заболевании наблюдать трудно. Знание общих черт генных болезней поможет заподозрить наследственную болезнь даже в спорадическом случае.
Ниже приведены 3 главные характеристики генных болезней и их биологические основы: особенности клинической картины, клинический полиморфизм, генетическая гетерогенность.
Особенности клинической картины
К таким особенностям относятся многообразие проявлений, разный возраст начала болезни, прогредиентность клинической картины и хроническое течение, тяжесть течения, обусловливающая инвалидность с детства и меньшую продолжительность жизни.
Симптоматика каждой генной болезни очень многообразна. Как правило, патологическим процессом затрагивается не одна система или орган, а несколько органов уже на первичных этапах формирования болезни. Это касается болезней, проявляющихся в нарушении процессов эмбрионального развития (врожденные пороки развития), наследственных болезней обмена веществ и комбинированных болезней. Биологической основой многообразия проявлений генных болезней служит генный контроль первичных механизмов обмена или морфогенетических процессов.
Для некоторых групп болезней вовлечение в патологический процесс многих органов и тканей обусловлено тем, что первичный дефект локализован в клеточных или межклеточных структурах многих органов. Например, при наследственных болезнях соединительной ткани нарушен синтез специфического для каждой болезни белка той или иной волокнистой структуры. Поскольку соединительная ткань есть во всех органах и тканях, то и многообразие клинической симптоматики при этих болезнях - следствие аномалии соединительной ткани. Так, при синдроме Марфана в патологический процесс вовлечены скелетно-мышечная система, глаза, сердечно-сосудистая система, наружные покровы, легкие, ЦНС; при синдроме Элерса-Данло - кожа, суставы, глаза, сердце, сосуды, грудная клетка, мозг, зубы.
Наряду с понятными механизмами многообразных проявлений генных болезней имеются примеры необычайно широкого клинического полиморфизма с пока неизвестными механизмами. Нейрофиброматоз I типа проявляется пигментными пятнами, кожными, подкожными и плексиформными нейрофибромами, костными изменениями, опухолями нервных стволов и головного мозга, снижением способности к обучению. Эти многообразные проявления пока не удается связать в единый патогенетический комплекс, хотя уже известны структура гена и его первичный продукт. Не исключается, что в этом и других подобных случаях речь идет о первичной плейотропии, т.е. множественных эффектах гена в разных органах.
Другая черта клинической картины генных болезней, помимо многообразия проявлений, - разный возраст начала болезней. В целом, для наследственной патологии возраст начала практически не ограничен: от ранних стадий эмбрионального развития (врожденные пороки развития) до пожилого возраста (хорея Гентингтона, болезнь Альцгеймера). Из всех генных болезней 25% развиваются
внутриутробно, т.е. как врожденная патология. За первые 3 года жизни проявляется еще почти 50% генных болезней (в сумме с внутриутробным формированием - 70% всех болезней). На конец пубертатного периода приходится 99%.
Возраст дебюта различен при многих генных заболеваниях. Например, хорея Гентингтона (аутосомно-доминантное заболевание) может начинаться в любом возрасте: от детского (описаны случаи начала заболевания в 6-летнем возрасте) и до 60-летнего, средний возраст начала заболевания 38 лет. Клиническая картина миотонической дистрофии (аутосомно-доминантное заболевание) может возникать внутриутробно - врожденная форма, в юношеском возрасте - ювенильная, у взрослых - классическая. Возможна мягкая форма с поздним началом.
Возраст начала заболевания различен и при рецессивных болезнях. Муковисцидоз может развиваться внутриутробно (мекониальный илеус), в грудном возрасте или после 3-7 лет жизни.
Биологическая основа разного возраста начала в целом для группы генных болезней заключается в строгих временных закономерностях онтогенетической регуляции экспрессии генов. Функционирование каждого гена в норме начинается и заканчивается в строго определенное в отношении онтогенеза время и в строго определенных клетках. Это правило относится и к мутантному гену.
Причинами разного возраста начала одной и той же болезни могут быть индивидуальные характеристики генома больного. Действие других генов на эффект мутантного гена (взаимодействие генов) может менять время развития болезни. Какие-то комбинации генов будут способствовать более раннему проявлению действия патологических генов, какие-то - тормозить его. Небезразличны для времени проявления патологических генов и условия среды в онтогенезе индивида, особенно во внутриутробном периоде.
Все это больше гипотетические предположения о причинах разного возраста начала конкретной генной болезни. Вместе с тем молекулярно-биологические исследования позволяют конкретизировать биологические основы клинических проявлений отдельных форм генных болезней в разном возрасте. Так, установлено, что сроки развития хореи Гентингтона могут быть связаны с импринтингом соответствующего гена у отца (унаследовавшие ген с увеличением числа повторов от отцов заболевают раньше), а при миотонической дистрофии - с числом тринуклеотидных повторов, определяемых
в мейозе у женщин (чем больше повторов, тем раньше развивается болезнь и тем тяжелее она протекает).
Генным болезням свойственны прогредиентность клинической картины, а также затяжное течение с рецидивами.
При многих болезнях клиническая картина и тяжесть течения усиливаются по мере развития патологического процесса. Приведем несколько примеров.
Нейрофиброматоз I типа начинается с возникновения безобидных пигментных пятен цвета кофе с молоком, веснушек в подмышечных и паховых областях. Затем появляются единичные нейрофибромы, опухоли или костные изменения и т.д. При фенилкетонурии прогрессируют умственная отсталость, гипомеланоз кожи и волос. Нарушение свертываемости крови при гемофилии с возрастом не ослабевает, а усиливается. При GM2-ганглиозидозе с 6-месячного возраста начинает развиваться демиелинизация нервных волокон, что продолжается вплоть до летального исхода в возрасте 2-4 лет. Затяжное или хроническое течение имеют многие генные болезни (муковисцидоз, болезнь Рандю-Ослера-Вебера, гепатолентикулярная дегенерация и др.).
Как видно из приведенных примеров, прогредиентность клинической картины и хроническое течение наблюдаются при генных болезнях с разными типами наследования. Первичная биологическая основа этой характеристики - непрерывность функционирования патологического гена (либо отсутствие его продукта). К этому присоединяются вторичные процессы (воспаление, дистрофия, нарушенный обмен веществ, гиперплазия и т.д.), которые усиливают первично запущенный патологический процесс.
Естественно, что прогредиентность присуща не всем болезням. При развитии некоторых болезней к определенному возрасту достигается конечный фенотип. Например, при ахондроплазии болезнь полностью формируется по мере роста костей (нарушен хондрогенез) пропорционально возрасту. Развитие болезни как бы запрограммировано без прогредиентности.
Течение большинства генных болезней тяжелое, что приводит к инвалидизации в детском возрасте и сокращению продолжительности жизни. Тяжесть течения болезни не всегда связана с врожденным характером заболевания. Такие тяжелые формы, как хорея Гентингтона, гепатолентикулярная дегенерация, миотоническая дистрофия, наследственные кардиомиопатии, развиваются у взрослых.
Чем важнее место, которое занимает моногенно детерминируемый процесс в обеспечении жизнедеятельности, тем клинически тяжелее проявляется мутация в соответствующем локусе.
Клинический полиморфизм и его причины
Клиническая генетика всегда опиралась в своих принципах на закономерности, установленные экспериментальной генетикой. Это в полной мере относится к анализу клинического полиморфизма. В 1934 г. Н.В. Тимофеев-Ресовский в статье «Связь между геном и внешним признаком (феноменология проявления генов)» писал «...лишь первый шаг к генетической физиологии развития, а именно к так называемой феноменологии проявления генов. Этим я обозначаю расчленение и классификацию всеобщих явлений в чудовищно многогранной и изменчивой области проявления самых различных наследственных признаков». Он обратил внимание и проиллюстрировал на конкретных экспериментальных материалах «общие феномены проявления генов», среди них такие, как гетерогенные гены, полифенные (плейотропные) гены и константно и вариабельно повторяющиеся гены. Он проявил интерес к концептуальности и практическому использованию таких знаний «прежде всего в области наследственной патологии человека». Спустя почти 40 лет В. Маккьюсик эти феномены проявления генов у человека, не меняя сути в их интерпретации, назвал «принципами клинической генетики»: клинический полиморфизм, генетическая гетерогенность и плейотропизм. Еще раньше в России, как уже отмечалось, С.Н. Давиденков, практикуя в клинике нервных болезней, в 1930-е годы ими широко пользовался.
Одна из основных и наиболее старых аксиом клинической медицины сводится к тому, что болезнь любой этиологии (инфекционной, травматической, алиментарной, гормональной и др.) проявляется неодинаково у разных индивидов, поэтому нужно лечить не болезнь, а больного. В ряде случаев клиническая картина одного и того же заболевания варьирует от стертых форм до тяжелейших клинических проявлений. Формирование клинической картины связывают с особенностями действия этиологических факторов (например, вирулентность возбудителя), исходного состояния организма (иммунный статус, обмен веществ), сопутствующих условий (стресс, температура). Кроме того, признается роль врожденных характеристик организма в патогенезе и клинической картине болезней.
Казалось бы, можно ожидать более или менее унифицированной клинической картины какой-либо нозологической формы генных болезней, поскольку этиологический фактор для всех больных с этой формой одинаков (мутация в соответствующем гене), а патогенез развертывается на фоне жестко детерминированного контроля генной активности. Такой вывод подсказывал общегенетический взгляд на моногенно детерминируемые события. Однако клиническая практика показала, что симптоматика наследственных болезней различна. При накоплении наблюдений одних и тех же нозологических форм оказалось, что клинический полиморфизм генных болезней выражен не меньше, чем ненаследственной патологии.
При многих заболеваниях, достаточно хорошо изученных на клиническом, генетическом и молекулярном уровнях, нет строгой корреляции между генотипом и фенотипом. Неясно, почему заболевания, вызванные строго доказанными одними и теми же мутациями на молекулярном уровне, имеют разные клинические проявления иногда даже у идентичных близнецов. Нужны дальнейшие исследования с анализом генных сетей, активности факторов транскрипции, транспортных белков и других модификаторов экспрессии генов.
Клинический полиморфизм генных болезней проявляется в разных сроках начала заболевания, полноте и тяжести симптоматики (глубина патологического процесса), продолжительности болезни, степени инвалидности, толерантности к терапии, в сокращении продолжительности жизни. Вместе с тем следует подчеркнуть, что генные болезни не имеют плавных переходов от нормы к патологии. Даже самая легкая форма болезни обязательно имеет минимальные диагностические критерии. Генетическое правило гласит: нормальный генотип детерминирует нормальный фенотип, а мутантный генотип детерминирует мутантный фенотип (болезнь).
Генетической причиной полиморфизма может быть явление взаимодействия главного гена и генов модификаторов (эпистаз, особенности инактивации и дозовая компенсация Х-хромосомы, цитоплазматический геном), с другой стороны - это могут быть и факторы внешней среды, в которых осуществляется развитие индивида.
На рис. 4.6 изображено влияние названных двух групп факторов (генетических и внешнесредовых) на фенотипы моногенных болезней. Так, фенилкетонурия в пространстве двух обозначенных координат занимает срединное положение, отражая заметное влияние средовых и случайных факторов, а также эффектов других генов
на клинические проявления болезней. В то же время для болезни Тея-Сакса влияние этих факторов менее выражено, а в клинических проявлениях недостаточности Г6ФДГ преобладающим модифицирующим фактором является внешняя среда.
К настоящему времени накопился огромный фактический материал по феноменологии клинического полиморфизма отдельных форм и факторам, его определяющим. В первую очередь следует рассматривать значение характера мутации в конкретном локусе для проявления болезни или формирования фенотипа (мутантного). Первично возникшие и унаследованные от предыдущих поколений мутации имеют достаточно сходное фенотипическое проявление, т.е. длительность унаследования мутации не отражается на клиническом полиморфизме генных болезней. Как подчеркивалось выше, десятки и даже сотни разных мутаций (и даже разных типов) в одном и том же локусе ведут к одной и той же болезни. В большинстве случаев характер мутации не определяет клиническую
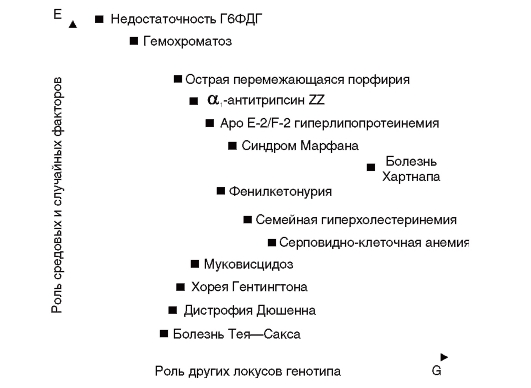
Рис. 4.6. Потенциальное влияние генетических (G) и негенетических (E) факторов на фенотип некоторых моногенных болезней (по Ч. Скрайверу и П. Уотерс)
картину болезни. Фенотип определяет первичный эффект гена (нет продукта или мало продукта).
Однако накапливается все больше данных о зависимости фенотипа (клинической картины болезни) от генотипа (разных мутаций в одном и том же локусе). Такие формы болезней, при которых мутации не полностью блокируют выработку первичного продукта, уже известны. Расшифровка корреляций между гено- и фенотипом стала возможной благодаря молекулярно-биологическим исследованиям структуры генов, мутаций и их первичных продуктов.
Мутации в одном и том же локусе, ответственные за синтез дистрофина, приводят к двум клиническим формам: миопатии Дюшенна (тяжелой) и миопатии Беккера (легкой). Установлено, что миопатия Дюшенна развивается при полной блокаде, а Беккера - при частичной блокаде синтеза РНК для дистрофина (при миопатии Беккера делеции гена меньшего размера).
Сплайсинговые мутации, как правило, не полностью блокируют образование мРНК, поэтому соответствующие формы болезни бывают мягкими по клинической картине и течению.
Четкая корреляция между генотипом (характером мутации) и фенотипом (клинической картины болезни) отмечена пока лишь при одном виде мутаций - экспансии тринуклеотидных повторов в гене.
Чем больше повторов в мутантном аллеле, тем тяжелее протекает болезнь. Особенно четко это проявляется при синдроме Мартина- Белл и миотонической дистрофии. Поскольку экспансия повторов формируется в мейозе у одного из родителей (у мужчины или женщины при различных болезнях), это обусловливает явление антиципации - более тяжелое течение наследственной болезни в последующих поколениях. До открытия этого типа мутаций (экспансии триплетов) и молекулярно-генетического прослеживания числа повторов в поколениях явление антиципации рассматривалось как артефакт наблюдений. Теперь стало ясно, что феномен антиципации существует. Для него уже обнаружена биологическая основа при небольшой группе болезней.
В последнее время появились данные, свидетельствующие о том, что развитие некоторых наследственных заболеваний может быть обусловлено резким изменением числа копий не только тринуклеотидных повторов, но и тандемных повторов ДНК большей протяженности (тетра- и пентануклеотидов, мини- и мегасателлитов), при
этом возможно как увеличение, так и, напротив, уменьшение числа копий повторяющегося элемента. Примеры наследственных заболеваний, связанных с так называемыми динамическими мутациями, приведены в табл. 4.2.
Таблица 4.2. Заболевания человека, обусловленные динамическими мутациями
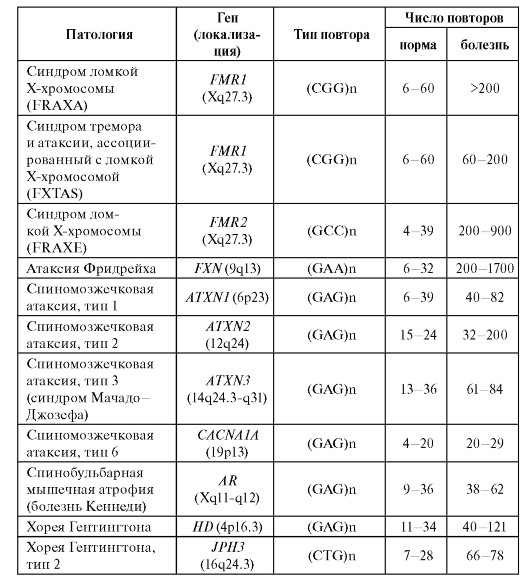
Окончание таблицы 4.2
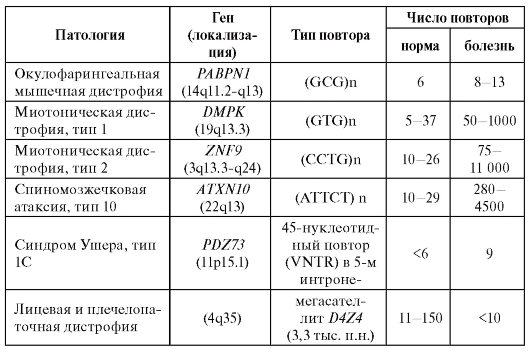
Примечание. FMR - fragile mental retardation; VNTR - полиморфизм по количеству копий (variable number of tandem repeats); п.н. - пар нуклеотидов.
Большинство болезней, обусловленных нестабильными нуклеотидными повторами, фенотипически (клинически) проявляются главным образом неврологической симптоматикой (атаксии, когнитивные нарушения, деменции, нистагм, паркинсонизм), хотя иногда в болезнь включаются и другие органы (семенники - макроорхидизм, дисплазия соединительной ткани, нарушение сердечной проводимости и др.).
Патогенетические механизмы болезней экспансии нестабильных повторов разнообразны. Их можно разделить на три класса.
- Класс 1: экспансии некодирующих повторов, вызывающие нарушение функции белков и транскрипции патологического гена преРНК (синдром Мартина-Белл, атаксия Фридрейха).
- Класс 2: экспансии некодирующих повторов, вызывающие появление новых свойств РНК (миотоническая дистрофия 1 и 2, синдром тремор/атаксии с ломкой Х-хромосомой).
- Класс 3: экспансии кодирующих повторов, приводящие к новым свойствам мутантного белка (хорея Гентингтона, спиномозжечковая атаксия).
К настоящему времени обнаружено несколько геновмодификаторов при моногенных заболеваниях человека. Приведем несколько примеров. Хорошо изучено улучшение состояния у гомозигот по мутации, приводящей к β-талассемии, унаследовавших также аллель α-талассемии. Последний выступает как ген-модификатор. Дисбаланс синтеза цепей глобина, обусловленный β-талассемией, улучшается за счет снижения синтеза α-цепей, вызванного мутацией при α-талассемии. Еще один установленный факт - пациенты с муковисцидозом, гомозиготные по наиболее частой мутации, имеют очень вариабельную патологию легких. Показано, что это связано с наличием по меньшей мере одного гена-модификатора.
Клиническая картина болезни может зависеть от «дозы» генов (числа аллелей). Так, гомозиготность (два аллеля) при аутосомнодоминантных болезнях определяет более тяжелую клиническую картину, а иногда даже внутриутробную гибель плода (ахондроплазия, синдром Элерса-Данло). Аутосомно-рецессивные болезни проявляются в полной мере при условии гомозиготного состояния по мутантному аллелю. Однако некоторые признаки заболевания могут проявляться и у гетерозигот (один аллель, легкая форма), они усиливаются до клинических проявлений при действии провоцирующих факторов. Например, симптомы кислородной недостаточности (при подъемах на большую высоту) проявляются у гетерозигот по серповидно-клеточной болезни; беременность у гетерозигот по β-талассемии приводит к развитию анемии.
Генетические причины клинического полиморфизма могут быть обусловлены не только патологическим геном, но и генотипом в целом, т.е. генотипической средой в виде генов-модификаторов. Геном в целом функционирует как хорошо скоординированная система. Вместе с патологическим геном индивид наследует от родителей комбинации других генов, которые могут усиливать или ослаблять действие патологического гена. В правильности этого положения не приходится сомневаться, хотя реально гены-модификаторы только начинают идентифицировать. Сравнение выраженности клинической картины у членов одной семьи и разных семей показывает, что межсемейные различия больше, чем внутрисемейные, но внутрисемейные тоже существуют.
Примером гена-модификатора может служить ген, локализованный в длинном плече хромосомы 1, кодирующий «повсеместный» транскрипционный фактор (upstream stimulatory factor 1, USF1), участвующий в метаболизме углеводов и липидов у больных с семейной комбинированной гиперлипидемией. Показано, что разные аллельные варианты этого гена определяют разный уровень его транскрипции и, как следствие, USF-факторы активируют или подавляют экспрессию многих генов, вовлеченных в липидный (АРОЕ, АРОА1, АРОА5) и углеводный (глюкокиназа, рецептор глюкагона) обмены, а также участвующих в регуляции уровня артериального давления (ренин, ангиотензиноген). Важно, что аллельные варианты USF1 вносят заметный вклад в риск сердечно-сосудистых заболеваний на популяционном уровне. Таким образом, модифицирующая функция гена USF1 в отношении генов, вовлеченных в регуляцию липидного и углеводного обменов, а также уровня артериального давления, делает этот ген ответственным за признаки, составляющие метаболический синдром.
Одним из факторов вариабельности фенотипа или разной экспрессивности может быть соматический мозаицизм.
На клиническом уровне явление индивидуальной модификации действия патологического гена изучал С.Н. Давиденков на примерах наследственных болезней нервной системы. Многие «мелкие», наследуемые независимо от основного патологического гена, признаки усиливают клинические проявления болезни. Это явный признак взаимодействия генов. С.Н. Давиденков (1947) высказал гипотезу «условного тропизма патологических нервных задатков»: «Помимо своего прямого влияния на развитие нервной системы, патологический задаток обладает еще способностью усиливать эффект от других наследственных факторов, обладающих сходно направленным тропизмом».
В развитии генной болезни, как и любого наследственного признака человека, имеет значение не только генотип, но и внешняя среда. Выше рассмотрена роль таких генетических факторов, как характер мутаций патологического аллеля и генотипическая среда (взаимодействия генов) в формировании клинического полиморфизма. В то же время не вызывает сомнений влияние окружающей среды в широком смысле слова на развитие болезни. Этому положению есть много доказательств из клинической практики и специальных исследований. Приведем несколько примеров.
Симптоматика фенилкетонурии у ребенка более тяжелая, если во время его внутриутробного развития в рационе матери было много продуктов, богатых фенилаланином. Обострение наследственных миодистрофий наблюдается после стрессов, охлаждений, переутомления. Клиническая картина гемофилии у ребенка усиливается с увеличением у него кровоизлияний от падений и травм. У женщин, больных нейрофиброматозом I типа, резко усиливается рост нейрофибром при беременности.
Генетическая гетерогенность
Понятие генетической гетерогенности означает, что клиническая форма генной болезни может быть обусловлена мутациями в разных локусах или разными мутациями в одном локусе (множественные аллели). Фактически это разные нозологические формы с этиологической точки зрения, объединенные в связи с клиническим сходством фенотипа.
Генетическую гетерогенность наследственных болезней впервые подметил (и ввел этот термин) С.Н. Давиденков в 1930-х годах. Некоторые высказывания о генетической гетерогенности болезней в 50-х годах ХХ в. принадлежат ряду авторов. В 1960-х годах В. Маккьюсик сформулировал принцип изучения болезней с точки зрения их генетической гетерогенности. Этот принцип оказался чрезвычайно полезным для изучения нозологии генных болезней и их фенотипических различий.
Явление генетической гетерогенности носит общий характер, его уже можно назвать правилом, поскольку оно распространяется на все белки организма, не только на патологические, но и на нормальные варианты. С молекулярно-генетической и биохимикогенетической точек зрения вполне объяснимо, что различные патологические гены могут иметь примерно одинаковый фенотип при клинической оценке. Конечный эффект поломки какого-либо процесса на клиническом уровне может быть обусловлен наследственным нарушением синтеза разных белков или разных вариантов одного и того же белка.
Выяснение степени генетической гетерогенности при любой наследственной болезни проходит через все этапы: описание проявлений на клиническом уровне, изучение типа наследования и локализации гена, выяснение первичного биохимического дефекта, установление молекулярной сущности мутации на уровне ДНК.
Генетическая гетерогенность, обусловленная мутациями в разных локусах, - межлокусная гетерогенность - отчетливо видна на примере синдрома Элерса-Данло (6 форм), нейрофиброматоза (по меньшей мере 6 форм), гликогенозов (более 10 форм), гипертрофической кардиомиопатии (11 форм), врожденной катаракты (29 генов), витамин- D-резистентного рахита и т.д. Гетерогенность в упомянутых формах прослеживается даже при применении клинико-генеалогического метода. В этих группах имеются и аутосомно-доминантные, и аутосомно-рецессивные, и сцепленные с Х-хромосомой варианты болезней, т.е. мутации в локусах, расположенных в разных хромосомах.
Источником генетической гетерогенности в том же локусе - внутрилокусной гетерогенности - могут быть множественный аллелизм и генетические компаунды. Разные мутантные аллели могут проявляться фенотипически неодинаково (например, разные β-талассемии, некоторые мукополисахаридозы).
Генетические компаунды, иногда неправильно называемые двойными гетерозиготами, - это сочетание двух разных патологических аллелей одного локуса у индивида. Фенотипы генетических компаундов отличаются от фенотипов гомозиготных форм по обоим унаследованным аллелям (например, фенотип при гемоглобинопатии HbSC отличается от фенотипов обеих мутантных гомозигот - HbSS и НЬСС).
Для некоторых групп болезней генетическая гетерогенность проявляется и на межлокусном, и на внутрилокусном уровнях (мукополисахаридозы, гликогенозы).
Расшифровка гетерогенности генных болезней интенсивно продолжается одновременно в клиническом и генетическом направлениях. Общая задача сводится к выявлению корреляции между генотипом и фенотипом.
Анализ фенотипа (клинической картины болезни) - первый этап в расшифровке генетической гетерогенности. Чем точнее изучен фенотип, тем больше возможностей в открытии новых форм болезней, в разделении изучаемой формы на несколько нозологических единиц.
С помощью классических клинических методов открыто несколько форм нервно-мышечных дистрофий, наследственных форм карликовости. Клинико-биохимическими методами разделены наследственные несфероцитарные анемии, гемоглобинопатии, гликогенозы. Иммунологическими методами дифференцированы пер-
вичные иммунодефицитные состояния. По результатам клиникофизиологических исследований описана гетерогенность гемофилии, цветовой слепоты.
Все перечисленные методы с некоторыми усовершенствованиями в параклиническом плане и сейчас применяются для расшифровки природы наследственных болезней.
Анализ фенотипа не должен ограничиваться организменным уровнем. Перспективное направление - изучение клеточного уровня, т.е. исследование клеток в культуре ткани (клеточная гибридизация, метаболическое кооперирование, физиологическая комплементация). Генетическая гетерогенность нескольких групп болезней была открыта с помощью методов культуры клеток (мукополисахаридозы, болезни репарации ДНК).
Генетические методы включают весь арсенал генетического анализа болезни от применения клинико-генеалогического метода до секвенирования гена. Накопление родословных по какому-либо заболеванию и их генетический анализ позволяют разделять ранее описанную одну болезнь на реально существующие формы, если в этой группе встречаются мутации с доминантным и рецессивным типами наследования. Так были разделены синдром Марфана (доминантное наследование) и гомоцистинурия (рецессивное наследование), имеющие сходную клиническую картину (высокий рост, подвывих хрусталика, деформация грудной клетки). Разные типы наследования обнаружены во многих гетерогенных группах болезней (синдром Элерса-Данло, мукополисахаридозы, витамин-D-резистентный рахит, амиотрофия Шарко-Мари).
Изучение аллелизма рецессивных мутаций дало возможность установить гетерогенность наследственной глухоты (у глухих родителей рождаются нормальные дети). Возможности этого подхода в настоящее время уже ограничены.
Наиболее полную информацию о гетерогенности клинической формы болезни дает применение современных методов анализа генов человека. Отнесение гена к одной или разным группам сцепления, локализация гена, его структура, сущность мутаций позволяют однозначно идентифицировать нозологические формы.
Концепция генетической гетерогенности генных болезней открывает много возможностей в понимании сущности отдельных форм и причин их клинического полиморфизма, что крайне важно для практической медицины (правильная диагностика, выбор методов
лечения, медико-генетическое консультирование). Если врач не принимает во внимание генетическую гетерогенность наследственных болезней, то он может дать пациенту неправильные или неоправданные советы и необоснованные прогнозы.
КЛИНИКА И ГЕНЕТИКА НЕКОТОРЫХ ГЕННЫХ БОЛЕЗНЕЙ
Нейрофиброматоз (болезнь Реклингхаузена)
Это тяжелая полисистемная болезнь с аутосомно-доминантным типом наследования. Наиболее тяжело поражается нервная система, поэтому болезнь считают неврологической. Термин «нейрофиброматоз» охватывает по меньшей мере две болезни: нейрофиброма-
тоз I типа и нейрофиброматоз II типа, сначала считавшиеся двумя формами одного и того же заболевания (периферический нейрофиброматоз и центральный нейрофиброматоз). Это яркий пример генетической гетерогенности наследственных болезней. Ниже будет описан только нейрофиброматоз I типа.
Симптоматика нейрофиброматоза I типа разнообразна, в патологический процесс вовлекается несколько систем, что подтверждает плейотропный эффект гена. Диагноз нейрофиброматоза I типа можно установить при наличии не менее двух из перечисленных ниже признаков, но при условии, что они не являются симптомами какойлибо другой болезни.
- Светло-коричневые пигментные пятна (рис. 4.7).
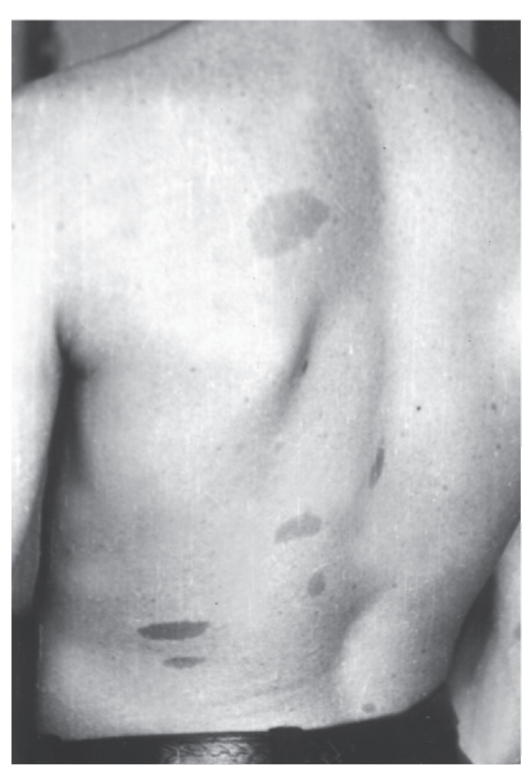
Рис. 4.7. Типичные светло-коричневые пятна у молодого мужчины с нейрофиброматозом I типа
У детей их должно быть не менее 5, а диаметр пятен не менее 5 мм. У взрослых число пятен должно быть не менее 6, а диаметр пятен не менее 15 мм. Для выявления пятен необходимо хорошее освещение. Пигментные пятна появляются обычно к 3 годам жизни, их число увеличивается с возрастом. Они встречаются у 95% больных. - Решающий признак - две нейрофибромы любого типа и более или одна плексиформная нейрофиброма (по данным анамнеза или клинического обследования) (рис. 4.8). Нейрофибромы могут возникать в любом участке тела, захватывая кожные нервы, часто располагаются по ходу нервных стволов, иногда захватывают крупные нервы и нервные сплетения (плексиформные нейрофибромы). В месте локализации нейрофибром больные часто ощущают зуд, жжение и боль. У одного больного могут быть тысячи нейрофибром, а их масса может достигать 15 кг и более, если своевременно не сделано их иссечение. У детей таких нейрофибром
мало. Их число увеличивается с возрастом, особенно у женщин при беременности. К 30-летнему возрасту нейрофибромы отмечаются у 95% больных.
- Множественные, похожие на веснушки пигментные пятна в подмышечной ямке (рис. 4.9), паховой области, на других участках тела со складками. Они обычно возникают в детстве, их число трудно определить. Пигментные пятна обнаруживают у 80% больных.
- Костные изменения (дисплазия крыла клиновидной кости, врожденное искривление или утончение длинных трубчатых костей, ложный сустав).
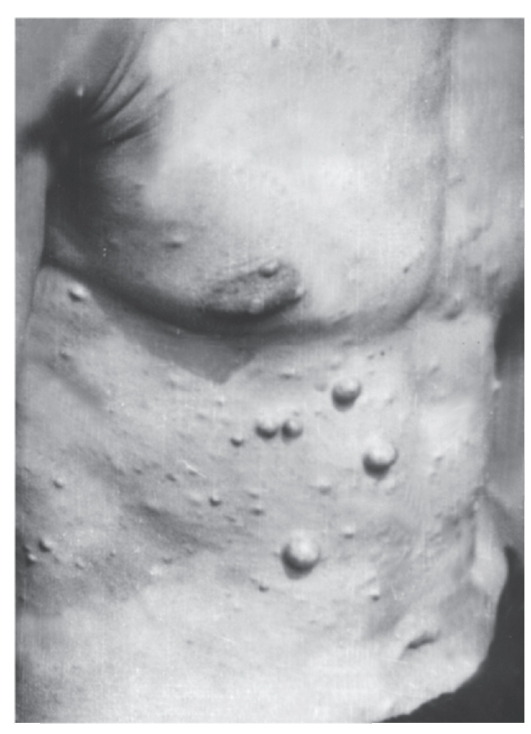
Рис. 4.8. Многочисленные нейрофибромы разных размеров у мужчины с нейрофиброматозом I типа
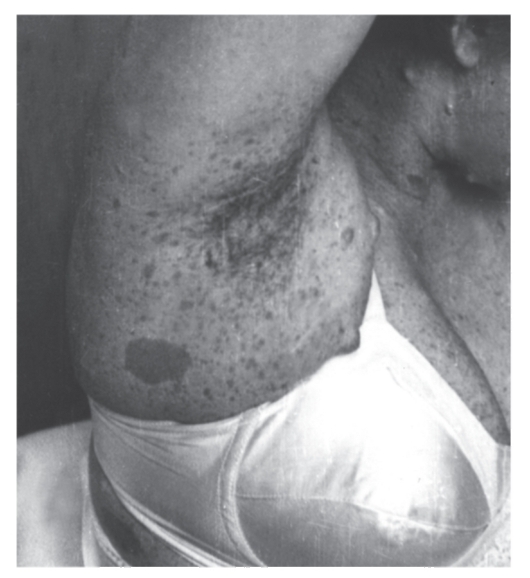
Рис. 4.9. Мелкие пигментные пятна и одно крупное пятно в подмышечной области у женщины с нейрофиброматозом I типа. Видны также нейрофибромы на шее и грудной клетке
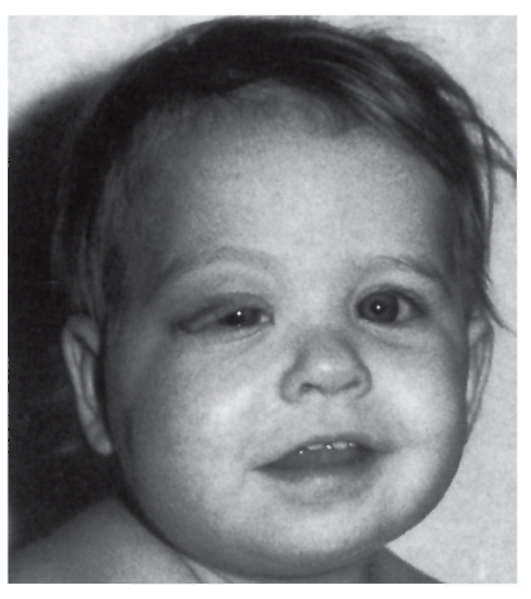
Рис. 4.10. Плексиформная орбитальная нейрофиброма у ребенка
Дисплазия глазницы сочетается с плексиформной нейрофибромой глазницы (рис. 4.10).
- Глиома зрительного нерва. Эта опухоль характерна для нейрофиброматоза I типа. С помощью КТ и МРТ (магнитнорезонансной томографии) обнаруживается утолщение зрительного нерва. Глиома зрительного нерва протекает обычно бессимптомно, но иногда может вызывать ухудшение зрения, косоглазие, зрачковые аномалии, проптоз и гипоталамическую дисфункцию.
- Узелки Лиша (два и более) на радужной оболочке. Узелки представляют собой гамартомные новообразования и не влияют на зрение. После полового созревания узелки Лиша наблюдаются практически у всех больных. Для обнаружения узелков глаза осматривают с помощью щелевой лампы.
- Нейрофиброматоз I типа по приведенным выше критериям у родственника I степени родства (родитель, сибс, потомок). Нейрофиброматоз I типа относится к полностью пенетрантным
аутосомно-доминантным болезням, поэтому нейрофиброматоз I типа у родственника можно использовать как диагностический критерий.
У большинства больных диагноз очевиден уже к 3 годам.
Течение заболевания прогрессирующее, с очень большим размахом клинической картины. Наряду с описанными выше симптомами (они либо врожденные, либо зависят от возраста) у 20-30% детей наблюдаются когнитивные нарушения (трудности в обучении). Наиболее опасными проявлениями становятся опухоли - иногда из-за злокачественности, иногда из-за места расположения (черепные нервы, малый таз, ЖКТ). Неблагоприятное течение нейрофиброматоза I типа отмечается у 30% больных.
Наряду с основными симптомами у больных нейрофиброматозом часто встречаются осложнения: низкий рост у 25-35% пациентов, плексиформные нейрофибромы у 20%, сколиоз у 10%, головные боли у 20%, нейрофиброматоз 1-ассоциированные злокачественные футлярные опухоли периферических нервов у 7-12%, глиома зрительного нерва у 7%. К более редким осложнениям нейрофиброматоза I типа относятся: эпилепсия у 1-2% пациентов, гидроцефалия у 2%, псевдоартроз у 3%, стеноз почечной артерии у 1-2%, ксантогранулемы - 1-2%, феохромоцитома - менее чем у 1%.
Патогенез заболевания пока недостаточно изучен, поскольку трудно объединить разнообразнейшие проявления: пигментные пятна, костные изменения, когнитивные нарушения и т.д. Осложнения могут возникать в любой системе организма, в состав которой входят ткани, происходящие из эктодермы, мезодермы и нервной трубки. Фенотипические проявления нейрофиброматоза I типа значительно различаются даже у членов одной семьи. Однако известно, что ключевую роль играют шванновские клетки, которые в результате соматической мутации теряют гетерозиготность. Так как этот ген является геном-супрессором опухолей, то после потери функции обоих аллелей развиваются опухоли и заболевание манифестирует.
Больные нейрофиброматозом I типа встречаются в практике врачей многих специальностей. Пациенты обращаются в первую очередь к дерматологам с жалобами на косметический дефект в виде пигментных пятен и нейрофибром. Наиболее часто больные нейрофиброматозом I типа являются пациентами невропатологов
и нейрохирургов. Определенная часть таких больных лечится у хирургов и ортопедов.
Генетика нейрофиброматоза I типа на генеалогическом уровне была ясна уже в начале прошлого столетия. В последние годы она изучена до уровня гена. Локус нейрофиброматоза I типа расположен в коротком плече хромосомы 17q11.2. Величина гена - 350 000 пар оснований, в нем 59 экзонов. Ген полностью секвенирован. Он относится к группе генов-супрессоров опухолей, чем и объясняются его опухолевые эффекты. Обнаружено более 500 мутаций (транслокации, делеции, вставки, точковые замены). Однако корреляция клинической картины с типом мутаций еще не установлена. Также обнаружены гены-модификаторы, в настоящее время идет их изучение. Первичный продукт гена назван нейрофибромином.
Распространенность нейрофиброматоза I типа равна примерно 1:3500-5000 новорожденных, одинакова у обоих полов, у всех рас и этнических групп. Аутосомно-доминантный тип наследования не нарушается ни в одной популяции. Доля спорадических случаев составляет 50-70%. Частота мутаций по этому гену высокая: 1 мутация на 10 000 гамет на поколение.
Лечение нейрофиброматоза I типа в основном хирургическое или симптоматическое. В настоящее время идет разработка патогенетической терапии (антифиброзные средства).
Миотоническая дистрофия
Синонимы: болезнь Штайнерта, дистрофическая миотония. Миотоническая дистрофия - аутосомно-доминантное многосистемное заболевание с сильно вариабельной экспрессией гена, обусловливающей клинический полиморфизм по началу заболевания и тяжести течения. Главные клинические проявления: миотония, мышечная слабость, катаракты, аритмии сердца, облысение со лба, нарушенная толерантность к глюкозе, умственная отсталость. Мышечные судороги особенно выражены в руках, челюстях, языке (в виде фибрилляции). Одновременно постепенно усиливается мышечная слабость в связи с дегенерацией отечных мышечных клеток и атрофией волокон. Миотония и мышечная слабость у пациентов сочетаются с нарушением речи и глотания. Начальные признаки миотонической дистрофии различны. Миотония сначала выявляется только при специальном тестировании. Мышечные подергивания и слабость
обычно асимметричны. В первую очередь в патологический процесс вовлекаются лицевые и височные мышцы (миотоническое лицо), затем шейные, плечевые, бедренные мышцы (рис. 4.11, 4.12).
В настоящее время известно 2 типа миотонической дистрофии. Миотоническая дистрофия 1-го типа составляет около 98% миотонической дистрофии и характеризуется началом мышечной слабости от дистальных мышц к проксимальным. При миотонической дистрофии 2-го типа мышечная слабость развивается, наоборот, от проксимальных к дистальным отделам.
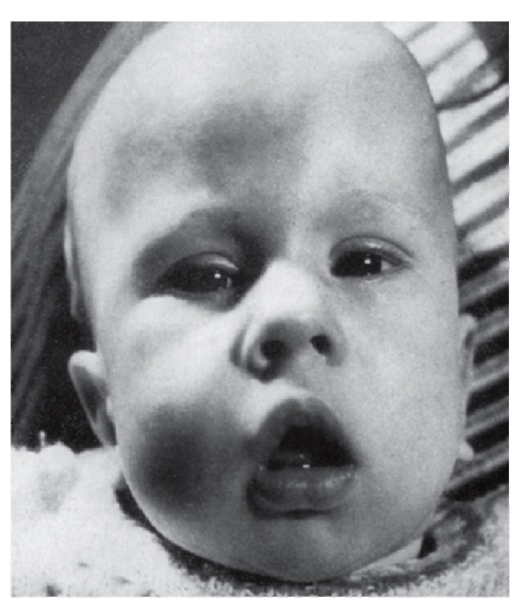
Рис. 4.11. Врожденная форма миотонической дистрофии (мышечная гипотония, амимичное лицо, рот треугольной формы - миотоническое лицо)
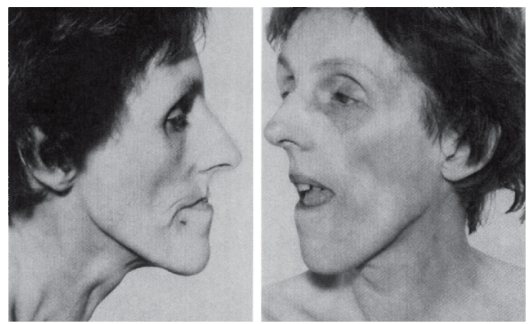
Рис. 4.12. Взрослая пациентка с миотонической дистрофией (птоз, слабость лицевых мышц, атрофия жевательных мышц)
Наряду с нервно-мышечными симптомами при миотонической дистрофии отмечаются катаракта (очень ранний симптом), гипогонадизм (атрофия семенников), аменорея, дисменорея, кисты яичника, облысение со лба, изменения проводимости сердца с аритмией, абдоминальные симптомы (на почве холелитиаза), прогрессирующая умственная отсталость.
Тяжесть клинических проявлений очень сильно различается даже в пределах одной семьи.
Начало миотонической дистрофии возможно от пренатального периода до возраста 50-60 лет. Выделяют 4 формы заболевания (в зависимости от возраста манифестации): врожденную, юношескую, классическую (20-30 лет) и минимальную (50-60 лет). Это объясняется различиями в числе тринуклеотидных повторов в локусе миотонической дистрофии (см. ниже).
Смерть при миотонической дистрофии наступает в возрасте 50-60 лет (при классической форме) вследствие пневмонии, сердечных осложнений или других интеркуррентных заболеваний.
Частота болезни может различаться в этносах и популяциях. Эффект родоначальника описан у канадцев французского происхождения. В Израиле распространенность болезни в среднем равна 1 : 6369 с различиями между общинами: у евреев-ашкенази 1 : 17 544, у евреев-сефардов 1 : 5000, у евреев-йеменитов 1 : 2114. Обобщенно распространенность миотонической дистрофии можно оценить как
1 : 7500-10 000.
Генетика миотонической дистрофии хорошо изучена на генеалогическом, формально-генетическом и молекулярно-генетическом уровнях. При миотонической дистрофии 1-го типа у пациентов всех стран обнаружена одна и та же мутация в гене протеинкиназы мышечной дистрофии (символ гена DMPK), локализованном в хромосоме 19q13.2-19q13.3. Суть мутации - экспансия (увеличение числа) нестабильных CTG-повторов в 3'-нетранслируемой области гена. В норме число CTG-повторов колеблется от 5 до 30. При миотонической дистрофии этот показатель значительно увеличивается и составляет 50-2000 и более. Обнаружена корреляция между тяжестью и числом тринуклеотидных повторов. Чем больше повторов, тем раньше начинается заболевание и тяжелее протекает болезнь. Клиническая картина у гомозигот более тяжелая. При миотонической дистрофии 2-го типа найдена другая мутация - в гене цинко-
вых пальцев (ZNF9), локализованном в хромосоме 3q21.3. Мутация представляет собой нестабильную экспансию CCTG-тетраплета с повторами от 75 до 11 000.
Во многих семьях с миотонической дистрофией в нескольких поколениях отмечается антиципация, т.е. более тяжелая манифестация болезни и начинающаяся в более молодом возрасте в каждом последующем поколении. Этот признак описан для миотонической дистрофии давно и рассматривался в 1940-х годах как статистический артефакт. Однако сведения о молекулярном дефекте указывают на возможность увеличения числа триплетов в поколениях.
Описаны семьи с более чем тремя поколениями с миотонической дистрофией: в 1-м поколении - только катаракты, во 2-м поколении - умеренная слабость мышц, в 3-м поколении - врожденная форма.
При миотонической дистрофии выражен импринтинг. Пациенты, рожденные от больных матерей, имеют более тяжелую форму болезни с более ранним началом, чем пациенты, рожденные от больных отцов. Врожденная форма миотонической дистрофии наблюдается только у детей от больных матерей. Механизм импринтинга выяснен: экспансия триплетов происходит в мейозе у женщин, а при сперматогенезе она отсутствует.
Уменьшение длины мутантного повтора (почти до нормы) у потомков с легкой клинической картиной или бессимптомным заболеванием наблюдается при передаче гена от отца. Одним из объяснений этого может быть селекция против длинных аллелей в мужском гаметогенезе.
Семейная гиперхолестеринемия
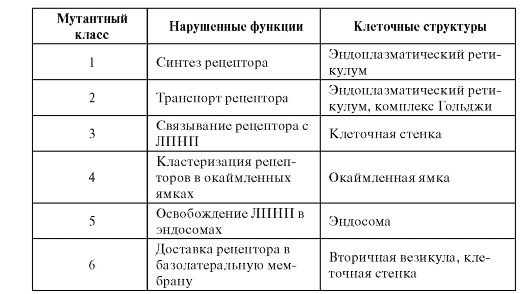
Эта генетически гетерогенная аутосомно-доминантная болезнь клинически выражается в чрезвычайно высокой гиперхолестеринемии. Следовательно, клинически это единая нозологическая форма. Заболевание связано с наследованием мутантных генов, кодирующих рецептор ЛПНП. Ген локализован в хромосоме 19р13.2.
В настоящее время идентифицировано 6 классов мутаций рецептора ЛПНП. В результате этих мутаций происходит нарушение одной из следующих функций:
• синтез рецепторов;
• транспорт рецепторов из эндоплазматического ретикулума в комплекс Гольджи;
• связывание ЛПНП рецептором;
• кластеризация рецепторов в окаймленных ямках;
• рециклизация рецептора (неспособность освобождать ЛПНП в эндосомах);
• доставка рецептора в базолатеральную мембрану (табл. 4.3).
Таблица 4.3. Генетическая гетерогенность семейной гиперхолестеринемии
Мутации в гене APOB, кодирующем ароВ-100, могут вызывать гетерозиготный фенотип семейной гиперхолестеринемии (не часто). Кроме того, обнаружен ген ARH, вызывающий гомозиготный фенотип семейной гиперхолестеринемии, и ген PCSK9, определяющий гетерозиготный фенотип семейной гиперхолестеринемии. Однако эти ЛПНП-рецепторные гены редко бывают причиной болезни. В справочнике OMIM указано еще много генов, влияющих на обмен липидов.
Таким образом, семейная гиперхолестеринемия - наглядный пример генетической гетерогенности наследственной патологии. Более того, по каждому классу мутаций, которые фактически являются отдельными нозологическими формами с генетической точки
зрения (разная этиология), обнаружены уже десятки мутантных аллелей с патологическим действием.
Клиническая картина полной (гомозиготной) формы семейной гиперхолестеринемии включает необычно высокую гиперхолестеринемию и появление уже в детском возрасте ксантом на коже, на сухожилиях. Возникает липоидная дуга на роговице. В период полового созревания формируются атероматозное поражение устья аорты, а также стеноз венечных артерий сердца, что проявляется систолическим шумом на аорте и ангиографически определяемым сужением корня аорты и стенозом коронарных артерий. Клинически развивается типичная картина ишемической болезни сердца. До применения современных методов лечения пациенты с семейной гиперхолестеринемией внезапно умирали в возрасте до 30 лет в связи с острой коронарной недостаточностью. Патологоанатомически обнаруживается массивный атероматоз аортального клапана и восходящей части дуги аорты. В крупных артериях наблюдаются сходные, но менее выраженные изменения.
Тяжесть клинической картины и возраст, в котором развивается болезнь, определяются состоянием рецепторов ЛПНП. По этому биохимическому признаку всех больных можно разделить на рецепторнегативных и рецептордефицитных. Наиболее ранняя форма ишемической болезни сердца отмечается у рецепторнегативных гомозигот. В возрасте до 10 лет у 65% из них развивается ишемическая болезнь сердца, 20% больных умирают в возрасте до 25 лет. У рецептордефицитных гомозигот ишемическая болезнь сердца развивается после 10 лет, а к 25 годам умирают 4% больных.
Поражаемость атеросклерозом и ишемической болезнью сердца лиц обоего пола при гомозиготной семейной гиперхолестеринемии одинакова.
Гетерозиготная форма семейной гиперхолестеринемии часто остается невыявленной до взрослого состояния, пока не развивается сердечно-сосудистая недостаточность. У таких больных отмечаются гиперхолестеринемия, липоидная дуга на роговице, ксантелазмы или холестериновые отложения в коже (рис. 4.13, 4.14), ксантомы сухожилий (тыльная поверхность кисти, локтевой сустав, пяточные и коленные сухожилия). К 50 годам у 50% мужчин-гетерозигот по семейной гиперхолестеринемии развивается ишемическая болезнь сердца (на 20 лет раньше, чем в попу-
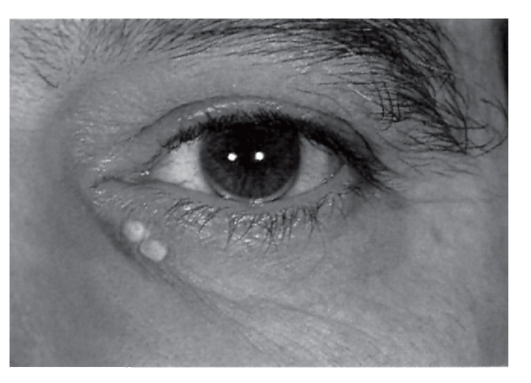
Рис. 4.13. Липоидная дуга на роговице и ксантелазмы у 47-летнего мужчины с гетерозиготной формой гиперхолестеринемии
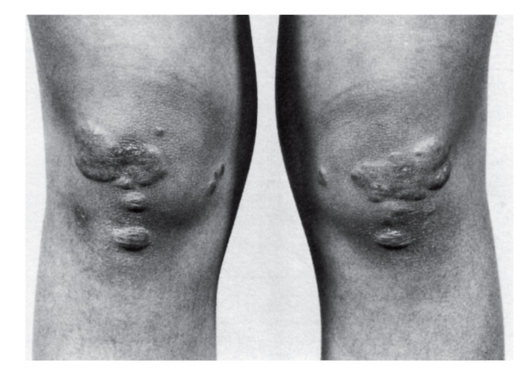
Рис. 4.14. Множественные ксантелазмы на ногах у мужчины с гомозиготной формой семейной гиперхолестеринемии
ляции). У женщин-гетерозигот по семейной гиперхолестеринемии первые признаки ишемической болезни сердца появляются на 9-10 лет раньше, чем у мужчин.
Лечение гомозиготных больных семейной гиперхолестеринемией - необычайно трудная задача. Диета и лекарственные препараты неэффективны. Эффективной мерой может быть плазмаферез с двухнедельным интервалом. Более радикальная мера - пересадка печени.
При гетерозиготных формах семейной гиперхолестеринемии применяют лекарственную терапию, направленную на снижение уровня холестерина, исключение факторов риска ишемической болезни сердца (особенно курения), позже выполняют шунтирование.
Распространенность обеих форм семейной гиперхолестеринемии составляет в большинстве популяций 0,2%. В основном наблюдается гетерозиготная форма. Частота мутантных аллелей по всем 6 классам нарушения рецепторов ЛПНП равна 1 : 500, а гомозиготная форма встречается с частотой 1 : 250 000 или даже реже. Подробные сведения по диагностике семейной гиперхолестеринемии в России можно найти на компакт-диске и в статье М.Ю. Мандельштама др. «Диагностика семейной гиперхолстеринемии в России: достижения и проблемы».
Синдром Марфана
Синдром Марфана - наследственная аутосомно-доминантная болезнь соединительной ткани. Синдром клинически идентифицировал В. Марфан в 1886 г. Причиной синдрома Марфана являются
мутации в гене фибриллина (локализация в хромосоме 15q21). Уже выявлено несколько типов мутаций (в основном миссенс), ведущих к нарушению синтеза фибриллина. Обнаружение связи гена фибриллина с синдромом Марфана дает возможность проводить молекулярно-генетическую диагностику, в том числе пренатальную. Симптоматика синдрома Марфана многосистемная и разнообразная: от легких форм, трудноотличимых от нормы, до инвалидизирующего течения.
Наиболее специфичны для синдрома Марфана нарушения скелета, вывих хрусталика, сердечно-сосудистые изменения, эктазия твердой мозговой оболочки.
- Мышечно-скелетная система: арахнодактилия, долихостеномелия, высокий рост, длинные конечности, деформация позвоночника (сколиоз, грудной
лордоз, гиперкифоз),деформация передней стенки грудной клетки (вдавленная грудь, «куриная» грудь или оба варианта), ненормальная подвижность суставов (гиперподвижность, врожденные контрактуры или оба варианта), плоская стопа, высокое арковидное нёбо, недоразвитие вертлужной впадины, мышечная гипотония (рис. 4.15- 4.17).
- Глаза: вывих хрусталика, миопия, отслоение сетчатки, большая роговица, удлиненная ось глазного яблока, уплощение роговицы.
- Сердечно-сосудистая система: аортальная регургитация, аневризма восходящей части аорты, расслоение аорты, митраль-
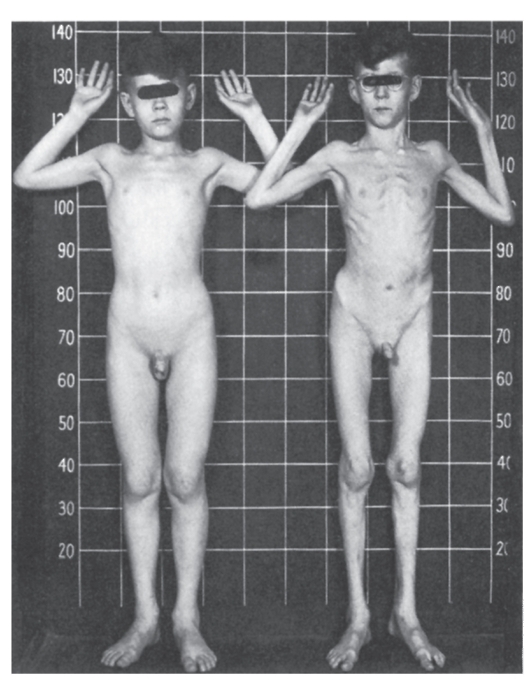
Рис. 4.15. Два брата: слева - нормальный мальчик 10 лет; справа - 8-летний мальчик с синдромом Марфана (подвывих хрусталика, в связи с чем ребенок в очках и у него наблюдается высокий рост, отсутствие подкожной клетчатки, сколиоз, деформация грудной клетки)
ная регургитация, застойные сердечные нарушения, пролапс митрального клапана, кальцификация митрального отверстия, аритмия.
- Наружные покровы: паховые грыжи, атрофические стрии.
- Легочная система: спонтанный пневмоторакс.
- Нервная система: эктазия твердой мозговой оболочки, включая поясничнокрестцовое менингоцеле, аномалии развития нервной системы.
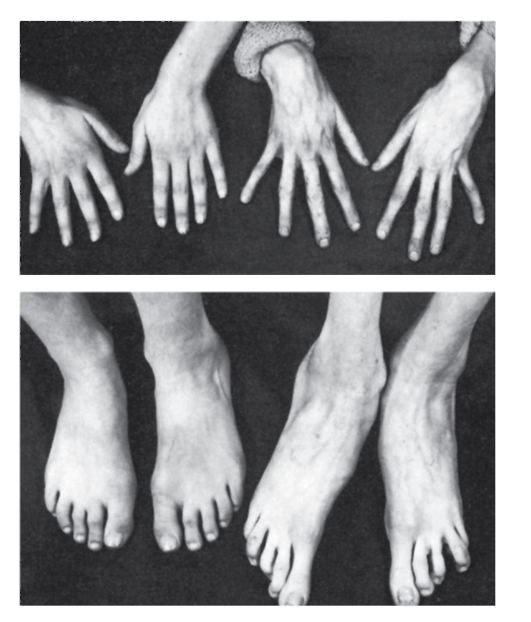
Рис. 4.16. Кисти и стопы в норме (слева) и при синдроме Марфана (справа)
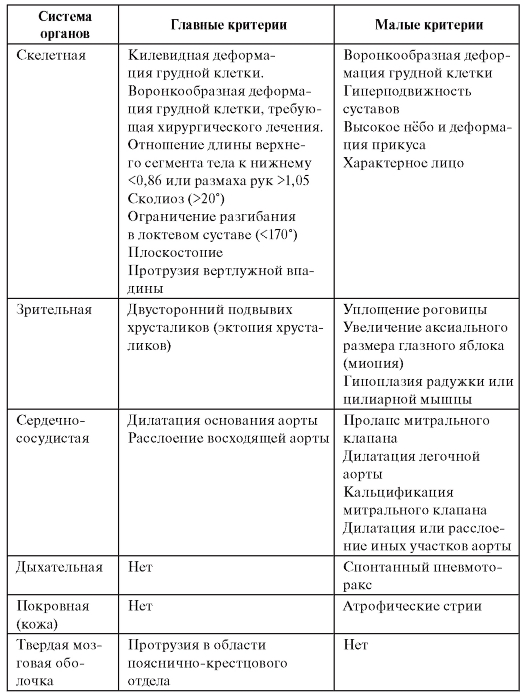
Система диагностических признаков синдрома Марфана приведена в табл. 4.4. Для постановки диагноза синдрома Марфана
необходимо по меньшей мере по одному главному критерию в двух системах и одного малого - в третьей системе органов.
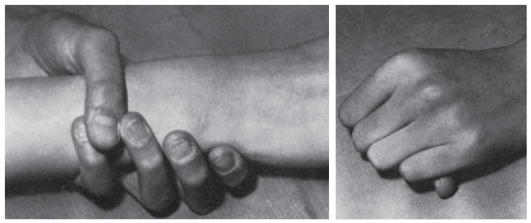
Рис. 4.17. Диагностические критерии арахнодактилии при синдроме Марфана
Таблица 4.4. Диагностические признаки синдрома Марфана
Окончание таблицы 4.4

Диагностические критерии синдрома Марфана должны строго учитываться, поскольку за синдром Марфана могут быть приняты некоторые другие врожденные дисплазии соединительной ткани (наследственной или еще невыясненной природы).
При несомненном синдроме Марфана у родственника I степени родства этот диагноз можно установить у пробанда, имеющего проявления болезни в двух системах (органах) и более. Наиболее специфичными проявлениями для диагностики считаются вывих хрусталика, расширение аорты, расслоение аорты, эктазия твердой мозговой оболочки.
При синдроме Марфана ростовые скачки и закрытие зон роста скелета наблюдаются на 2,4 года раньше у лиц мужского пола и на 2,2 года раньше у лиц женского пола. Рост взрослых мужчин равен в среднем 191 см, женщин - 175 см.
Частота синдрома Марфана в популяции равна 1 : 10 000-15 000. Популяционных и этнических различий в частоте и клинической картине болезни не отмечено.
Синдром Марфана - типичная аутосомно-доминантная болезнь, хорошо изученная в клинико-генетическом плане. Клинический полиморфизм выражен очень ярко, но его причины неясны. Разницы в клинической картине случаев, унаследованных от больных родителей, и спорадических случаев нет. С увеличением возраста отца (особенно после 35 лет) повышается вероятность рождения ребенка с синдромом Марфана.
Синдром Элерса-Данло
Синдром Элерса-Данло - гетерогенная группа наследственных болезней соединительной ткани с разными типами наследования,
но общими клиническими признаками: гипермобильность суставов, повышенная растяжимость кожи, скелетные изменения, повышенная ранимость кожи, проявления со стороны внутренних органов. Частичное клиническое описание этого синдрома впервые было сделано еще в 1657 г. голландским хирургом Д. ван Мекреном с зарисовкой внешнего вида больного. Первая документальная фотография (больной позировал, сильно растянув кожу на груди) относится к 1880 г. (рис. 4.18). В России синдром подробно описал А.Н. Черногубов (1891), назвав его генерализованным нарушением соединительной ткани. Однако он не продолжил этих наблюдений, а за границей был опубликован только реферат его работы. Позже этот синдром был описан Э. Элерсом (1901) и Х.А. Данло (1908) и назван по имени этих ученых.
Синдром Элерса-Данло проявляется врожденной гиперрастяжимостью соединительной ткани в связи с нарушениями синтеза кол-
лагена, обусловленными мутациями в разных генах коллагена и других белков экстраклеточного матрикса. Клинически, биохимически, молекулярногенетически идентифицировано 6 типов синдрома Элерса-Данло, которые с клинико-генетической точки зрения должны считаться самостоятельными нозологическими формами. Их симптомы перекрываются, поэтому целесообразно познакомиться со всем комплексом признаков, встречающихся при различных типах данного заболевания.

Рис. 4.18. Фотография больного с синдромом Элерса-Данло с выставки (1885)
- Кожа: сверхрастяжимость (щеки, под наружными концами ключиц, локти, колени), бархатистость, хрупкость, кровоточивость, темно-коричневые веснушки (более 20), рубцы (множественные, типа
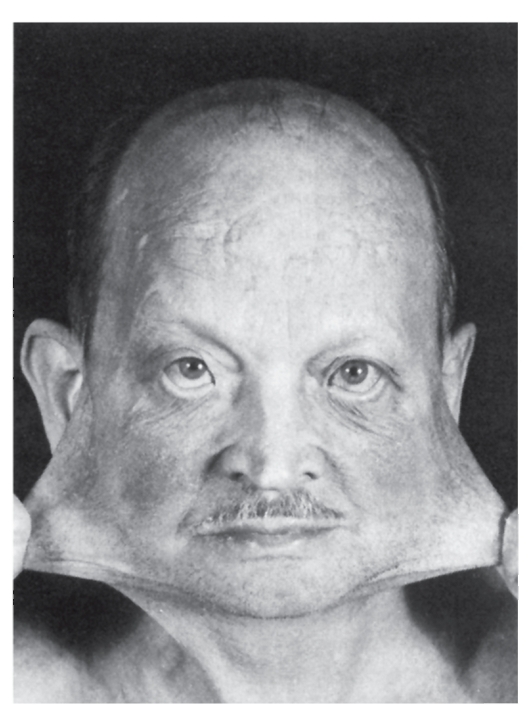
Рис. 4.19. Повышенная растяжимость кожи лица, рубцы на лбу
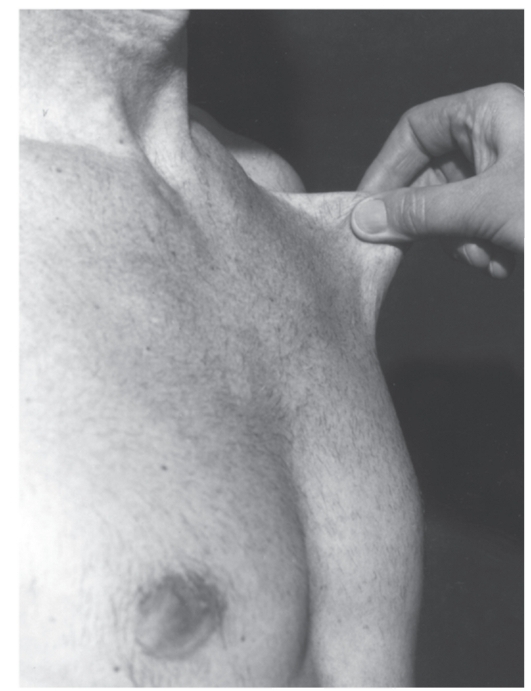
Рис. 4.20. Повышенная растяжимость кожи под ключицей
папиросной бумаги, келоидные), стрии в области поясницы, просвечивающие вены, расхождение послеоперационных швов (рис. 4.19, 4.20).
- Суставы: пассивное разгибание мизинца на 90° и более, приведение большого пальца кисти к предплечью, переразгибание локтевого сустава на 10° и более, переразгибание коленного сустава на 10° и более, свободное касание ладонями пола при несогнутых коленях, переразгибание межфаланговых, запястных, голеностопных и других суставов, привычный вывих суставов, плоскостопие (рис. 4.21, 4.22).
- Глаза: птоз, избыточное развитие периорбитальной клетчатки, отслойка сетчатки, остатки эпиканта, разрыв глазного яблока.
- Уши: сверхрастяжимость.
- Зубы: частичная адентия, сверхкомплектные зубы, опалесцирующая эмаль, пародонтоз, множественный кариес.
- Грудная клетка: сколиоз, кифоз, лордоз, плоская спина, вдавление грудины.
- Живот: грыжи (пупочная, белой линии, паховая, диафрагмальная), спонтанная перфорация кишечника.
- Конечности: варикозные вены, подкожные подвижные узелки на голенях, плоскостопие.
- Сердце: пролапс митрального клапана, аритмии, вегетососудистая дистония.
- Внутренние органы: птоз желудка, почек и матки.
- Мозг: аневризмы сосудов мозга, субарахноидальное кровоизлияние.
- Стремительные роды.
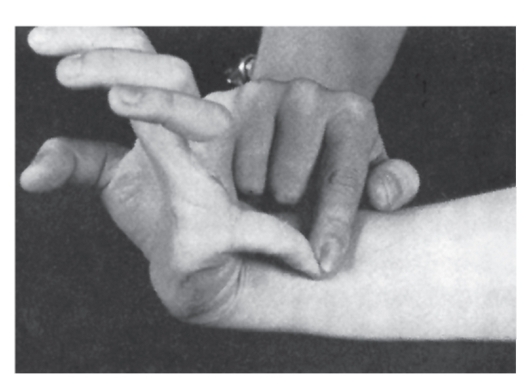
Рис. 4.21. Переразгибание V пальца
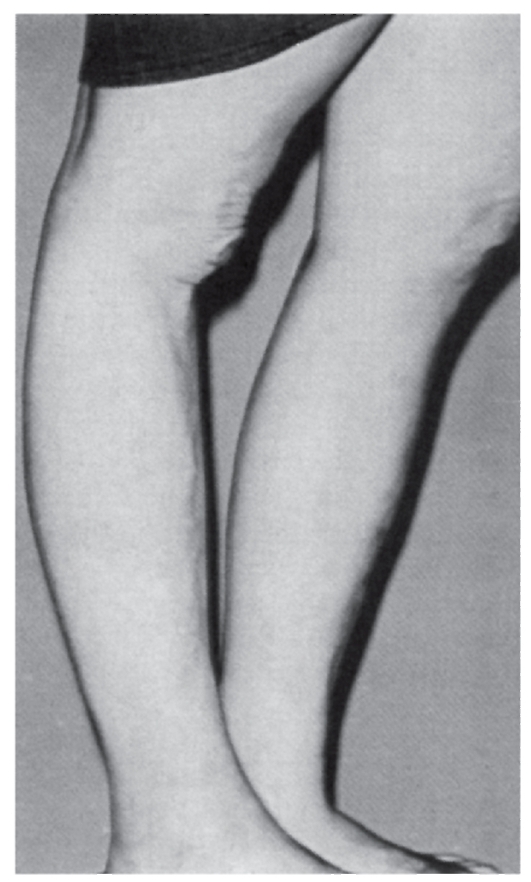
Рис. 4.22. Переразгибание коленного сустава. Рубцы в области коленных суставов
Как видно из перечня симптомов, при синдроме Элерса- Данло имеются нарушения (первичные или вторичные) во всех системах организма. Наиболее важные диагностические признаки: гиперэластичность кожи, подкожные узелки (сферулы), легче прощупываемые на передней поверхности голени; переразгибание суставов; повышенная ранимость тканей; симптомы геморрагического диатеза; пролапс митрального клапана.
Клиническая картина разных типов синдрома Элерса-Данло перекрывается почти по всем симптомам. Для дифференциальной диагностики учитывают сочетания выраженности сим-
птомов и осложнений. Для этого нужен опыт лечения больных с врожденными нарушениями соединительной ткани.
С клинико-генетической точки зрения наиболее приемлема последняя классификация синдрома (1997), основанная в целом на этиологическом принципе (табл. 4.5). Следует подчеркнуть, что клиническая диагностика синдрома Элерса-Данло осложняется из-за перекрывания клинических признаков различных типов данной патологии и других соединительнотканных заболеваний.
Таблица 4.5. Классификация синдрома Элерса-Данло
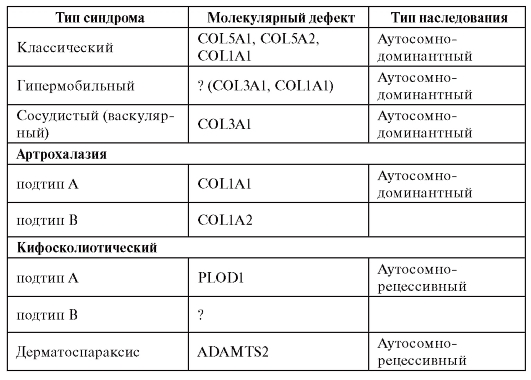
Основные клинические характеристики разных типов синдрома Элерса-Данло представлены ниже.
Классический тип. Основные диагностические критерии: гиперрастяжимость кожи, атрофичные («папиросные») рубцы, гипермобильность суставов. Кожа гладкая, бархатистая, тонкая. Атрофичные рубцы развиваются на местах, наиболее подвергающихся механическим воздействиям (колени, локти, лоб). Патологическая подвижность суставов проявляется повторными вывихами и подвывихами
суставов (особенно часто плечевых, височно-челюстных и надколенников). Характерны сколиоз, плоскостопие, грыжи, опущение внутренних органов (гениталий у женщин и выпадение прямой кишки у детей). У беременных с классическим типом синдрома Элерса-Данло наблюдается преждевременный разрыв плодных оболочек, преждевременные роды.
Гипермобильный тип. Этот тип более доброкачественный, чем другие. В нем доминирует выраженная гипермобильность суставов. Скелетные изменения отсутствуют. Кожные проявления относительно незначительны (растяжимость вариабельна). Атрофичные рубцы нехарактерны. Наиболее характерны боли в суставах и мышцах. Они начинаются в подростковом возрасте и носят хронический и тяжелый характер.
Сосудистый (васкулярный) тип. Наиболее опасный тип синдрома Элерса-Данло в связи с разрывами стенок сосудов среднего и крупного калибра, а также стенок полых органов (кишечника, матки, мочевого пузыря). Чаще всего разрываются артерии среднего калибра. В случае внезапной смерти необходимо исключить синдром Элерса-Данло, особенно в семейных случаях. Отмечаются следующие дополнительные признаки этого типа патологии: артериовенозные каротидно-кавернозные фистулы, пневмоторакс, атрофия краев десен.
Артрохалазия. При этой форме отмечается сильно выраженная гипермобильность суставов с повторными вывихами или подвывихами, кифосколиоз, врожденный вывих бедра, низкий рост, который является следствием кифосколиоза. Подтипы выделены на основании молекулярно-генетических исследований (см. табл. 4.5).
Кифосколиотический тип. Для этого типа характерна мышечная гипотония, задержка моторного развития, прогрессирующий сколиоз с рождения. В последующем развивается кифосколиоз, из-за которого после 20-30 лет больной теряет способность к самостоятельному передвижению. Характерным признаком является глазная патология: разрывы глазного яблока, хрупкость склер, миопия, микрокорнеа. Подтипы клинически не различаются.
Дерматоспараксис («рвущаяся кожа»). Основные критерии диагностики следующие: хрупкая, отслаивающаяся, «избыточная» кожа, большие грыжи (пупочные и паховые). Избыток кожи на лице напоминает cutis laxa, но отличается тем, что для cutis laxa не характер-
ны синяки и хрупкость кожи. Дерматоспараксис описан только у шести пациентов.
Синдром Элерса-Данло - типичный пример разнолокусной гетерогенности. Все локусы, мутации в которых вызывают синдром, имеют отношение к синтезу белков волокнистых элементов соединительной ткани (главным образом коллагена). Коллагеновые волокна имеют неправильную форму и расположены неупорядоченно (рис. 4.23).
Наличие синдрома Элерса- Данло мало отражается на репродуктивной функции, хотя у больных снижено количество потомков. Имеются изоляты с выраженным эффектом родоначальника на протяжении нескольких поколений, в которых больные с синдромом Элерса-Данло составляют 10% всего населения.
Рис. 4.23. Структура пучка коллагеновых волокон при синдроме Элерса-Данло и в норме (правый нижний угол). У больных волокна неправильной формы, разных размеров и расположены неупорядоченно
Фенилкетонурия
Классическая фенилкетонурия - аутосомно-рецессивная болезнь аминокислотного обмена. Фенилкетонурия клинически выделена в самостоятельную форму А. Фелингом в 1934 г. Патологические проявления связаны с недостаточностью печеночного фермента фенилаланингидроксилазы. Мягкая форма фенилкетонурии и доброкачественная гиперфенилаланинемия обусловлены мутациями других генов, также затрагивающих обмен фенилаланина. С генетической точки зрения это самостоятельные формы.
Недостаточность фермента ведет к нарушению процесса гидроксилирования фенилаланина в тирозин. Вследствие этого происходят накопление фенилаланина в крови (фенилаланинемия), обра-
зование избыточного количества фенилпировиноградной кислоты, которая выделяется с мочой, нарушение формирования миелиновой оболочки вокруг аксонов в ЦНС.

Рис. 4.24. Больной фенилкетонурией. Слабая пигментация кожи, волос, радужной оболочки глаз; умеренная степень олигофрении
Дети с фенилкетонурией рождаются здоровыми, но в первые месяцы в связи с поступлением фенилаланина с молоком матери развиваются клинические проявления: повышенная возбудимость, гиперрефлексия, повышенный тонус мышц, тремор, судорожные эпилептиформные припадки, характерный «мышиный» запах. Позже отмечают умственную отсталость, микроцефалию. Поскольку нарушение обмена фенилаланина ведет к снижению уровня тирозина, одно из фенотипических проявлений фенилкетонурии - снижение уровня или прекращение
образования меланина, поэтому уменьшена пигментация кожных покровов, волос, радужной оболочки глаз (рис. 4.24). Течение болезни прогредиентное. Без лечения умственная отсталость может достигать тяжелой степени. Диагноз устанавливают на основании клинической картины и результатов биохимического исследования мочи (фенилпировиноградная кислота) или крови (фенилаланинемия).
Ранняя диагностика фенилкетонурии и профилактическое лечение (искусственное вскармливание) предупреждают развитие клинической картины болезни (см. главу 11).
Генетика фенилкетонурии хорошо изучена. Уже через год после клинического описания болезни Л. Пенроуз доказал аутосомнорецессивный характер наследования.
Локус фенилкетонурии (фенилаланингидроксилазы) расположен в длинном плече хромосомы 12 (12q22-24). Ген секвенирован. Для большинства семей существует возможность выполнения
молекулярно-генетической пренатальной диагностики и выявления гетерозигот.
Популяционная генетика фенилкетонурии, как и большинства аутосомно-рецессивных болезней, сложная. Частота фенилкетонурии в большинстве европейских стран в среднем составляет, по-видимому, 1 : 10 000 новорожденных, в России 1 : 8000. Однако по этому показателю имеются значительные различия между популяциями: 1 : 2600 в Турции, 1 : 4500 в Ирландии, 1 : 6000 в Белоруссии, 1 : 16 000 в Китае, 1 : 30 000 в Швеции, 1 : 119 000 в Японии. Частота гетерозигот в большинстве европейских популяций составляет 1 : 100. Механизмы накопления гетерозигот в популяциях неизвестны. Вероятно, это обусловлено селективным преимуществом гетерозигот, потому что гомозиготы, т.е. больные, без лечения ранее не оставляли потомства, а доказательств повышенного мутационного процесса в соответствующем локусе нет.
Муковисцидоз
Синоним: кистозный фиброз. Это аутосомно-рецессивная болезнь, в основе патогенеза которой лежит нарушение транспорта ионов хлора и натрия через клеточные мембраны. Ген муковисцидоза детерминирует синтез белка, называемого муковисцидозным трансмембранным регулятором проводимости.
Патогенез болезни обусловлен тем, что при отсутствии синтеза первичного продукта гена (трансмембранного регулятора) нарушается транспорт хлоридов в эпителиальных клетках. Это приводит к избыточному выведению хлоридов. Следствием становится гиперсекреция густой слизи в клетках эндокринной части поджелудочной железы, эпителии бронхов, слизистой оболочке ЖКТ. Выводные протоки поджелудочной железы закупориваются, слизь не выводится, образуются кисты (отсюда второе название муковисцидоза - кистозный фиброз) (рис. 4.25). Ферменты поджелудочной железы не поступают в просвет кишечника. Гиперпродукция слизи в бронхиальном дереве ведет к закупорке мелких бронхов и последующему присоединению инфекции (рис. 4.26). Подобные процессы развиваются в придаточных пазухах, в канальцах семенников. В потовой жидкости повышена концентрация ионов натрия и хлора, это основной диагностический лабораторный признак.
Клинически болезнь проявляется в 4 формах (иногда в одной и той же родословной по несколько форм) с большим клиническим
Рис. 4.25. Патогистологическая картина поджелудочной железы (кисты и расширенные протоки)
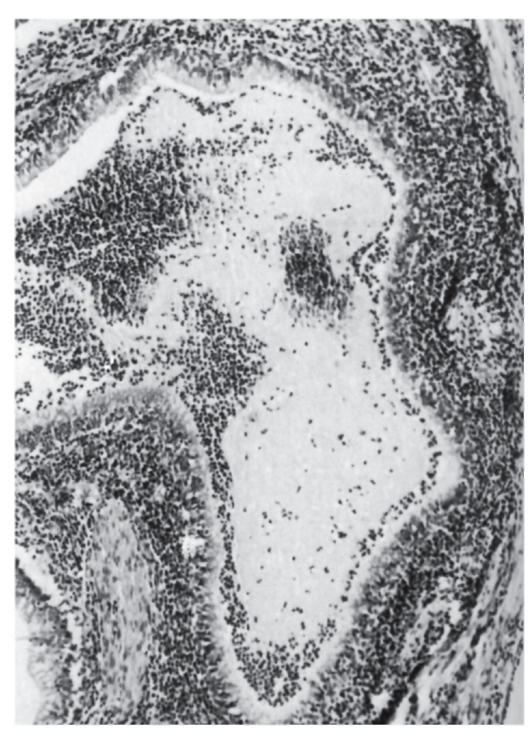
Рис. 4.26. Патогистологическая картина легкого при муковисцидозе (бронхоэктазы со слизистым воспалительным экссудатом в просвете)
полиморфизмом - от врожденных состояний до легких форм у взрослых.
- Мекониевый илеус новорожденных - врожденная форма болезни с избыточным заполнением кишечника густым меконием к моменту рождения. В первые дни внеутробной жизни болезнь проявляется признаками полной кишечной непроходимости, которая трудно разрешается без оперативного вмешательства. Врожденная форма встречается редко - не более 1% всех случаев.
- Кишечная форма начинается в раннем детском возрасте, часто после перевода ребенка на искусственное вскармливание из-за недостаточности панкреатических ферментов. Нарушение пищеварения ведет к сниженному питанию, отставанию в развитии, обильному зловонному стулу, светлому, с большим количеством жира. Живот у ребенка всегда вздут. Со временем в
патологический процесс вовлекается печень (жировая инфильтрация, холестатический гепатит, цирроз). Частота кишечной формы составляет 5-10% всех больных муковисцидозом. - Бронхолегочная форма обусловлена гиперпродукцией вязкого секрета в бронхолегочной системе. Первые клинические признаки появляются на фоне острой респираторной инфекции. Вязкий секрет приводит к обструктивному синдрому, присоединению вторичной инфекции. Рецидивирующий хронический инфекционно-воспалительный процесс осложняется гнойнообструктивным бронхитом, тяжелыми пневмониями, возникающими несколько раз в год. К вторичным изменениям относятся бронхоэктазы, эмфизема, пневмосклероз, легочное сердце (рис. 4.27). В бронхиальном содержимом в основном выявляются синегнойная палочка, золотистый стафилококк и гемофильная палочка, нередко в ассоциации. Флора часто устойчива к антибиотикам. Дети умирают от тяжелой дыхательной и сердечной
недостаточности. Бронхолегочная форма встречается у 15-20% всех больных муковисцидозом. - Смешанная (легочно-кишечная) форма - наиболее распространенная (65-75% всех больных муковисцидозом). При этом варианте клинической картины отмечается сочетание кишечных и бронхолегочных симптомов с разной выраженностью то одних, то других.
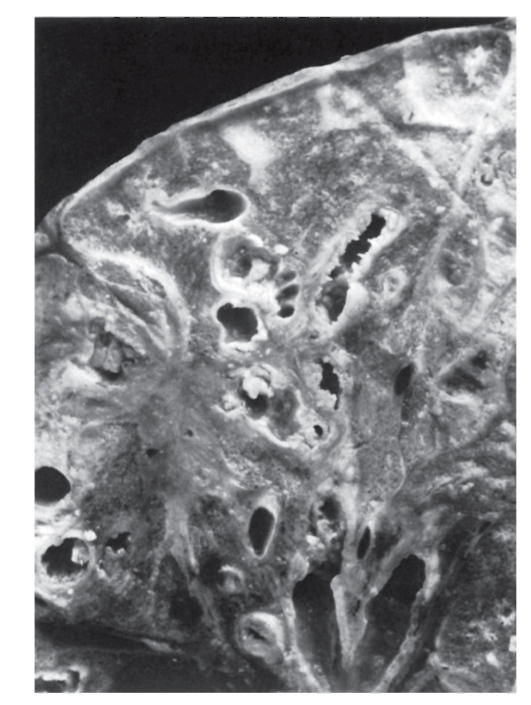
Рис. 4.27. Муковисцидоз. Легкое на аутопсии (бронхоэктазы и пневмосклероз)
Тяжесть клинической картины зависит от типа мутаций. Также отмечен тот факт, что одни и те же мутации могут приводить к разной клинической картине заболевания. В настоящее время это объясняется мутациями генов, модулирующих степень
поражения легких (например, гены HLA класса II, фактора некроза опухолей α (ФНОα), глутатион-D-трансферазы М1, трансформирующего фактора роста β1 и др.).
Не все мутации в гене муковисцидоза приводят к соответствующей клинической картине. Очевидно, что многие мутации нейтральны, как и по ряду других генов (глобины, рецептор ЛПНП и др.). Вместе с тем установлено, что более 10 мутаций, не приводящих к клинической картине муковисцидоза, способствуют развитию диссеминированных бронхоэктазов неизвестной природы, цирротических процессов в печени. Возможна также связь с эмфиземой легких. Гетерозиготность по патологическим мутациям встречается в 2 раза чаще у больных хроническим панкреатитом. У 4% больных муковисцидоз диагностируется во взрослом состоянии.
Прогноз при муковисцидозе всегда серьезный. Требуются пристальное внимание со стороны врача и большое терпение со стороны пациента (или родственников). Почти 50 лет назад, когда был описан муковисцидоз, больные умирали в первые годы жизни. Сейчас продолжительность жизни существенно увеличилась. В настоящее время основными направлениями лечения больных с муковисцидозом являются: назначение микросферических панкреатических ферментов с рН-чувствительной оболочкой, антибиотикотерапия (раннее начало, длительное лечение, применение с профилактической целью), прием урсодезоксихолевой кислоты. Также в настоящее время активно ведутся разработки в области генной терапии заболевания и поиск веществ, способных стимулировать синтез, транспорт и функции неполноценного кистофиброзного трансмембранного регулятора (CFTR) [например, фенилбутират стимулирует цАМФ-зависимый хлоридный поток (цАМФ - циклический аденозинмонофосфат)]. Благодаря такому подходу в развитых странах в последние годы отмечается рост числа больных муковисцидозом подросткового, юношеского возраста и взрослых, что свидетельствует о постепенной его трансформации из фатального заболевания детского возраста в хроническую патологию взрослых. Средняя продолжительность жизни больных муковисцидозом в развитых странах в начале XXI столетия составила 32 года, в России - 25 лет.
Диагностика муковисцидоза основана на клинической картине, результатах биохимического определения ионов натрия и хлора в поте (натрий более 70 ммоль/л, хлор более 60 ммоль/л). По непонятным причинам у 1-2% пациентов с муковисцидозом концентрация
хлоридов в поте бывает нормальной. В затруднительных случаях для диагностики используют молекулярно-генетическую технологию. Для просеивающей преклинической диагностики муковисцидоза лучший метод - измерение уровня иммунореактивного трипсина в каплях высушенной на фильтровальной бумаге крови, которое позволяет судить об активности трипсиногена. Разработаны специальные наборы для такой диагностики. Однако по поводу массовых обследований на муковисцидоз есть две точки зрения: проводить в 1-й месяц жизни и не проводить совсем. Вторая точка зрения аргументируется тем, что раннее выявление муковисцидоза ничего не дает для больных. Лечение начинается по существу только при обострении болезни, которое легко распознается клинически.
Во многих западных странах проводится скрининг на муковисцидоз, и расходы на него считаются оправданными. В России скрининг на муковисцидоз введен с 2006 г. в рамках национального проекта «Здоровье». На 4-й день жизни ребенка в роддоме забирают кровь на тест-бланк и передают (или пересылают) в лабораторию. Ответ о результатах первичного обследования поступает в лечебнопрофилактическое учреждение к 9-10 дню. Подробности неонатального скрининга на муковисцидоз см. также в гл. 11.
Генетика муковисцидоза (формальная, клиническая, молекулярная, популяционная) всесторонне изучена. Ген муковисцидоза локализован в хромосоме 7 (7q31-32), его размер составляет 250 000 пар оснований, ген включает 27 экзонов. Зрелая мРНК состоит из 6500 оснований, кодирует полипептидную цепь длиной 1480 аминокислотных остатков. Физиологическая роль и строение первичного белка (трансмембранного регулятора проводимости) хорошо изучены, что и позволило расшифровать многие стороны патогенеза муковисцидоза. Экспрессия гена ограничена главным образом эпителиальными клетками, она проявляется в наибольшей степени в экзокринных железах (слюнных, поджелудочной и потовых), семенниках и кишечнике. В эпителии легких ген функционирует, но слабо, и поэтому дефект хлоридного транспорта там четко выражен.
В гене муковисцидоза обнаружено более 1500 мутаций, из них около 300 дают патологический эффект (миссенс, делеции, нонсенс, сдвиг рамки считывания, нарушения сплайсинга). Наиболее частая мутация (до 70% всех случаев) - делеция трех пар оснований, ведущая к отсутствию аминокислотного остатка в 508-м положении (отсюда название этой мутации: F508del) полипептидной цепи.
В ряде работ показано, что наиболее тяжелая и ранняя манифестация заболевания наблюдается у гомозигот именно по этой мутации.
Географические и этнические различия в частоте муковисцидоза и вариантах мутаций гена муковисцидоза очень значительны.
Данные по частоте муковисцидоза в Европе представлены в табл. 4.6.
Таблица 4.6. Частота муковисцидоза по данным неонатального скрининга в Европе (2007)

Что касается частоты муковисцидоза в России, то величины сильно колеблются - от 1 : 4000-5000 (Приморский край, Томская область) до 1 : 17 000 (Северо-Запад). Это можно объяснить популяционными различиями или малыми выборками, с одной стороны, или, несовершенством скрининговых программ - с другой (на муковисцидоз они введены в России только в 2006 г.). Наиболее строгие критерии диагностики и достаточные величины в выборках выполнены в Москве (1 : 11 500) и Северо-Западном регионе (1 : 17 000). В любом случае можно говорить о существенно меньшей частоте муковисцидоза в России по сравнению с европейскими странами.
Муковисцидоз редко встречается в восточных популяциях и у африканского чернокожего населения (1 : 100 000). Причина таких популяционных различий неясна. Частота гетерозигот в Европе очень высока (до 5% населения), что можно объяснить селективным преимуществом гетерозигот. В чем заключается это преимущество, еще не выяснено. Не исключено, что гетерозиготы по муковисцидозу устойчивы к туберкулезу. В ряде популяций описан эффект родоначальника как причина высокой концентрации мутантных аллелей.
Молекулярно-генетическая диагностика муковисцидоза и носительства соответствующего гена возможна для большинства мутаций на основе полимеразной цепной реакции (ПЦР). Пренатальная диагно-
стика муковисцидоза вошла в широкую практику. Диагностическая панель праймеров для 15-86 мутаций (из 200-300) позволяет распознавать 60-80% носителей мутаций среди населения европеоидной расы. В России такие лаборатории есть в Москве, Санкт-Петербурге, Томске, Ростове-на-Дону и Уфе.
Адреногенитальный синдром
Синонимы: врожденная вирилизирующая гиперплазия коры надпочечников; врожденная надпочечниковая гиперплазия; врожденная дисфункция коры надпочечников. Адреногенитальный синдром относится к группе наследственных нарушений биосинтеза стероидных гормонов. Как известно, процесс образования стероидов многоступенчатый. Каждая ступень катализируется соответствующим ферментом. Известны, по меньшей мере, 5 разновидностей наследственных дефицитов ферментов, обеспечивающих синтез стероидов (21-гидроксилаза, 3-β- гидроксистероиддегидрогеназа, 11-β-гидроксилаза, 17-α-гидроксилаза, 20-α-холестеролгидроксилаза), а также транспортного StAR-протеина, осуществляющего перенос холестерина на внутреннюю мембрану митохондрий, где начинается ферментативное превращение холестерина в стероиды. Все варианты этих наследственных нарушений наследуются по аутосомно-рецессивному типу.
Наиболее распространена форма адреногенитального синдрома (90-95% всех случаев), обусловленная дефицитом фермента 21-гидроксилазы (цитохром CYP21), катализирующего превращение прогестерона в дезоксикортикостерон и 17-гидроксипрогестерона в 11-дезоксикортизол. Это заболевание очень разнообразно по клиническим проявлениям (более подробно об этом можно прочитать в статье Н.С. Осиновской «Врожденная гиперплазия коры надпочечников (ВГКН) как дефицит α1-гидроксилазы: современные представления и генетическая диагностика» на компакт-диске. Известны два классических клинических варианта этой болезни - сольтеряющая и простая вирильная формы.
Сольтеряющая форма характеризуется полным дефицитом фермента и проявляется в нарушении солевого обмена (дефицит минералокортикоидов). В патологический процесс вовлечена ренинальдостероновая система. Клиническая картина возникает в первые дни после рождения. У новорожденного отмечаются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела. Обезвоживание вызывает

Простая вирильная форма
сопровождается прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. У новорожденных девочек при кариотипе 46,ХХ отмечается маскулинизация различной выраженности -
от умеренной гипертрофии клитора (рис. 4.28) до полного срастания губно-мошоночных складок с формированием мошонки и пениса. Внутренние половые органы сформированы правильно по женскому типу. У мальчиков вирильная форма адреногенитального синдрома при рождении обычно не распознается (гениталии нормальные). Диагноз устанавливают лишь на 5-7-м году жизни при появлении первых признаков преждевременного полового развития.
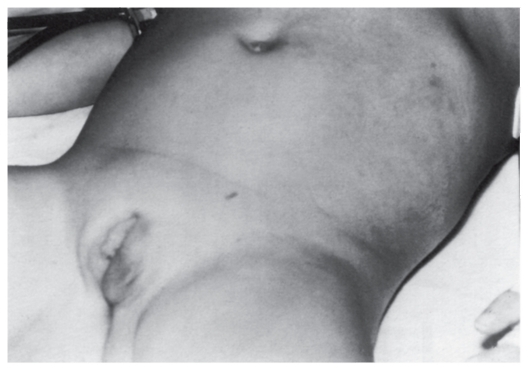
Рис. 4.28. Вирилизация наружных половых органов у девочки с адреногенитальным синдромом
Неклассическая форма дефицита 21-гидроксилазы, или поздняя форма, вызвана снижением активности фермента на 40-80% по сравнению с нормальными значениями и клинически проявляется только в подростковом возрасте. У девочек наблюдаются умеренное увеличение клитора, раннее развитие молочных желез, более раннее закрытие зон роста скелета, нарушение менструального цикла, гирсутизм, аспе, у женщин - нарушение менструального цикла и бесплодие. Симптомом избытка андрогенов у мальчиков может быть ускоренное закрытие зон роста костей, олигоспермия с последующим снижением фертильности у мужчин. У детей обоего пола наиболее частым симптомом неклассической формы заболевания является раннее оволосение на лобке и под мышками (адренархе).
Латентная форма не имеет клинических проявлений, но в сыворотке крови умеренно повышен уровень предшественников кортизола. Некоторые авторы не выделяют такие состояния в отдельную форму болезни.
Средняя частота дефицита 21-гидроксилазы составляет 1 : 13 500 новорожденных (в Швеции - 1 : 9800, в США - 1 : 15 981, в Европе -
1 : 14 970, в Японии - 1 : 18 000), а в некоторых изолированных популяциях она в 5-10 раз выше. В России частота адреногенитального синдрома составляет 1 : 8000-1 : 10 000. Адреногенитальный синдром относят к группе заболеваний, подлежащих просеивающей диагностике у новорожденных. Чем раньше установлен диагноз адреногенитального синдрома, тем более эффективной оказывается терапия глюкокортикоидами и минералокортикоидами, хотя не все проявления заболевания поддаются коррекции. Лучше всего лечится неклассическая форма. Более целесообразно проведение пренатальной диагностики и пренатального лечения дефицита 21-гидроксилазы в семьях, где оба родителя гетерозиготны и, как правило, уже имеют больного ребенка. Это позволяет родителям решить вопрос о сохранении беременности. Если беременность решено сохранить, то проводят внутриутробную терапию глюкокортикоидами (девочка избавляется от хирургической коррекции гениталий). У мальчиков своевременно диагностируется сольтеряющая форма, диагностика которой в постнатальном периоде часто задерживается.
Ген стероид-21-гидроксилазы (CYP21A2) локализован в коротком плече хромосомы 6 (6р21.3), это часть гена цитохрома Р450. Его мутантные формы вызывают недостаточность 21-гидроксилазы. При разных клинических формах адреногенитального синдрома, как правило, имеются свои спектры мутаций. Однако причины широкой вариабельности клинических симптомов адреногенитального синдрома полностью не ясны. Строгой корреляции между генотипом и фенотипом (сольтеряющая или простая вирильная форма) нет. Одни и те же мутации, идентифицированные на молекулярногенетическом уровне, имеют разные клинические проявления. Так, было проведено ДНК-обследование более 200 больных с адреногенитальным синдромом с описанием степени вирилизации женских наружных половых органов; секреции андрогенов в ответ на стимуляцию адренокортикотропным гормоном (АКТГ); реакции на бессолевую диету путем оценки дефицита альдостерона и потери соли. Больные были разделены на 26 групп с полностью идентичными мутациями. Оказалось, что в половине групп фенотипические проявления и генотип не коррелируют.
Однако неклассическая форма дефицита 21-гидроксилазы в 90% случаев обусловлена двумя миссенс-мутациями в гене цитохрома QYP21.
Среди причин клинического полиморфизма наряду с особенностями мутаций можно назвать также и модифицирующее влияние
других генов. Так, тяжелая сольтеряющая форма адреногенитального синдрома ассоциируется с антигенами системы HLA: A3, Bw47, DR7.
Миодистрофия Дюшенна-Беккера
Это одна из частых форм многочисленных наследственных нервномышечных заболеваний. Мышечные дистрофии характеризуются прогрессирующими дегенеративными изменениями в поперечнополосатой мускулатуре без первичной патологии периферического мотонейрона.
Миодистрофия Дюшенна-Беккера вызвана мутацией в гене, ответственном за синтез белка дистрофина. Этот белок находится в большом количестве в области сарколеммы, поддерживая, по-видимому, целостность мембраны (рис. 4.29). Структурные изменения в сарколемме приводят к дегенерации цитоплазматических компонентов, усиленному входу Са2+ внутрь волокон, что вызывает гибель миофибрилл.
Генетически единая форма миодистрофии Дюшенна-Беккера клинически разделяется на миодистрофию Дюшенна и миодистрофию Беккера.
Рис. 4.29. Гистохимическая картина мышц в норме (а) и при миопатии Дюшенна (б)
Миодистрофия Дюшенна встречается с частотой 1 : 5000 живорожденных мальчиков. Генетически она относится к Х-сцепленным рецессивным летальным нарушениям. Болезнь проявляется рано. Первые симптомы появляются в возрасте до 2 лет: дети позднее начинают ходить, не умеют бегать и прыгать. Симптомы становятся выраженными в 2-3-летнем возрасте. Это изменения походки («утиная» походка), псевдогипертрофия икроножных мышц (рис. 4.30). Процесс атрофии мышц постепенно приобретает восходящее направление: мышцы бедра тазовый пояс плечевой пояс руки. Наблюдается псевдогипертрофия не только икроножных мышц, но и ягодичных, дельтовидных, мышц живота, языка. У детей развиваются поясничный лордоз, крыловидность лопаток (рис. 4.31). Из наклоненного положения больные с трудом распрямляются, опираясь на колени
(рис. 4.32).
Атрофический процесс развивается и в сердце (кардиомиопатия). Острая сердечная недостаточность - причина летальных
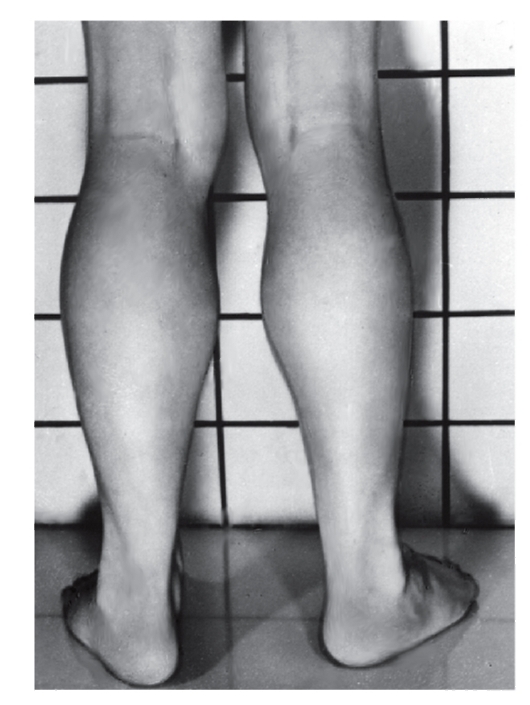
Рис. 4.30. Псевдогипертрофия икроножных мышц
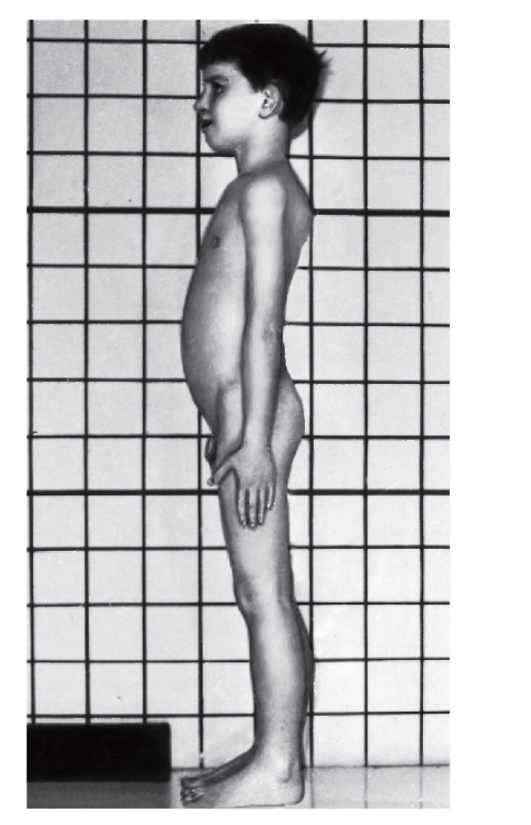
Рис. 4.31. Лордоз, псевдогипертрофия икроножных мышц, атрофия бедренных мышц
исходов. Нарушается моторика ЖКТ. Обнаруживаются вторичные изменения в костной системе. Интеллект у больных детей снижен. Корреляции между тяжестью мышечного дефекта и степенью снижения интеллекта нет. На самой последней стадии атрофия (слабость) захватывает мышцы лица, глотки и дыхательные мышцы. Больные умирают на 2-3-м десятилетии жизни.
Из биохимических показателей для миодистрофии Дюшенна наиболее характерен резко повышенный (в 10-100 раз) уровень креатинфосфокиназы в сыворотке крови. Активность этого фермента повышена в первые дни внеутробной жизни и, возможно, даже во внутриутробном периоде.
При клинической картине миодистрофии Дюшенна у девочек следует исключить моносомию по Х-хромосоме (синдром Тернера). Редкая возможность миодистрофии Дюшенна у девочек с кариотипом 46,ХХ не исключается из-за инактивации Х-хромосомы с нормальным аллелем во всех (или почти всех) клетках на ранних стадиях развития (16-32-клеточная бластоциста).
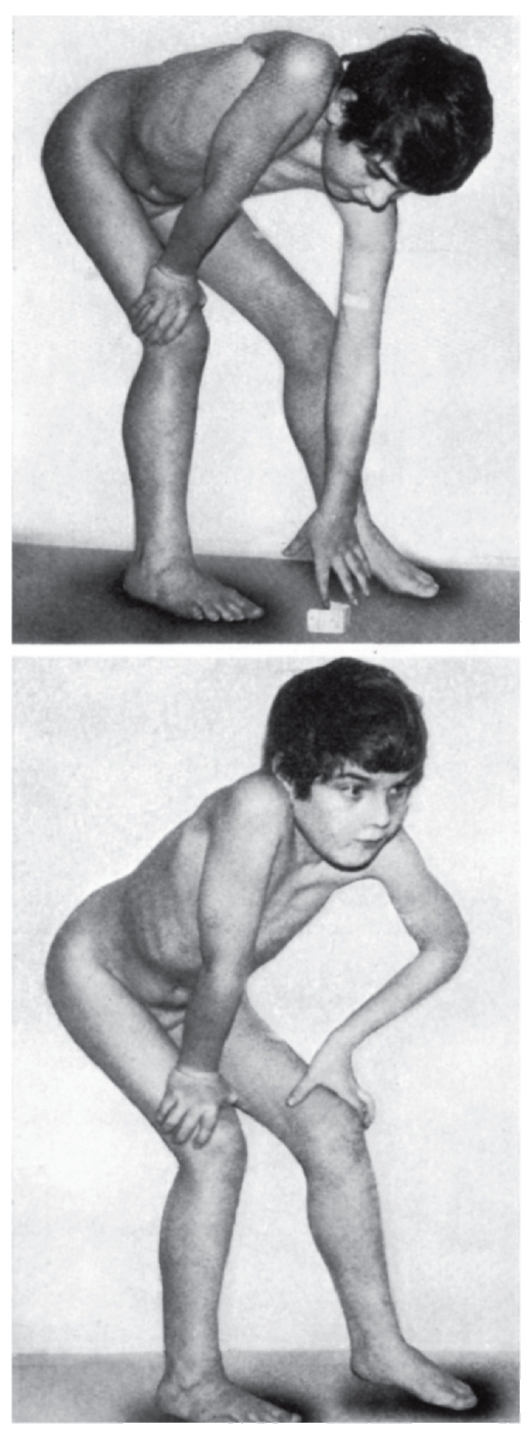
Рис. 4.32. Трудности при распрямлении после наклона
Гетерозиготные носительницы миодистрофии Дюшенна могут иметь субклинические симптомы: увеличенные икро-
ножные мышцы, повышенную утомляемость при физической нагрузке, изменения электромиограммы, увеличение уровня креатинфосфокиназы в крови. Более или менее выраженные симптомы миодистрофии Дюшенна отмечаются у 70% гетерозиготных носителей.
Миодистрофия Беккера - доброкачественная форма нервномышечных болезней. Частота миодистрофии Беккера у новорожденных мальчиков составляет 1:20 000. По клиническим симптомам миодистрофия Беккера напоминает миодистрофию Дюшенна, но в менее выраженной форме. Начало болезни не ранее 10-15 лет, течение мягкое, больные сохраняют работоспособность в возрасте 20-30 лет. Фертильность не снижена. Нарушения интеллекта и кардиомиопатии не отмечаются. Активность креатинфосфокиназы заметно повышена, но не в такой степени, как при миодистрофии Дюшенна.
Мягкая (или доброкачественная) форма миодистрофии Беккера объясняется тем, что, в отличие от миодистрофии Дюшенна, когда полностью прекращается синтез дистрофина, при миодистрофии Беккера синтез дистрофина детерминируется, но либо вырабатывается мало белка, либо продуцируется аномальный дистрофин.
В настоящее время разрабатывается терапия миодистрофии Дюшенна-Беккера: генная (заместительная), клеточная (миобласты, стволовые клетки), аминогликозидная терапия, применение ингибиторов протеосом и др.
Генетика миодистрофии Дюшенна и миодистрофии Беккера хорошо изучена (еще с 1930-х годов). Это типичный пример Х-сцепленного рецессивного наследования. Ген дистрофина локализован в коротком плече Х-хромосомы, он уже клонирован и секвенирован. Это самый длинный ген из всех изученных. В нем более 2х106 пар оснований (более 60 интронов); длина мРНК 16 000 пар оснований. В связи с большой длиной в гене часто наблюдаются перестройки, ведущие к мутациям. Доля свежих мутаций среди всех случаев миодистрофии Дюшенна-Беккера составляет 30%. Поскольку ген секвенирован, то молекулярно-генетическая диагностика геми- и гетерозиготных состояний возможна почти во всех семьях.
Миодистрофия Дюшенна-Беккера в 60-70% случаев вызывается большими делециями гена дистрофина (по меньшей мере одного экзона). Дупликация больших сегментов обнаруживается у 10% пациентов. Остальные 30% случаев обусловлены точковыми
мутациями (нуклеотидные замены и делеции одного нуклеотида). Сообщается также о случаях малых вставок и делеций (2-5 пар нуклеотидов). Делеции в экзонах размером более 5 нуклеотидов встречаются крайне редко.
Недавно был идентифицирован ген-модификатор миодистрофии Беккера - ген миогенного фактора-6 (MYF6), локализованный в хромосоме 12q21. Мутации в этом гене совместно с легкими изменениями гена дистрофина ведут к тяжелой клинической картине миодистрофии (о клиническом полиморфизме мышечной дистрофии Дюшенна см. также в статье О.В. Напалковой и др. на компакт-диске).
Синдром умственной отсталости с ломкой Х-хромосомой
Синоним: синдром Мартина-Белл. Ранее эту болезнь называли синдромом олигофрении с маркерной Х-хромосомой. Синдром Мартина-Белл - одна из наиболее часто встречающихся (после болезни Дауна) форм умственной отсталости. Популяционная частота заболевания составляет 1:2000-5000 живорожденных. Больных мальчиков в 2-3 раза больше, чем девочек, мальчики болеют тяжелее. Частота носительства от 1:165 до 1:1540.
Внешность больных неспецифична, но определенное диагностическое значение имеют удлиненное лицо, высокий выступающий лоб, макро- и долихоцефалия, гипоплазированная средняя часть
лица (особенно в сравнении с выступающими и часто увеличенными щеками), выступающий подбородок (рис. 4.33). Нёбо обычно дугообразное, губы толстые, нижняя губа часто вывернута. Отмечаются также увеличенные оттопыренные ушные раковины, большие кисти и стопы. Одно из типичных проявлений синдрома - макроорхидизм. При рождении у детей яички нормальных размеров, но с наступлением пубертатного периода они существенно увеличиваются в результате избыточного роста соединительной
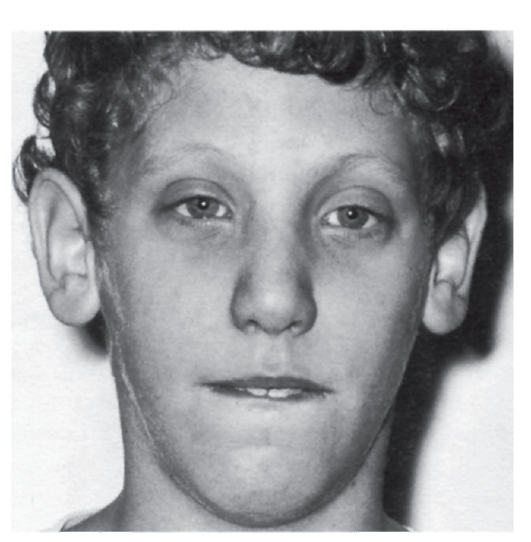
Рис. 4.33. Лицо подростка с синдромом Мартина-Белл
ткани и накопления жидкости. Однако половая активность таких больных минимальна, к половой жизни способны единицы.
Возможны повышенная растяжимость кожных покровов, слабость связочного аппарата суставов (что приводит к самопроизвольным вывихам, обычно пальцев кисти), плоскостопие. У многих больных формируется пролапс митрального клапана. Все эти симптомы обусловлены врожденной дисплазией соединительной ткани.
Умственная отсталость, типичная для синдрома Мартина-Белл, как правило, умеренная или выраженная, хотя у 10-15% больных обнаруживаются глубокая олигофрения и еще у такого же процента больных - мягкая умственная отсталость. Большинство пациентов социально адаптированы, выполняют несложную физическую работу. Психологи оценивают их как контактных и доброжелательных. Лишь в тяжелых случаях таких больных приходится помещать в специализированные интернаты. Неврологические симптомы включают мышечную гипотонию и в отдельных случаях судороги.
Пороки развития и нарушения других органов сравнительно редки; это расщелины нёба, нистагм, страбизм, птоз, катаракта, кривошея в сочетании со сколиозом, кифоз, дефект межпредсердной перегородки.
Помимо определенного комплекса клинических аномалий, синдром Мартина-Белл сопровождается характерной цитогенетической картиной: ломкостью в дистальной части длинного плеча Х-хромосомы (в зоне Xq), что внешне напоминает «спутник» длинного плеча (рис. 4.34). Эта ломкость выявляется лишь при культи-
Рис. 4.34. Ломкая Х-хромосома при синдроме Мартина-Белл: слева - набор Х-хромосом женщины (одна хромосома ломкая); справа - мужской набор (ломкая Х-хромосома)
вировании лимфоцитов в условиях дефицита фолиевой кислоты, поэтому для обнаружения ломкости нужно либо использовать культуральные среды, лишенные фолиевой кислоты, либо вводить в культуральную среду антагонисты фолиевой кислоты. Однако даже и при этих условиях ломкость Х-хромосомы выявляется не во всех клетках (до 60%).
Долго считали, что синдром наследуется по Х-сцепленному рецессивному типу. Однако в родословных отмечались случаи тяжелой болезни у женщин и легкой у мужчин. У женщин-гетерозигот отмечали некоторое снижение интеллекта, чаще оцениваемое как пограничная умственная отсталость. У некоторых из них в небольшом проценте клеток обнаруживали ломкую Х-хромосому. Таким образом, наследование синдрома Мартина-Белл не укладывалось в строгие рамки Х-сцепленного рецессивного наследования. Более того, в родословных отмечалась антиципация. Этиология этого заболевания была выяснена с помощью методов молекулярно-генетического анализа. Обнаружена экспансия нестабильных тринуклеотидных повторов (CGG) в 5'-нетранслируемой области гена FMR1 (fragile mental retardation). В норме в этом гене число повторов варьирует от 6 до 42. Частота экспансии премутационных триплетных повторов в гене FMR1 в полную мутацию в оогенезе является функцией длины премутационного аллеля, который имеется у гетерозиготной женщины (рис. 4.35). Хромосомы, в которых имеется 50-200 повторов, считают премутацией. Для этого состояния характерны незначительные проявления: задержка развития, признаки аутизма, атаксия. В следующем поколении число повторов может увеличиться (экспансия) до 1000 и более, что и обусловит выраженную клиническую картину, зависящую от числа повторов. Огромное влияние оказывает также цитозиновое метилирование повторов. Такое состояние называют полной мутацией. Соответственно если женщина унаследовала большое число повторов, то она будет больной. Обнаружены еще два синдрома [FXTAS (синдром тремора и атаксии, ассоциированный с ломкой Х-хромосомой) и FRAXE (синдром ломкой Х-хромосомы)], обусловленные динамическими мутациями в локусе Xq27.3, но эти гены (FMR1 и FMR2) являются причиной болезни намного реже. При этом клиническая картина сходна с синдромом Мартина-Белл, но не идентична.
Диагноз ставят на основании клинической картины и по результатам клинико-генеалогического и цитогенетического исследований.
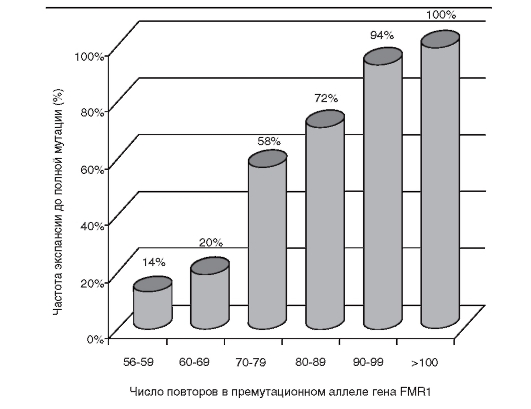
Рис. 4.35. Зависимость экспансии триплетных повторов в гене FMR1 от длины премутационного аллеля у гетерозиготной женщины
Наиболее точный метод - молекулярно-генетическая диагностика, в том числе с использованием методов оценки метилирования.
В настоящее время возможна пренатальная диагностика данного синдрома. Этиотропной терапии пока не существует.
ЭПИДЕМИОЛОГИЯ
Клинико-генеалогические наблюдения свидетельствуют о стандартном или своеобразном поведении генов в семьях, что и рассматривалось в предыдущих главах. Эти, безусловно, важные сведения должны быть дополнены знанием судьбы генов в популяциях, реальных закономерностей их распространения среди населения. Это особенно важно в настоящее время, потому что происходят интенсивные социальные процессы, с которыми человек ранее почти не сталкивался: массовая миграция населения, ломка этнических, национальных, религиозных, классовых, географических границ браков, увеличение численности населения планеты (неравномерное в разных популяциях), планирование деторождения (сокращение числа детей), улучшение медицинской помощи. Все эти факторы
могут влиять на распространенность генных болезней в ближайшем и отдаленном будущем.
Эпидемиология генных болезней включает сведения о распространенности этих болезней, частотах гетерозиготного носительства и факторах, их обусловливающих. Первичная основа возникновения наследственных болезней - мутационный процесс, а распространенность болезней (или частота больных) в популяции определяется уже популяционными закономерностями: интенсивностью мутационного процесса, давлением отбора (скоростью элиминации), который определяет плодовитость мутантов и гетерозигот в конкретных условиях среды, миграцией населения, изоляцией, дрейфом генов.
Наиболее объективная оценка распространенности наследственных болезней в разных популяциях - определение их частоты среди новорожденных, включая мертворожденных. В последующем частота детей с наследственными болезнями меняется в связи с повышенной смертностью в раннем возрасте. Смертность детей от генных болезней может различаться в разных странах и в разные годы, поскольку этот показатель зависит от многих факторов (уровня медицинской помощи, социально-экономических условий, культурных традиций и т.п.). Оценка частоты определенных форм наследственных болезней среди разных контингентов (в детских больницах, в специализированных школах-интернатах для инвалидов, на амбулаторном приеме и т.д.) прямого отношения к эпидемиологии генных болезней не имеет. Такую оценку можно использовать только для косвенных расчетов распространенности болезни. Социальное перераспределение больных в разные группы населения, безусловно, зависит от экономического уровня, религии, государственного устройства и других факторов.
В связи с большим числом нозологических форм генных болезней, их редкой встречаемостью, неполной клинической и патологоанатомической диагностикой наследственной патологии данные по распространенности наследственных болезней в целом еще отрывочные. Однако по формам, по которым проводятся массовые диагностические или профилактические программы, собран убедительный материал для суждения об эпидемиологии генных болезней.
Общая частота новорожденных с генными болезнями в популяциях в целом составляет примерно 1%, из них с аутосомно-доминантным типом наследования - 0,5%, с аутосомно-рецессивным - 0,25%, с Х-сцепленным - 0,25%; Y-сцепленные и митохондриальные болезни встречаются крайне редко. Распространенность отдельных форм болез-
ней колеблется от 1:500 (первичный гемохроматоз) до 1 : 100 000 и ниже (гепатолентикулярная дегенерация, атаксия-телеангиэктазия и др.).
Распространенность генной болезни условно можно считать высокой, если 1 больной встречается на 10 000 новорожденных и чаще, средней - 1 : 10 000-40 000, низкой - очень редкие случаи. В группу распространенных входит не более 15 генных болезней, но они обусловливают почти 50% общей частоты больных с наследственной патологией. Данные по распространенности некоторых наиболее изученных генных болезней представлены в табл. 4.7.
Таблица 4.7. Распространенность генных болезней
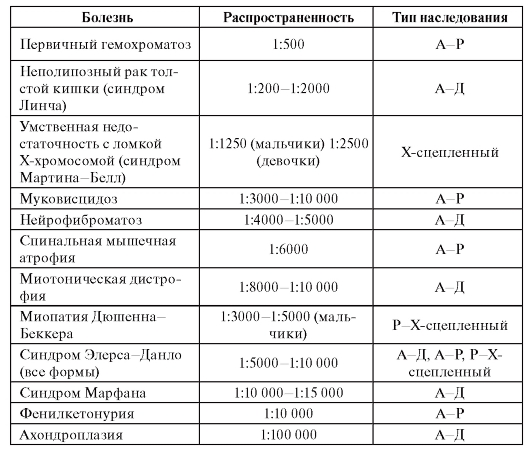
Примечание. А - аутосомный, Д - доминантный, Р - рецессивный.
Как видно из табл. 4.7, распространенность разных форм генных болезней сильно различается независимо от типа наследования. Причины таких различий во многом неясны. Распространенность
генных болезней определяется частотой вновь возникающих мутаций и числом детей у больных родителей и гетерозигот. Биологические основы распространенности доминантных и рецессивных болезней неодинаковые.
Распространенность многих доминантных болезней определяется в основном новыми мутациями. Практически все доминантные болезни ведут к снижению фертильности, а некоторые - даже к стерильности. Репродуктивная функция у больных в целом снижена по биологическим или социальным причинам. Так, только 10% случаев ахондроплазии относятся к семейным. Семейные формы нейрофиброматоза I типа составляют 50-70%. Снижена репродуктивная способность при синдроме Марфана. Поздно начинающиеся аутосомнодоминантные болезни (хорея Гентингтона, болезнь Альцгеймера) не отражаются на репродуктивной способности (число детей). К началу болезни (после 35-40 лет) деторождение уже заканчивается.
Распространенность рецессивных болезней определяется частотой гетерозигот в популяциях. Популяционные закономерности распределения генов и генотипов таковы, что частота гетерозигот во много раз выше частоты гомозигот по мутантному аллелю.
Накопление гетерозигот в популяциях обусловлено их репродуктивным преимуществом (оставление потомства) по сравнению с гомозиготами по нормальному и патологическому аллелям. Если преимущества никакого нет и не было в истории популяции, то частота гетерозигот, казалось бы, должна приближаться к частоте мутационных событий (равное число появляющихся и элиминирующихся мутаций). На самом деле по всем рецессивным мутациям сохраняется повышенная частота гетерозигот, как бы подхватываемая отбором для размножения. Популяции не только человека, но и всех живых существ отягощены грузом рецессивных мутаций. Эта общебиологическая закономерность была открыта русским генетиком С.С. Четвериковым в 1926 г.
Распространенность генных болезней в общем плане определяется популяционными закономерностями поведения генов (см. ниже).
Мутационный процесс - одна из биологических характеристик любого вида. Он постоянно протекает у человека в зародышевых и соматических клетках и является основой возникновения и поддержания генетического разнообразия человека. В то же время это первичный источник наследственных болезней. По разным оценкам, частота возникновения мутаций (спонтанный уровень) у человека
ориентировочно составляет 1х10-5-1х10-7 гена на поколение, т.е. мутационные события в каждом гене достаточно редки. Лишь в нескольких генах мутации возникают с повышенной частотой (1 на 104 гамет). Эти гены отличаются от других необычайно большими размерами (360 000 пар оснований в гене нейрофиброматоза и 2х106 - в гене миопатии Дюшенна-Беккера).
Таким образом, текущий мутационный процесс на генном уровне в одном поколении не может обеспечивать наблюдаемую высокую частоту патологических аллелей в популяциях. По приблизительным косвенным оценкам (а точные прямые пока невозможны) общий вклад мутационного процесса в распространенность наследственных болезней составляет около 20%.
Отбор в любых популяциях обусловлен дифференциальной смертностью и плодовитостью особей с разными генотипами, что и приводит через какое-то число поколений к различной концентрации аллелей в популяциях. Поскольку отбор (его направленность и интенсивность) тесно связан с условиями окружающей среды, на этой основе возникают разные концентрации аллелей в различных популяциях. Элиминация или преимущественное размножение зависят от дифференциальной приспособленности гетерозигот, нормальных или мутантных гомозигот к условиям окружающей среды.
В качестве примера этой закономерности можно привести факторы распространения аутосомно-рецессивных гемоглобинопатий (серповидно-клеточная анемия, талассемия, аномальные гемоглобины - гемоглобин С, НиЕ) в регионах с высокой заболеваемостью малярией. Малярия у гомозигот по гену нормального гемоглобина сокращает жизнь и репродукцию у здоровых людей. Гомозиготы по мутантным аллелям (больные гемоглобинопатиями) умирают от наследственной болезни, не оставляя потомства. Гетерозиготы выживают, потому что малярийный плазмодий не поражает людей с аномальным гемоглобином, а сама гетерозиготность не вызывает патологического процесса в нормальных условиях. Преимущественное размножение гетерозигот приводило к высокой частоте носительства гена среди населения в прошлом «малярийных» регионов (Юго-Восточная Азия, Африка, Италия, Греция, Кипр, Азербайджан, Узбекистан). Такая частота сохраняется и до настоящего времени.
Отбор, безусловно, продолжает действовать и сейчас в популяциях человека не только во внутриутробном, но и в постна-
тальном периоде онтогенеза, несмотря на социальную и медицинскую помощь больным. В детском возрасте умирают больные GM2-ганглиозидозом, миопатией Дюшенна, с моногенными врожденными пороками развития. Репродукция понижена у больных гемофилией, ахондроплазией, нейрофиброматозом I типа и т.д. В то же время необходимо обращать внимание на снижение давления отбора в современных условиях (разное для разных болезней), которое в популяциях человека идет двумя путями. Во-первых, улучшение медицинской и социальной помощи больным (особенно лечение наследственных болезней) приводит к тому, что гомозиготы (например, больные фенилкетонурией, муковисцидозом), ранее не доживавшие до репродуктивного периода, теперь не только живут 30-50 лет и более, но и вступают в брак, имеют детей. Следовательно, популяции пополняются гетерозиготами по патологическим генам. Во-вторых, планирование семьи (произвольное сокращение рождаемости, чаще всего 1-2 или 3 ребенка) изменяет действие отбора в связи с репродуктивной компенсацией.
Суть этого явления в современных популяциях заключается в следующем. Наследственно отягощенные супружеские пары, у которых повышена смертность детей из-за наследственных болезней, в результате большего числа беременностей по сравнению с таковыми у наследственно неотягощенных пар «достигают» того же числа детей. Это положение иллюстрирует табл. 4.8, составленная по данным Л.П. Большаковой (1986).
Популяционные последствия репродуктивной компенсации очевидны, хотя развиваться они будут медленно (десятки, а для некоторых генов сотни поколений). Патологические аллели в этих случаях будут иметь большую вероятность сохранения и увеличения частоты, чем при естественной репродукции индивидов с разными генотипами.
Таблица 4.8. Репродуктивная компенсация в разных популяциях
Окончание таблицы 4.8
Дрейф генов - это случайное повышение частоты какого-то аллеля в результате нескольких совпадающих событий, имеющих стохастический характер (соответствующий брак, большая семья, унаследование детьми патологических генов, снова «подходящие» браки этих детей, хорошее материальное положение и т.п.). Это явление наблюдается для редко встречающихся признаков (или болезней), которые не отметаются отбором. Благодаря дрейфу генов патологические гены могут долго сохраняться в роду или в небольшой популяции, особенно в изоляте (популяция из 500-1500 человек, в которой практически отсутствует миграция).
Неравномерное распределение патологических генов в популяциях, а точнее, их высокие частоты, могут быть обусловлены так называемым эффектом родоначальника. Это явление по популяционногенетическому смыслу близко к дрейфу генов - накоплению какой-либо генной болезни (или многих), унаследованной от одного или нескольких индивидов, переехавших в другое место. Хорошо документированных историческими материалами примеров эффекта родоначальника в генетике человека уже много.
В XVII в. иммигранты из Европы (Голландии, Дании, Германии) прибыли в Южную Африку (современная ЮАР). Среди них были носители генов порфирии (мягко текущее аутосомно-доминантное заболевание), хореи Гентингтона (аутосомно-доминантная болезнь с поздним началом), семейного полипоза толстой кишки (аутосомнодоминантная болезнь с поздним началом), липопротеиноза (аутосомно-рецессивная болезнь). Семьи иммигрантов были большими (более 10 детей), поэтому людей с этими болезнями в ЮАР теперь во много раз больше, чем в Голландии и Дании. Родословная лиц с аутосомно-доминантными болезнями прослеживается до одного брака иммигрантов, а ген липопротеиноза - до брата и сестры,
прибывших в 1652 г. в теперешнюю ЮАР. Они, их дети и внуки имели большие семьи, что и способствовало увеличению частоты этого рецессивного гена.
Китайский иммигрант, прибывший в Южную Африку, имел 7 жен. Он страдал аутосомно-доминантным заболеванием - дисплазией костей и зубов, вызывающей полную потерю зубов к 20 годам, и передал этот ген 70 из 356 прослеженных потомков в 4-х поколениях.
В штате Пенсильвания (США) живут изолированно амиши (переселенцы из Европы), переехавшие туда в XVIII в. В 60-х годах XX в. в их поселении обнаружены 82 человека с аутосомно-рецессивной болезнью (карликовость и 6 пальцев), все эти люди - потомки одной супружеской пары.
Естественно, имеется тенденция к элиминации патологических генов из популяций путем естественного отбора, поэтому эффект родоначальника сам по себе не может объяснить долгое существование патологического гена в популяции.
Миграция населения по регионам или странам с оседлостью в новом месте - теперь уже неизбежный спутник многих социальных процессов (беженцы, переезд в поисках работы и т.д.). Миграция может отражаться на эпидемиологии генных болезней. Она уменьшает или увеличивает частоту носителей патологических генов в «донорских» или «реципиентных» популяциях вплоть до эффекта родоначальника или уравнивания частот в популяциях.
Кровнородственные браки имеют особенно большое значение в распространенности рецессивных генных болезней. Такие браки встречаются в западных странах с частотой около 1% на уровне двоюродных братьев и сестер. Однако в ряде этнических групп частота кровнородственных браков на уровне двоюродных родственников составляет 10-20% и даже до 30%, если включается троюродный уровень.
В кровнородственных браках существенно повышается вероятность рождения потомства, гомозиготного по патологическому рецессивному гену, по сравнению с таковой в потомстве от неродственного брака. Многие редкие рецессивные болезни встречаются в основном у детей от кровнородственных браков (табл. 4.9). Более того, постнатальная смертность существенно выше у детей, рожденных в кровнородственных браках (рис. 4.36). Особую опасность представляют инцестные браки, т.е. браки между родителями и детьми или между братом и сестрой.
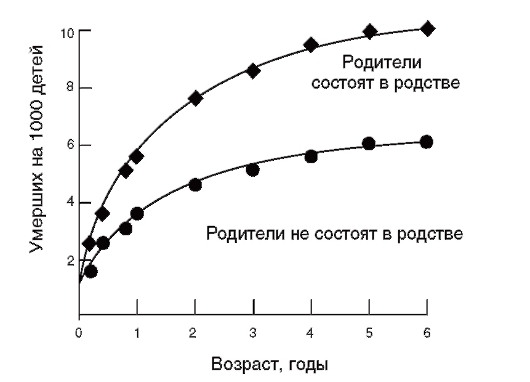
Рис. 4.36. Постнатальная смертность у двоюродных братьев и сестер в родственных и неродственных браках
Таблица 4.9. Частота редких аутосомно-рецессивных болезней в Японии
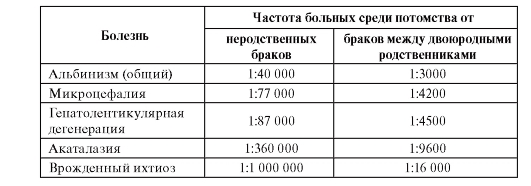
Вариации распространенности генных болезней (этнические, географические, популяционные) подтверждены во многих исследованиях. Выше были рассмотрены факторы, влияющие на распространенность генных болезней. Каждый из них может действовать отдельно в конкретной популяции в отношении конкретной нозологической формы или действует в комбинации с другими факторами. Накопилось много доказательств того, что распространенность множества генных болезней различна в разных популяциях. Высокая частота редких генных болезней в отдельных этнических группах объясняется эффектом родоначальника и дрейфом генов в изолированных популяциях. Отбор в таких случаях имеет меньшее значение.
В таблице 4.10. приведены примеры высоких частот наследственных болезней в связи с длительной генетической изоляцией.
Таблица 4.10. Генетические изоляты с высокой частотой аутосомных болезней
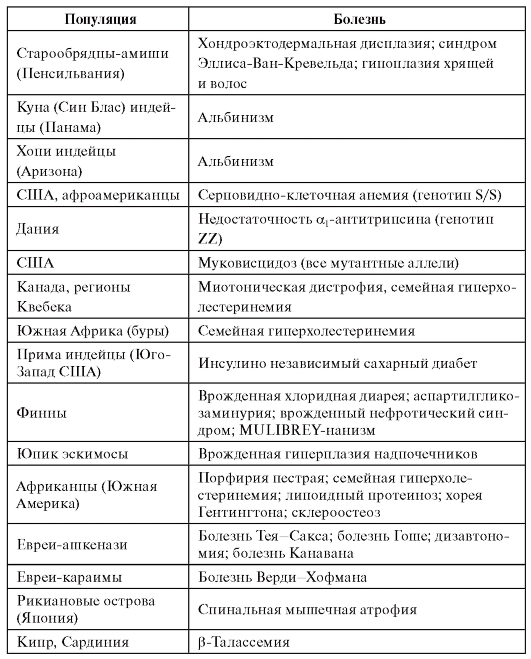
Примечание. MULIBREY-нанизм - MUscle-LIver-BRain-Eye-нанизм.
Редкие наследственные болезни относительно часто (в 10-100 раз чаще, чем в других популяциях) обнаруживаются в ряде популяций. У армян независимо от места их проживания отмечается высокая встречаемость семейной средиземноморской лихорадки (периодическая болезнь). В Финляндии часто встречается несколько редких болезней: врожденный нефроз, лизинурическая непереносимость белка, аспартилгликозаминурия, липофусциноз (детский тип) и др. У евреев-ашкенази, в нескольких поколениях постоянно проживающих в США, Европе и Израиле, отмечается высокая частота болезней накопления (GМ2-ганглиозидоз, болезнь Нимана-Пика, болезнь Гоше), синдрома Блума, семейной дизавтономии, абеталипопротеинемии, брахидактилии и др. В некоторых областях Японии часто встречаются акаталазия, болезнь Огаши, наследственный дисхроматоз. В Азербайджане в отдельных районах отмечается высокая частота синдрома Элерса-Данло (классический тип), хореи Гентингтона, гемофилии. Среди канадцев французского происхождения много больных тирозинемией, GM2-ганглиозидозом.
Как видно из приведенных примеров (подобной информации много и по другим популяциям), во всех случаях существует достаточно выраженная брачная изоляция, иногда даже вне географических ограничений (евреи-ашкенази, армяне).
Различия в распространенности касаются не только редко, но и часто встречающихся форм генных болезней. В последнем случае возможно как уменьшение, так и увеличение частоты болезни. Объяснения колебаний распространенности часто встречающихся генных болезней надо искать, скорее всего, в условиях отбора, как уже упоминалось для гемоглобинопатий. Например, две широко распространенные аутосомно-рецессивные болезни - фенилкетонурия и муковисцидоз - встречаются с неравной частотой в разных географических зонах или больших популяциях. Муковисцидоз встречается в среднем у 1 из 2000-3000 новорожденных в Европе и США, а среди чернокожих африканцев и японского населения его частота составляет 1:100 000. Частота фенилкетонурии в большинстве популяций равна 1:10 000, а в Финляндии, Израиле (для евреев-ашкенази), Японии болезнь встречается крайне редко (1:100 000-1:200 000). В Белоруссии и Ирландии ее частота составляет 1:6000, в Шотландии 1:5300.
В заключение следует подчеркнуть, что понимание эпидемиологии генных болезней необходимо врачу любой специальности, поскольку он может столкнуться с «пучковостью» редкой наслед-
ственной болезни в пределах обслуживаемого им района или контингента. Знание закономерностей и механизмов распространения генных болезней поможет своевременно разработать меры профилактики (обследование на выявление гетерогенности, генетическое консультирование и др.).
КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯ
Антиципация Болезни накопления
Болезни с экспансией триплетных повторов Виды генных мутаций у человека Внутрилокусная гетерогенность Возраст начала болезни Генетическая гетерогенность Генетические компаунды Генокопии Дрейф генов
Импринтинг на генном уровне Классификация генных болезней Корреляция генотип-фенотип Кровнородственные браки Летальный эффект мутаций Межлокусная гетерогенность Мутационный процесс и болезни Нормокопии
Определение генных болезней
Особенности клинической картины генных болезней
Первичные эффекты мутантных аллелей
Полные и мозаичные формы болезней
Примеры клеточного уровня патогенеза
Причины клинического полиморфизма
Прогредиентность клинической картины
Проявления клинического полиморфизма
Репродуктивная компенсация
Соматический и гонадный мозаицизм
Уровни патогенеза генных болезней
Фенокопии
Фенотипические эффекты мутаций
Эпидемиология генных болезней Эффект родоначальника
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Валиев Р.Р., Хусаинова Р.И., Хуснутдинова Э.К. Синдром Марфана: клинические и молекулярно-генетические аспекты. Медицинская генетика. - 2003. - Т. 2. - № 11. - С. 450-458.
Васильев В.Б. Генетические основы митохондриальных болезней. - СПб.: Изд-во «Нестор-История», 2006. - 146 с.
Вахарловский В.Г., Романенко О.П., Горбунова В.Н. Генетика в практике педиатра: руководство для врачей. - СПб.: Феникс, 2009. - 288 с.
Генетический паспорт - основа индивидуальной предиктивной медицины / под ред. В.С. Баранова. - СПб.: Изд-во Н-Л, 2009. - 527 с.
Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткани: руководство для врачей. - СПб.: Элби-СПб, 2009. - 704 с.: ил.
Капранов Н.И., Каширская Н.Ю., Петрова Н.В. Муковисцидоз: достижения и проблемы на современном этапе // Медицинская генетика. - 2004. - Т. 3. - № 9. - С. 398-412.
Козлова С.И., Демикова Н.С. Наследственные синдромы и медикогенетическое консультирование: атлас-справочник. - 3-е изд., доп. и перераб. - М.: Т-во научных изданий КМК; Авторская академия,
2007. - 448 с.: 236 ил.
Краснопольская К.Д. Наследственные болезни обмена веществ. Справ. пос. - М.: РОО «Центр социальной адаптации и реабилитации детей «Фохат», 2005. - 364 с.: ил.
Курникова М.А., Блинникова Е.О., Мутовин Г.Р. и др. Современные представления о синдроме Элерса-Данлоса // Медицинская генетика. - 2004. - Т. 3. - № 1. - С. 10-17.
Наследственные болезни в популяциях человека / под ред.
Е.К. Гинтера. - М.: Медицина, 2002. - 304 с.
Ferner R.E. Neurofibromatosis 1. // European Journal of Human Genetics. - 2007. - N 15. - P. 131-138.
Forest M.G. Recent advances in the diagnosis and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. // Human Reproduction Update. - 2004. - Vol. 10. - N 6. - P. 469-485.
Sibley C, Stone N.J. Familial hypercholesterolemia: A challenge of diagnosis and therapy. // Cleveland Clinic Journal Of Medicine. - 2006. -
Vol. 73. - N. 1. - P. 57-64.