Клиническая генетика : учебник / Н. П. Бочков, В. П. Пузырев, С. А. Смирнихина ; под ред. Н. П. Бочкова. - 4-е изд., доп. и перераб. - 2011. - 592 с. : ил.
|
|
|
|
Глава 6. БОЛЕЗНИ С НАСЛЕДСТВЕННОЙ ПРЕДРАСПОЛОЖЕННОСТЬЮ
ОБЩАЯ ХАРАКТЕРИСТИКА
Наряду с болезнями, этиологически строго детерминированными наследственностью (генные и хромосомные) или факторами среды (травмы, ожоги), есть большая группа болезней, развитие которых определяется взаимодействием определенных наследственных факторов (мутаций или сочетаний нормальных аллелей разных генов) и факторов среды. Их называют болезнями с наследственной предрасположенностью или многофакторными заболеваниями. Термины «болезни с наследственной предрасположенностью» и «многофакторные заболевания» означают одно и то же. В русской литературе чаще пользуются термином многофакторные (или мультифакториальные) заболевания.
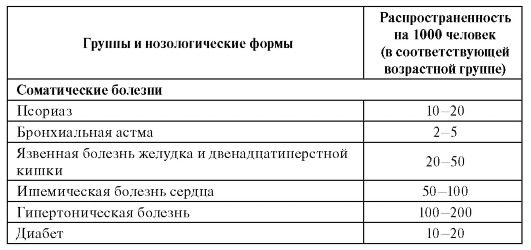
Многофакторные болезни могут возникать внутриутробно (врожденные пороки развития) или в любом возрасте постнатального развития. При этом чем старше индивид, тем больше вероятность развития у него многофакторного заболевания. В отличие от моногенных, многофакторные болезни относятся к распространенным заболеваниям (табл. 6.1). Большинство многофакторных болезней с генетической точки зрения являются полигенными, т.е. в их формировании участвуют несколько генов. Болезни с моногенной наследственной предрасположенностью (недостаточностью Г6ФДГ, аномальной холинэстеразой, злокачественной гипертермией и др.) обычно рассматриваются как моногенные болезни.
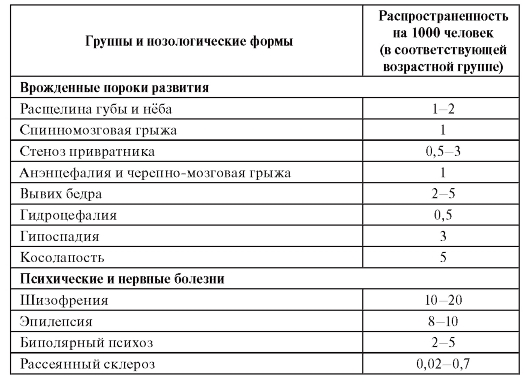
Врожденные пороки развития, такие, как расщелина губы и нёба, анэнцефалия, гидроцефалия, косолапость, вывих бедра и другие, формируются внутриутробно к моменту рождения и, как правило, диагностируются в самые ранние периоды постнатального онто-
* Исправлено и дополнено при участии д-ра биол. наук И.Н. Лебедева
генеза. Их развитие - результат взаимодействия многочисленных генетических факторов с неблагоприятными материнскими факторами или факторами среды (тератогены) в период развития плода. Они встречаются в популяциях человека по каждой нозологической форме нечасто (см. табл. 6.1), но суммарно - у 3-5% популяции.
Психические и нервные болезни, а также соматические болезни, относящиеся к группе многофакторных заболеваний, являются полигенными (генетически гетерогенными), но развиваются во взаимодействии с факторами внешней среды в постнатальном периоде онтогенеза у взрослых индивидов. Эта группа относится к социально значимым распространенным болезням (в английской транскрипции они обозначаются как «commone diseases»): сердечно-сосудистые (инфаркт миокарда, артериальная гипертензия, инсульт), бронхолегочные (бронхиальная астма, хронические обструктивные заболевания легких), психические (шизофрения, биполярный психоз), злокачественные новообразования, инфекционные болезни и др.
Таблица 6.1. Примеры болезней с наследственной предрасположенностью
Окончание таблицы 6.1
Для многофакторных болезней можно предложить следующую схему причин их развития (рис. 6.1).
Схема включает несколько положений. Во-первых, многофакторные заболевания представляют собой результат сложного взаимодействия генетических и внешнесредовых факторов, причем и те, и другие многочисленны. Во-вторых, в популяции эти факторы распределяются и комбинируются случайным образом. В-третьих, эти факторы действуют аддитивно, т.е. их суммарный эффект на болезнь теоретически равен сумме эффектов каждого из них в отдельности, хотя в действительности это более сложный процесс взаимодействия факторов, до конца не изученный.
Передача многофакторных болезней в семьях не соответствует законам Менделя. Распределение таких заболеваний в популяции и
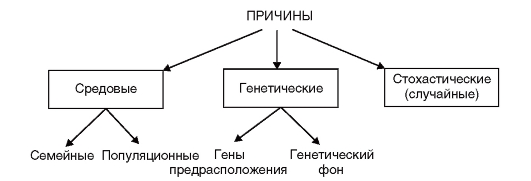
Рис. 6.1. Причины болезней с наследственной предрасположенностью
сегрегация (распределение больных и здоровых) в семьях принципиально отличается от моногенных (менделирующих) болезней.
- Риск развития заболевания у ребенка зависит от состояния здоровья родителей. Так, если один из родителей больного ребенка также страдает бронхиальной астмой, вероятность развития заболевания у ребенка колеблется от 20 до 30%; если больны оба родителя - она достигает 75%. В целом считается, что риск возникновения бронхиальной астмы у ребенка, родители которого имеют признаки атопии, в 2-3 раза выше, чем в тех семьях, в которых родители не имеют этих признаков. Риск в семье, когда болеют оба родителя, значительно превышает индивидуальные риски каждого из родителей и, что очень важно, не является простой суммой рисков каждого из родителей. При сравнении потомков здоровых людей и потомков больных бронхиальной астмой оказалось, что риск для ребенка заболеть бронхиальной астмой в 2,6 раза выше, если болеет мать, в 2,5 раза выше, если болен отец, и в 6,7 раза выше, если болеют оба родителя. Эти результаты могут быть объяснены аддитивным взаимодействием многих генов предрасположенности к развитию бронхиальной астмы у ребенка.
- Если в популяции многофакторные заболевания встречаются с разной частотой среди лиц разного пола (половой диморфизм), то риск заболевания выше в семьях пробандов реже поражаемого пола (и наоборот). Так, инфаркт миокарда в 30-40 лет случается чаще у мужчин, чем у женщин. Однако если речь идет о прогнозе, например, инфаркта миокарда для потомков больной матери инфарктом миокарда в этом же возрасте, то для них риск поражения будет выше, чем для потомков больного отца.
- Если в двух сравниваемых популяциях многофакторные заболевания встречаются с разной частотой, то риск для родственников выше, если семья принадлежит к более поражаемой популяции. Для моногенных болезней, как известно, риск для родственников не зависит от популяционной частоты. Для муковисцидоза, например, встречающегося среди европейского населения чаще, чем среди африканского, вероятность поражения для детей в браке здоровых родителей-гетерозигот одна и та же - 25%. Другое дело для многофакторных болезней, например, артериальной гипертензии. Ее частота среди
сибирских народностей составляет не более 5%, в то время как у белого населения Южной Африки соответствующего возраста - 25-30%. В отношении этой патологии риск выше для родственников из семей, принадлежащих к южно-африканской популяции.
- Риск заболеть для родственников зависит от степени тяжести болезни у пробанда. Если заболевание манифестирует в раннем возрасте, протекает более тяжело и за короткий срок приводит к необратимым изменениям, риск для следующих детей существенно возрастает, а наследственная отягощенность проявляется в этих семьях большим процентом лиц с субклиническими проявлениями болезни. В целом генетический риск для родственников в отношении моногенной патологии, как правило, выше, чем в случае многофакторной. Частота многофакторного заболевания среди родственников больного 1-й степени родства приблизительно равна
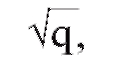
где q - частота болезни в популяции.
Это подтверждается расчетами с использованием коэффициента риска (λ), указывающего на степень генетического вклада в болезнь. Если λ >2, то утверждается, что генетический вклад в болезнь или любой исследуемый признак значителен. Некоторые примеры и формулы для расчета риска болезни приведены в табл. 6.2.
Таблица 6.2. Коэффициенты риска (λ) для многфакторных и моногенных заболеваний

ПОДХОДЫ К ИЗУЧЕНИЮ НАСЛЕДСТВЕННОЙ ПРЕДРАСПОЛОЖЕННОСТИ К БОЛЕЗНЯМ
ЧЕЛОВЕКА
Среди основных подходов к оценке роли наследственных факторов в этиологии и патогенезе широко распространенных многофакторных заболеваний следует назвать три: клиникогенеалогический, близнецовый и популяционный. Долгое время они были главными способами доказательства существования наследственной предрасположенности. Этот период исследования болезней с наследственной предрасположенностью называют формально-генетическим, так как названные три подхода надежно указывали на значение наследственных факторов в возникновении и развитии таких болезней, давали возможность измерить степень участия их в этих процессах, но не позволяли идентифицировать конкретные гены, составляющие основу наследственной предрасположенности. До недавнего времени возможность описания структуры наследственной компоненты многофакторного заболевания в терминах задействованных генов практически отсутствовала. Она появилась с осуществлением международного проекта «Геном человека» - 90-е годы ХХ в. открыли «охоту за генами» болезней, в том числе и многофакторных. У медицинских генетиков появился, по выражению В. Маккьюсика (1997), «свой» объект исследования - геном.
Однако прежние методы изучения наследственной предрасположенности человека к многофакторным заболеваниям (клиникогенеалогический, близнецовый, популяционный) не утратили своего значения. Более того, они являются базовыми в идентификации генов подверженности болезням при использовании современных генетических технологий (полногеномные SNP-микрочипы для генотипирования, полногеномные экспрессионные микрочипы, высокопроизводительное секвенирование и др.).
Клинико-генеалогические доказательства наследственной предрасположенности
Эмпирическое наблюдение за семьями с сердечно-сосудистыми, желудочно-кишечными, психическими, аллергическими заболеваниями давно уже побуждало врачей предполагать роль наслед-
ственности в возникновении этих болезней. Более строгие доказательства можно получить любым из трех способов анализа генеалогических данных.
- Всех опрашиваемых разделяют на группы в зависимости от наличия больных в родословной. При этом способе анализа пробандами являются только больные, от которых получены генеалогические сведения в отношении изучаемой болезни у родственников определенной степени родства. Например, у лиц с гипертонической болезнью собирают сведения о наличии гипертонии у живых или умерших родителей. Затем всех пробандов разделяют на группы: а) имеющих здоровых родителей (без гипертонической болезни); б) имеющих одного больного родителя; в) имеющих двух больных родителей. В заключение проводится сравнение величин выборок этих трех групп. При болезнях с более ранним проявлением (например, бронхиальная астма) для разделения пробандов на группы за основу принимают наличие больных братьев и сестер.
- Частоту больных среди родственников больных пробандов сравнивают со специально подобранной контрольной группой, т.е. с группой лиц, не страдающих изучаемой болезнью (с учетом пола, возраста, этнической принадлежности, бытовых условий, производственных вредностей и т.д.). Всю работу по сбору клинико-генеалогических данных проводят строго идентично для родственников соответствующей степени родства. Например, из большого числа студентов отбирают две примерно равные по величине группы: больные язвенной болезнью и здоровые. Далее собирают клинико-генеалогические данные о наличии язвенной болезни у родителей, братьев, сестер. Затем сравнивают частоту язвенной болезни у родственников здоровых и больных студентов.
- Заболеваемость в семьях больных (обобщенно) можно сравнивать с популяционной частотой этой же болезни. Такой подход - фактически комбинация клинико-генеалогического и популяционно-статистического методов. Его широко применяли для изучения наследственной природы шизофрении, аллергии, ревматизма и др.
Генетический анализ родословных на основе менделевского, т.е. дискретного разделения потомства по фенотипу для болезней с
наследственной предрасположенностью, можно применять только в ограниченных пределах и на больших выборках, чтобы в них вошел материал по всему многообразию клинического проявления данного заболевания. При этом, по-видимому, следует ограничивать исследование данными о родственниках II степени родства (в крайнем случае, не далее III степени).
При клинико-генеалогическом изучении болезней с наследственной предрасположенностью анализировать родословные следует особенно тщательно. Наряду с получением обычной для моногенных форм информации (наличие болезни) необходимо обращать особое внимание на следующие методические вопросы.
- Поскольку диагностика стертых форм многофакторной патологии трудна, нужно в полной мере обеспечить точность диагностики заболевания у всех членов семьи.
- При оценке выраженности болезни у разных членов семьи следует оценивать сходство фенотипического проявления среди различных пораженных родственников (течение болезни, возраст начала, тяжесть, реакция на лекарства и т.д.). Большое сходство клинической картины обычно указывает на существенное значение генетической компоненты и, следовательно, на повышенный риск развития заболевания у близких кровных родственников.
- Степень кровного родства среди больных членов семьи должна быть установлена точно. При полигенном характере болезни это важно знать для оценки риска, так как степень риска у близких родственников резко возрастает.
- Необходимо собирать подробные и точные сведения о действии факторов среды в пре- и постнатальном периодах. Это позволяет существенно уточнить прогноз.
Особый способ клинико-генеалогического изучения предрасположенности к болезням - изучение заболеваемости среди биологических и приемных родственников больных или метод приемных детей. В таких семьях имеется своеобразный контроль - генетически неродственные индивиды, имеющие с пробандом общие семейные средовые влияния. Анализ этих семей оказался особенно полезным для доказательства генетической предрасположенности к шизофрении и алкоголизму: заболеваемость в 2-3 раза выше у биологических родственников по
сравнению с таковой у членов семьи, не имеющих кровного родства. В частности, частота алкоголизма у приемных детей коррелирует с этим показателем у биологических родителей. У приемных детей масса тела и явная полнота коррелируют с таковыми у биологических родителей, а не у членов семей, в которых выросли приемные дети. Такая же закономерность отмечается при сравнении супругов. Например, частота желчнокаменной болезни среди сибсов больных выше по сравнению с общепопуляционной, а у супругов ниже, чем у пробандов.
Близнецовые исследования
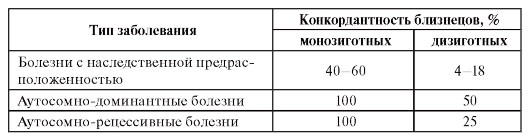
При изучении многофакторных болезней большие надежды возлагались на близнецовый метод как достаточно объективный и чувствительный. Исследования многофакторной патологии проводились путем сравнения конкордантности моно- и дизиготных близнецов. Второй вариант близнецового метода - сравнение конкордантности монозиготных близнецов, выросших вместе и порознь, не применялся из-за трудности подбора соответствующих групп, хотя для изучения болезней с наследственной предрасположенностью он был бы более чувствительным. Обобщенные результаты использования близнецового метода для понимания природы разных заболеваний представлены в табл. 6.3.
Таблица 6.3. Конкордантность близнецовых пар для разных групп болезней
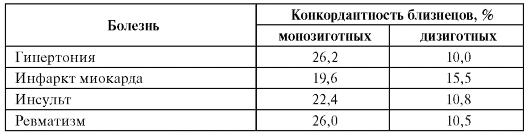
С помощью близнецового метода проведены многочисленные исследования природы предрасположенности ко многим заболеваниям, в том числе к сердечно-сосудистым болезням. В таблице 6.4. представлены некоторые обобщенные результаты изучения генетической компоненты в происхождении болезней этой группы. Как видно из данных табл. 6.4, во всех случаях конкордантность монозиготных близнецов выше, чем дизиготных.
Таблица 6.4. Результаты исследований предрасположенности к сердечнососудистым болезням близнецовым методом
С помощью близнецового метода изучали роль наследственности в происхождении опухолей. Несмотря на низкую конкордантность монозиготных близнецов, в 30-40-х годах ХХ в. был сделан вывод об определенном значении наследственной предрасположенности для возникновения злокачественных опухолей. В целом на основании близнецовых исследований было сделано заключение, что значение внешних факторов в возникновении рака намного больше, чем наследственных. Теперь ясно, что подобная постановка вопроса в данном случае неправомерна. Главным итогом близнецовых исследований природы рака стало привлечение внимания к генетической детерминации злокачественных новообразований. В дальнейшем для этого уже использовали клинико-генеалогический, цитогенетический и молекулярно-генетический методы, которые и определили прогресс в расшифровке природы злокачественных новообразований.
Близнецовый метод применяли для изучения наследственной природы такого сложного явления, как аллергия. Было показано, что конкордантность монозиготных близнецов по разным проявлениям аллергических реакций выше, чем дизиготных.
Близнецовым методом также показана наследственная предрасположенность к некоторым инфекционным болезням (полиомиелиту, туберкулезу). Конкордантность монозиготных близнецов по этим заболеваниям в несколько раз больше, чем дизиготных.
Выше были приведены доказательства наследственной предрасположенности к широко распространенным заболеваниям, полученные клинико-генеалогическим и близнецовым методами по отдельности. Доказательства генетической предрасположенности к широко распространенным болезням, полученные этими методами для одних и тех же нозологических форм, совпадают.
Популяционные исследования
Популяционные подходы к исследованию вопросов генетики многофакторных заболеваний используются в двух аспектах. Во-первых, они дают дополнительные доказательства существования наследственной предрасположенности в отношении конкретных заболеваний. Во-вторых, популяционно-статистические методы вместе с клиникогенеалогическим анализом составляют основу для картирования и идентификации генов подверженности болезням. Их объединение вылилось в самостоятельное научное направление медицинской генетики - генетическая эпидемиология, краткая характеристика которой представлена на компакт-диске в разделе «Модели исследования и наследственность многофакторных болезней. Идентификация генов предрасположенность многфакторным заболеваниям».
ГЕНЕТИЧЕСКИЕ АССОЦИАЦИИ
Исследования генетических ассоциаций осуществляют с использованием двух принципиально различных подходов. Один из них основан на том, что в качестве генетического маркера выступает аллельный вариант гена, белковый продукт которого участвует в патогенезе исследуемого заболевания. То есть выбор гена и его полиморфизмов определяется исходя из конкретной гипотезы развития заболевания, и ген этот называют «геном-кандидатом» подверженности многофакторному заболеванию. Задача исследования состоит в получении доказательств участия его первичного продукта в развитии заболевания и измерении (оценке) силы его эффекта. Такой «кандидатный» подход был первым вариантом исследования генетических ассоциаций. С его помощью проверяют гипотезу о патогенетической основе возможной связи полиморфизма гена и болезни.
Другой подход предполагает сканирование генома с плотным набором ОНП (SNP), вплоть до 1 млн в одном исследовании для поиска функциональных вариантов генов. Такое полногеномное ассоциативное исследование является масштабным по охвату включенности генома человека при отсутствии сведений о функции и расположении связанных с болезнью генов на хромосоме.
Оба подхода оценивают степень риска развития болезни у носителя определенных аллелей гена в сравнении с индивидами, не являющимися носителями такового. Существует много показателей и методик расчета эпидемиологических характеристик риска. Один
из них - показатель отношения шансов (OR - odds ratio, отношение шансов). Его получают путем сравнения частот двух сравниваемых показателей (М1 и М2) у больных (с - «случай») и здоровых (к - «контроль»).
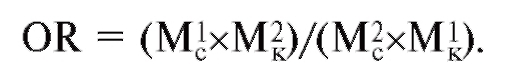
Отношение шансов принимает значение от 0 до бесконечности и равен 1 только при отсутствии влияния одного показателя (маркера) на другой. Отношение шансов - надежная мера ассоциации генетического маркера и болезни, а также хорошая оценка относительного риска. Термины «отношение шансов» и «относительный риск» (OR) имеют сходный смысл и в исследованиях типа «случай-контроль» часто бывают взаимозаменяемыми. Они показывают, во сколько раз увеличивается (или уменьшается) вероятность быть здоровым (или стать больным), если индивид является носителем конкретного аллеля соответствующего гена по сравнению с индивидом-неносителем данного генетического маркера.
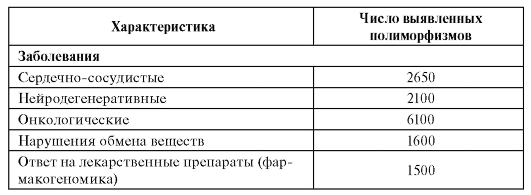
К настоящему времени проведено большое число исследований по генотипированию, т.е. изучению генетических ассоциаций с многофакторными заболеваниями, и получены доказательства связей конкретных генетических маркеров как с фенотипами болезней в целом, так и с разнообразными их клиническими проявлениями, а также с ответом пациентов на лекарственные средства (фармакогеномный анализ). Результаты таких исследований представлены в табл. 6.5.
Таблица 6.5. Число выявленных полиморфизмов генов для отдельных групп болезней (к 2009 г.)
Следует констатировать, что OR, пригодные уже сегодня для использования оценки риска, определены для ограниченного числа
многофакторных заболеваний и признаков. В таблице 6.6 представлены OR для полиморфизмов (все они являются ОНП и обозначены в таблице как «rs» - номер в базе данных, reference site) генов в отношении болезней, для которых OR достигает 20. Например, у индивидов, являющихся носителями рискового аллеля G (rs 3825942) гена LOXL1, вероятность иметь тяжелую форму глаукомы (эксфолиативная глаукома) в 20 раз выше, чем у индивидов, имеющих другие полиморфизмы этого гена.
Таблица 6.6. Показатели относительного генетического риска для некоторых многофакторных заболеваний по данным полногеномных исследований
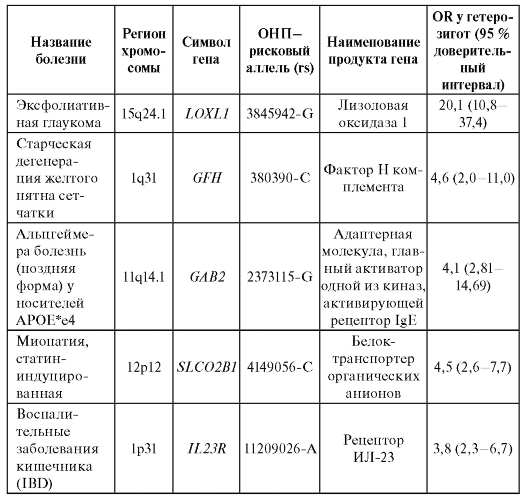
Окончание таблицы 6.6
Примечание. IBD - Inflammatory bowel disease; IgE - иммуноглобулин Е; АТФ - аденозинтрифосфат.
В таблице 6.7. приведены примеры OR в отношении некоторых признаков, а для одного из признаков - частоты рекомбинации, которая ассоциирует с полиморфизмом rs 3796619-Т гена RNF212. Уровень рекомбинации как у мужчин, так и у женщин существенно определяется тем, присутствует ли в их геномах данный аллель гена RNF212. OR достигает величин 70,7 и 88,2 у мужчин и женщин, соответственно.
Таблица 6.7. Признаки с высоким и умеренным относительным генетическим риском по данным полногеномных исследований
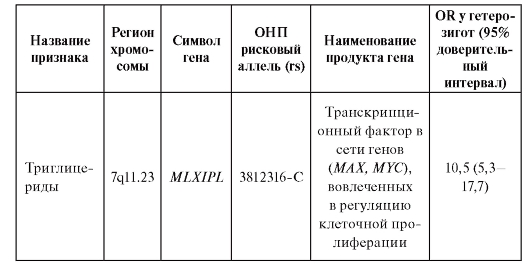
Окончание таблицы 6.7
В целом следует указать на три основные особенности обнаруживаемых генетических ассоциаций.
Во-первых, большая часть генетических ассоциаций вызывает эффекты небольшой величины (риски 1,1-1,5, т.е. 10-15% увеличения вероятности развития заболевания). Любой отдельный полиморфизм гена обычно объясняет только 1-8 % от общего риска заболевания в популяции, т.е. добавочный предсказательный эффект отдельных генетических полиморфизмов небольшой. Однако исследования в этом направлении только начаты и в специальных исследованиях (когортные, проспективные) риски уточняются. Подтверждается целесообразность введения генетической рискометрии.
Во-вторых, у индивидов-носителей комбинаций (ансамблей) аллельных вариантов генов отмечается заметное увеличение риска болезней и их осложненного течения. Расчеты показывают, что аддитивный эффект нескольких таких полиморфизмов может составлять до 20-70% общего риска, обусловленного генетическими факторами.
Речь идет о совместном действии генов и их аллельных вариантов (аддитивное, эпистатическое), взаимодействии между полиморфизмами генов и модифицирующими факторами, такими, как возраст, пол, лечение и многие средовые факторы.
В-третьих, для человека характерно наличие общих генов, вовлеченных в патогенез разных, но нередко сочетающихся болезней. Вопросы, касающиеся идентифицируемых генов подверженности распространенным болезням человека, освещены в одноименной статье В.П. Пузырева на компакт-диске.
Одним из феноменов хронических заболеваний человека является полипатия, множественность болезней. У одного пациента сочетается несколько болезней. Около 40% лиц в возрасте 15-75 лет имеют одновременно две болезни, а четыре болезни встречаются почти у каждого пятого терапевтического больного. Такие сочетания болезней могут носить случайный характер - это «соседство болезней». Однако нередко болезни, сочетающиеся у одного пациента, встречаются и у его ближайших родственников. Их обозначают как «семейство болезней». В отношении последней категории болезней немецкими патологами М. Пфаундлером и Л. фон Зехтом в 1921 г. предложен термин «синтропия». В современном определении его подчеркивается, что синтропия - это природно-видовое явление сочетания двух и более патологических состояний (нозологий, синдромов) у индивидуума и его ближайших родственников, неслучайное и имеющее эволюционно-генетическую основу. Однако есть и такие патологические явления, которые редко сочетаются у одного человека, «упорно» не ассоциируют, «взаимно отталкиваются». Такой феномен авторы назвали дистропией.
Наиболее известным примером синтропии является метаболический синдром («квартет»): гипертония, гиперхолестеринемия, гипергликемия и ожирение. Они встречаются у одного пациента, а также у родственников в таких же сочетаниях или отдельно. Известны и другие синтропии - пикквикский синдром, включающий ожирение и нарколепсию; сахарный диабет I типа, аутоиммунный тиреоидит и целиакия; бронхиальная астма, атопический дерматит и высокий уровень иммуноглобулина Е. Антагонистические взаимоотношения (дистропии) известны, например, для туберкулеза легких и бронхиальной астмы. Пролиферативные процессы на базе лимфоидного и миелоидного типов кроветворения не ассоциируются, что также может быть примером стойкой дистропии. Отмечают, что сахарный диабет I типа редко сочетается с язвенной болезнью.
Неслучайность сочетания отдельных патологических форм, объединенных сходством патогенеза у индивидуума и его ближайших родственников, указывает на участие общих генетических факторов. Гены, вовлеченные в развитие синтропий, названы синтропными генами. Более точно - синтропные гены есть набор функционально взаимодействующих корегулируемых генов, локализованных во всем пространстве генома человека, вовлеченных в общий для данной синтропии биохимический или иной путь. В том случае, когда регуляторные связи складываются так, что их особенностью являются альтернативные отношения, объясняющие взаимоисключения на клиническом уровне таких фенотипов (дистропии), такие гены называют дистропными в отношении соответствующих фенотипов.
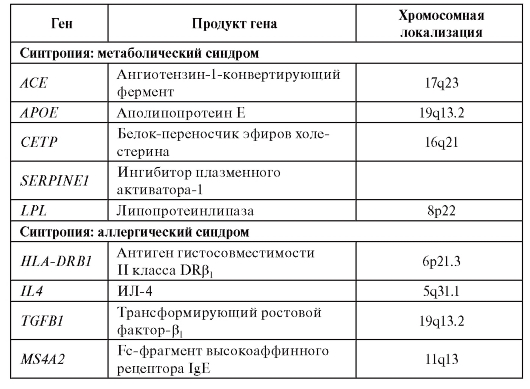
В качестве примеров назовем некоторые синтропные гены, которые вовлечены в синтропию «метаболический синдром» и синтропию «аллергические заболевания» (табл. 6.8).
Таблица 6.8. Синтропные гены для двух вариантов синтропий
ГЕНЫ ПОДВЕРЖЕННОСТИ НЕКОТОРЫМ МНОГОФАКТОРНЫМ ЗАБОЛЕВАНИЯМ
Достижения в геномике человека определили возможность обнаружения множества генетических вариантов, ассоциированных с риском предрасположенности к заболеванию. Такой набор генетических маркеров называют «геномным профилем». Идентификация совокупности генов, которые в комбинации с негенетическими факторами приводят к патологическому фенотипу, сталкивается с необъятностью генома и высокой степенью индивидуальной изменчивости. Такого рода вопросы относятся к общей патологии.
Систематическое накопление данных о связи полиморфных вариантов генов с многфакторными заболеваниями, анализ их воспроизведения в разных популяциях и этнических группах, обобщение всей известной информации о патогенетической и функциональной значимости конкретных генных вариантов - современные задачи клинической генетики. Результаты этих исследований будут кратко изложены для сердечно-сосудистых, иммунозависимых, инфекционных болезней и злокачественных новообразований.
Сердечно-сосудистые заболевания
Сердечно-сосудистые заболевания продолжают оставаться главной причиной смертности населения индустриально развитых стран Европы (в том числе России) и США. Ишемическая болезнь сердца и артериальная гипертония, их осложнения - инфаркт миокарда, инсульт и внезапная смерть составляют основную долю среди всех сердечно-сосудистых заболеваний. В патогенезе этих болезней большое значение отводится так называемым факторам риска - нарушению липидного обмена (дислипидемии), сахарному диабету, ожирению, курению и др. После почти 20-летних дискуссий и специальных исследований получены убедительные доказательства концепции сердечно-сосудистого континуума (Dzau V. et al., 2006). Суть ее состоит в том, что сердечно-сосудистые заболевания рассматриваются как цепь событий, вызванных многочисленными связанными и несвязанными между собой факторами риска, прогрессирующими через вовлечение многих физиологических, мета-
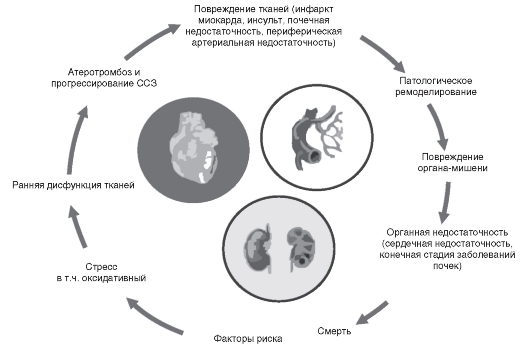
Рис. 6.2. Сердечно-сосудистый и почечный патофизиологический континуум (Dzau V. et al., 2006)
болических путей и завершающихся развитием конечной стадии заболевания сердца (рис. 6.2).
Более того, было показано, что вмешательство в любое звено цепи событий, ведущих к сердечно-сосудистым заболеваниям, модифицирует течение этой группы заболеваний. Следует заметить, что болезни сердечно-сосудистого континуума соответствуют представлениям о синтропных заболеваниях и сердечно-сосудистый континуум может быть также обозначен как синтропия.
В специальном исследовании для 7 болезней, относящихся к сердечно-сосудистому континууму (гипертония, коронарная болезнь, дислипидемии, инсульт, ожирение, метаболический синдром и сахарный диабет 2-го типа), среди более 2,5 тыс. исследованных генов идентифицирован 21 ген, которые ассоциируют с этими семью болезнями и, следовательно, являются общими для данной синтропии. Их назвали синтропными генами сердечно-сосудистого континуума (табл. 6.9).
Таблица 6.9. Общие гены синтропии «сердечно-сосудистый континуум»
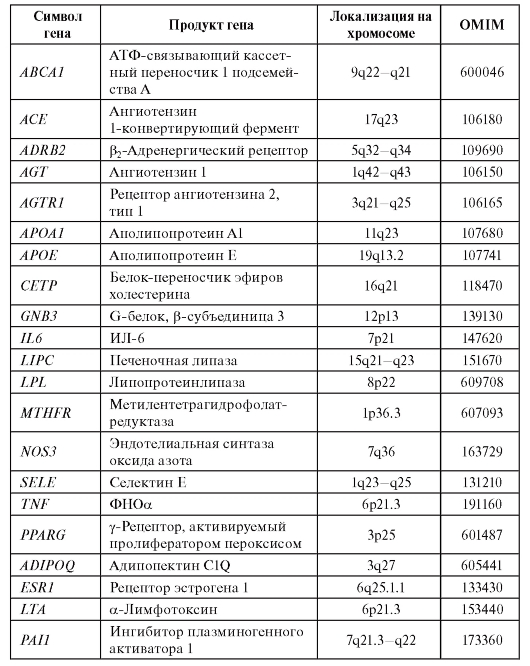
Примечание. АТФ - аденозинтрифосфат.
Эти синтропные гены, конечно, не «закрывают» всю наследственную компоненту подверженности этой группе заболеваний, в нее входит существенно больше генов. Однако сегодня общность для синтропии сердечно-сосудистого континуума доказана только для этой выборки генов из генома человека. Выявленные гены вовлечены в метаболизм липидов, функционирование ренин-ангиотензин-альдостероновой и симпатоадреналовой систем, воспаление, эндотелиальную дисфункцию. Именно они составляют основу патогенетики болезней сердечнососудистого континуума. О генетических факторах развития инфаркта миокарда у мужчин молодого возраста можно узнать из одноименной статьи С.Н. Пчелиной с соавт. на компакт-диске.
Иммунозависимые болезни
К данной группе болезней, условно названной иммунозависимыми, относятся неинфекционные заболевания человека, для которых отклонения в функционировании иммунной системы приобретают важное патогенетическое значение. Приобретенные иммунодефициты, аллергические, аутоиммунные и лимфопролиферативные заболевания, среди них атопии (бронхиальная астма, ринит), сахарный диабет 1 типа, болезнь Крона, ревматоидный артрит, хроническая обструктивная болезнь легких, коллагенозы и другие болезни - предмет изучения иммуногенетики.
Одна из наиболее активно развиваемых областей в генетике болезней с наследственной предрасположенностью - генетика атопических заболеваний человека, или атопий. Под атопией понимают индивидуальную или семейную предрасположенность к выработке IgE-антител в ответ на малые дозы аллергенов и развитию типичных симптомов астмы, риноконъюнктивита или дерматита/экземы.
Успехам в области генетики атопических заболеваний немало способствует достаточно полное понимание механизмов реализации IgE-опосредованных иммунных реакций (рис. 6.3).
Аллергены, попадая в организм, взаимодействуют с дендритными клетками. Результат этого - презентация доминирующего эпитопа аллергена с молекулами HLA-II на поверхности дендритных клеток. Связывание этого комплекса с рецепторами CD4 T-лимфоцитов стимулирует дифференцировку ThO-лимфоцитов в Th2 (Th - T-хелперы), способных к секреции цитокинов, функция которых тесно связана с гуморальным иммунным ответом: ИЛ-3, ИЛ-4, ИЛ-5, ИЛ-9, ИЛ-13, GM-CSF (granulocyte-macrophage colony stimulating factor - колониестимулирующий фактор гранулоцитов-макрофагов).
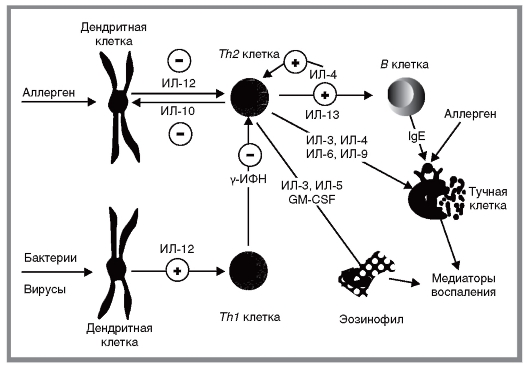
Рис. 6.3. Механизмы развития атопических реакций (Holgate S.T., 1999)
При действии антигенов микробактерий и некоторых вирусов CD4 клетки превращаются в Thl-лимфоциты, секретирующие ИЛ-2, γ-интерферон (γ-ИФН) и ФНО. Это приводит к активации макрофагов и элиминации с их помощью патогенных микроорганизмов. Сдвиг в сторону Тh2-ответа активируется ИЛ-4 и ингибируется ИЛ-12, γ-ИФН и α-ИФН. Thl-ответ требует высвобождения ИЛ-12 макрофагами и дендритными клетками и супрессируется
ИЛ-10.
Цитокины, высвобождаемые Тh2-клетками, главным образом ИЛ-4 и 13, взаимодействуют со своими рецепторами на В-лимфоцитах, активируют транскрипцию генного локуса тяжелой цепи типа ε иммуноглобулинов и индуцируют переключение изотопов с μ на ε IgE, синтезируемый активированными В-клетками, связываются с высокоаффинным (RcεRI) и низкоаффинным (RcεRII; CD23) рецепторами тучных клеток, инициируя высвобождение медиаторов воспаления и хемокинов: гистамина, простагландинов, лейкотриенов, фактора активации тромбоцитов, дегранулированных протеаз и др.
Медиаторы воспаления синтезируют также эозинофилы, активированные ИЛ-3, 5 и GM-CSF.
Действуя в совокупности, эти факторы приводят к микроизменениям сосудов стенок «шокового органа», сокращению гладкой мускулатуры, гиперсекреции слизи и другим симптомам, служащим основой клинических проявлений атопических заболеваний. Кроме того, цито- и хемокины ответственны за миграцию и активацию клеток воспаления, главным образом, эозинофилов, и, таким образом, вносят вклад в сохранение патологического процесса.
В отношении частой формы атопии - бронхиальной астмы, типичного многофакторного заболевания, в исследовании значения наследственных факторов предрасположенности был использован весь арсенал традиционных и новых исследовательских подходов, принятых в генетике многофакторных болезней: близнецовый, клинико-генеалогический, картирование (анализ ассоциаций и сцепления), полногеномные ассоциативные исследования, экспрессионные и транскриптомные методы, а также изучение на модельных животных.
К настоящему времени число генов, протестированных в связи с бронхиальной астмой или обнаруженных в полногеномных исследованиях, составляет около 550. Для 35 из них ассоциации с бронхиальной астмой продемонстрированы в независимых исследованиях не менее 10 раз (рис. 6.4).
Функционально ассоциирующиеся гены объединяются в четыре группы: гены врожденного иммунитета и иммунорегуляции (TLR2, TLR4, IL10); гены, связанные с дифференцировкой и функционированием Т-хелперов 2-го типа (IL4RA, IL4R, IL13); гены иммунитета слизистых оболочек и физиологии эпителия (CCL5, FLG); гены, ассоциированные с легочной функцией, ремоделированием дыхательных путей и бронхиальной гиперреактивностью (ADRB2, GSTM1, GSTP1, LTA, NOS1).
Как уже отмечалось, для многофакторныхных заболеваний характерен малый эффект отдельного генетического варианта в отношении фенотипа болезни. Это свойственно и для бронхиальной астмы. Однако в синергизме с другими аллельными вариантами (межгенные взаимодействия) этот эффект может заметно увеличиваться. Так, при исследовании более 1000 немецких детей в возрасте 9-11 лет было показано, что при пошаговом комбинировании генов ИЛ (IL4, IL13), гена кодирующего α-цепь рецептора ИЛ-4 (IL4RA) и гена внутриклеточного активатора транскрипции (STAT6) риск повышенного содержания IgE возрастал в 10,8 раза, а бронхиальной
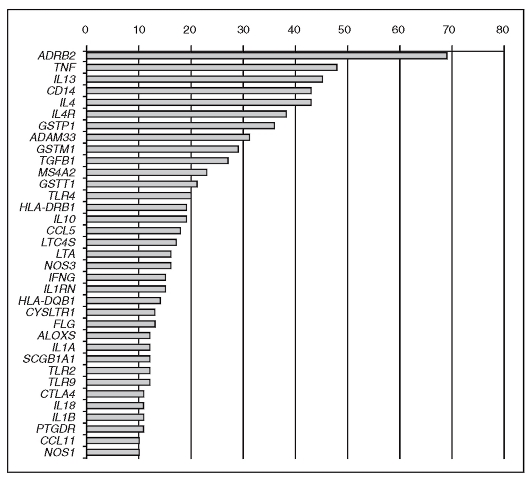
Рис. 6.4. Гены, для которых продемонстрированы ассоциации с бронхиальной астмой не менее 10 раз в независимых исследованиях. Горизонтальные столбики соответствуют количеству опубликованных работ о позитивных ассоциациях с заболеванием
астмы - в 16,8 раза по сравнению с суммарным эффектом отдельных полиморфизмов.
Наряду с анализом ассоциаций генов-кандидатов для идентификации генов подверженности бронхиальной астме, использовалось полногеномное ассоциативное исследование, которое, подтвердив некоторые ранее известные гены подверженности, обнаружило новые. Среди них: два близко расположенных гена на хромосоме 17q21 - ORMDL3 и GSDML, а также ген CH13L1, кодирующий хитиназаподобный протеин YKL-40.
Важнейший аспект в исследованиях генетики бронхиальной астмы - фармакогенетический, оценивающий индивидуальный ответ на наиболее часто применяемые противоастматические препараты (бронходилататоры, кортикостероиды и антилейкотриены). Так, показана ассоциация полиморфизмов гена ADRB2, кодирующего β2-адренергический рецептор, с бронходилататорным ответом на высокие дозы β-агонистов короткого действия, например, сальбутамола (альбутерола) при лечении приступов удушья: сальбутамол у гомозигот по Gly 16 лучше купирует и предупреждает приступ удушья, чем у гомозигот по Arg 16. Известен генетический маркер - вариант гена β-иммуноглобулинового рецептора тучных клеток (FCER), кодирующего низкоаффинный рецептор IgE, ассоциированный с повышенным риском приступа бронхиальной астмы у детей, принимающих ингаляционные кортикостероиды, несмотря на протективный эффект этих препаратов в лечении астмы.
Инфекционные болезни
Инфекционные болезни - одна из главных проблем здравоохранения во всем мире; они характеризуются высоким уровнем заболеваемости и смертности. С генетической точки зрения важно, что они отличаются от неинфекционных многофакторныхных болезней тем, что имеют специфическую причину - возбудителя болезни (патоген). Однако развитие болезни, характер течения инфекционного процесса, чувствительность организма к возбудителю определяются сложным взаимодействием факторов окружающей среды, патогена и наследственных факторов «хозяина» (рис. 6.5).
Основной взгляд на генетику подверженности распространенным инфекциям (малярии, ВИЧ/СПИДу, микобактериальным и вирусным инфекциям) обозначается как «одна инфекция - множество генов». Однако у лиц с первичными иммунодефицитами часто развиваются самые разнообразные инфекционные заболевания, которые рассматриваются как осложненное течение моногенного иммунодефицита. Такие случаи обозначаются как «один ген - многочисленные инфекции».
В настоящее время активно осуществляется поиск локусов (генных вариантов) чувствительности к инфекционным заболеваниям. Основной подход в этих исследованиях - скрининг генов-кандидатов и анализ их ассоциаций с инфекционными болезнями. К решению этой задачи в последнее время привлекаются и полногеномные ассоциативные исследования.
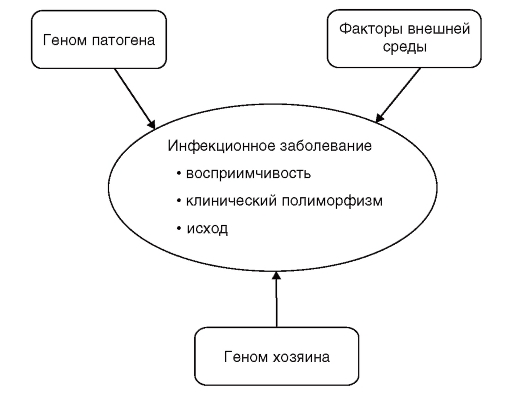
Рис. 6.5. Триединство факторов патогенетики инфекционного процесса
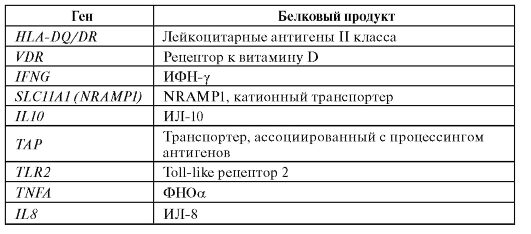
В отношении туберкулеза, наиболее активно исследуемого с генетических позиций заболевания, накоплена достаточно большая информация (табл. 6.10.).
Таблица 6.10. Гены, для которых показана ассоциация с туберкулезом
Окончание таблицы 6.10

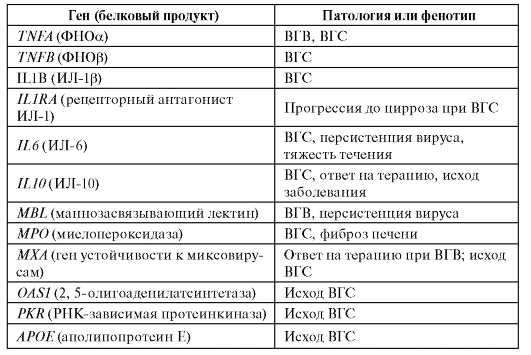
В таблице 6.11 приведены результаты ассоциативных исследований для вирусных гепатитов В и С.
Таблица 6.11. Гены, для которых показана связь с вирусным гепатитом В и С и ассоциированными клиническими фенотипами
Окончание таблицы 6.11
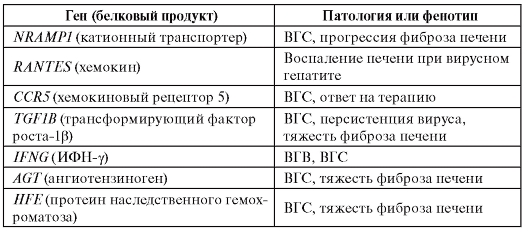
Примечание. ВГВ - вирусный гепатит B; ВГС - вирусный гепатит C.
Генетика инфекционных заболеваний, еще совсем недавно считавшаяся мало перспективной в силу небольших коэффициентов наследования (h2 - 10-20%) этой патологии, сегодня активно исследуется всем современным арсеналом генетических методов.
Злокачественные новообразования
Среди многочисленных и широко распространенных многофакторныхных болезней особую группу составляют злокачественные новообразования.
С позиций генетической классификации заболеваний человека большинство злокачественных новообразований относится к болезням, обусловленным мутациями в соматических клетках. Наряду с этим существуют и наследственные формы опухолей, обязательным компонентом развития которых является наследование генной мутации через половые клетки родителей (иногда их называют герминативными). Подробно о наследственной предрасположенности к онкологическим заболеваниям см. в статье Е.Н. Имянитова на компакт-диске.
Следует подчеркнуть, что злокачественные опухоли - болезни соматических клеток, способных к делению (стволовые клетки, клетки-предшественники). Опухоль возникает в результате накопления различных типов генетических нарушений, приводящих к потере геномного контроля над процессами деления, роста и естественной убыли клеток. Предполагается, что для индукции солидных типов рака необходимо накопление не менее 5-7 мутаций. Развитие
злокачественных опухолей протекает через 3 основные стадии: инициацию, промоцию и прогрессию, при этом скорость данных процессов определяется частотой возникновения мутаций, численностью клеточной популяции, скоростью пролиферации и преимуществом в размножении мутантных клеток.
Выделяют 3 основные группы факторов риска возникновения злокачественных новообразований. Первую из них составляют физические факторы, а именно ионизирующее и ультрафиолетовое излучение, повышающее частоту генных мутаций в соматических клетках.
Вторую группу составляют химические факторы, к которым относятся канцерогены - широкий спектр химических соединений органической и неорганической природы, способных вызывать мутации в соматических клетках (мутагены, или опухолевые инициаторы) или индуцировать опухолевый рост клеток, подвергшихся действию мутагена (митогены, или опухолевые промоторы). Рак возникает в результате последовательного действия инициатора и промотора, при этом риск злокачественной трансформации клеток зависит от дозы канцерогенов, продолжительности и периодичности их воздействия.
Следующей группой факторов риска являются биологические. Среди них особое место занимают вирусы. ДНК-содержащие вирусы, представители семейств паповавирусов (папилломавирус), гепаднавирусов (вирус гепатита B) и герпес-вирусов (вирус Эпстайна-Барр) способны к интеграции в геном клетки-хозяина и к индукции незапланированных циклов ее репликации. Многие вирусные онкогены способны при этом блокировать ключевые опухолесупрессорные механизмы клетки. РНК-содержащие вирусы семейства ретровирусов (вирус Т-лейкоза человека I типа и ВИЧ) имеют геном, представленный двумя молекулами одноцепочечной РНК. В геноме этих вирусов закодирован фермент обратная транскриптаза, необходимый для синтеза копий вирусной ДНК. Встраивание вирусной ДНК в геном клетки-хозяина - обязательный этап жизненного цикла ретровируса. Подобное встраивание может сопровождаться инсерционным мутагенезом, ведущим к появлению структурных мутаций в кодирующих последовательностях генов.
Среди биологических факторов риска отдельное место, несомненно, занимает наследственная предрасположенность к злокаче-
ственным новообразованиям, молекулярные основы которой будут рассмотрены ниже.
На каждом из этапов формирования опухоли развертываются скоординированные в патологическом направлении процессы сначала на молекулярном, а затем на клеточном уровне. Рассматривая канцерогенез как результат нарушения геномного контроля баланса численности клеточной популяции, можно выделить две группы генов, мутации которых этиологически связаны с развитием рака. Речь идет о протоонкогенах и опухолесупрессорах.
Протоонкогены (первая группа) - нормальные клеточные гены, контролирующие рост и размножение клеток. Продукты протоонкогенов осуществляют контроль роста и деления клеток и представлены ростовыми факторами и их рецепторами, внутриклеточными передатчиками сигналов, факторами транскрипции. Измененные в результате мутации протоонкогены или гены, вносимые в геном некоторыми вирусами, получили название онкогенов. Онкогены оказывают стимулирующие влияние на клеточную пролиферацию. Их действие носит доминантный характер, т.е. для развития рака достаточно мутации в одном из аллелей онкогена.
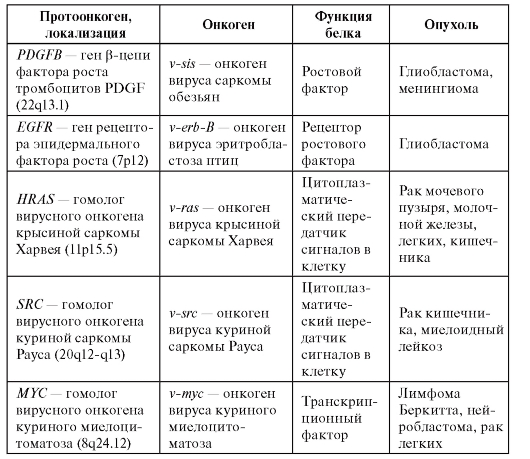
Было замечено, что некоторые вирусы приводят к развитию злокачественных опухолей у животных. Поскольку вирус привносит в клетку хозяина свой генетический материал, то предположили, что геномы вирусов содержат онкогены. В конце 70-х годов XX в. в РНК-содержащем вирусе куриной саркомы Рауса был выделен участок РНК, ответственный за злокачественную трансформацию клеток. Так был открыт первый онкоген - v-src. Позднее оказалось, что в нормальных клетках есть гены, по своей структуре близкие к онкогенам - протоонкогены. По всей видимости, в ходе эволюции вирусные онкогены возникли из нормальных клеточных генов в результате рекомбинации между предковым нетрансформирующим вирусом и ДНК клетки-хозяина. В нормальных клетках протоонкогены находятся под контролем других клеточных генов. Интеграция вируса в геном клетки-хозяина освобождает их от этого контроля, приводит к нерегулируемой активации и превращению в онкоген. К настоящему времени идентифицировано более 100 протоонкогенов и их число продолжает расти. Характеристика некоторых протоонкогенов и онкогенов приведена в табл. 6.12.
Таблица 6.12. Характеристика протоонкогенов и онкогенов
В клетке протоонкогены могут трансформироваться в онкогены через следующие механизмы.
- Рекомбинация с ретровирусной ДНК. Интеграция вирусной нуклеиновой кислоты в геном клетки-хозяина имеет два последствия. Во-первых, это нарушение структуры гена, в который произошло встраивание чужеродного генетического материала (инсерционный или вставочный мутагенез). Во-вторых, это стимулирование незапланированных циклов репликации ДНК и клеточного деления.
- Хромосомные транслокации. Перемещение участка хромосомы, содержащего протоонкоген, на другую хромосому может привести либо к изменению структуры гена, либо к нарушению
регуляции его активности. Например, транслокация t(9;22) (q34;q11), или так называемая «филадельфийская хромосома», встречается у 95% больных хроническим миелоидным лейкозом (рис. 6.6). Она возникает в клетках костного мозга еще до проявления основных симптомов заболевания и имеет важное прогностическое значение. При этой перестройке участок длинного плеча хромосомы 22 транслоцирован на длинное плечо хромосомы 9, а небольшой фрагмент хромосомы 9, содержащий ген ABL, присоединен к участку хромосомы 22, содержащему ген BCR. В результате транслокации на хромосоме 22 образуется химерный ген ABL-BCR, продукт которого (тирозиновая протеинкиназа) стимулирует непрерывное клеточное деление. Важно отметить, что расшифровка цитогенетических и молекулярных механизмов формирования химерного белка позволила создать новый лекарственный препарат (иматиниб), являющийся ингибитором тирозиновых протеинкиназ и демонстрирующий обнадеживающие результаты в лечении больных хроническим миелолейкозом. Другим примером, демонстрирующим роль хромосомных перестроек в активации протоонкогенов, является реципрокная транслокация t(8;14)(q24;q32), выявляемая при лимфоме Беркитта. Транслокация отделяет протоонкоген C-MYC (8q24.12) от его
нормального промотора. В новом месте он попадает под сильный регуляторный элемент гена иммуноглобулина H, расположенного на хромосоме 14. В результате перестройки в клетке резко повышается продукция белка C-MYC - транскрипционного фактора, обладающего онкогенными свойствами. - Амплификации протоонкогенов. Этот процесс существенно увеличивает число копий протоонкогена в клетке, что приводит к
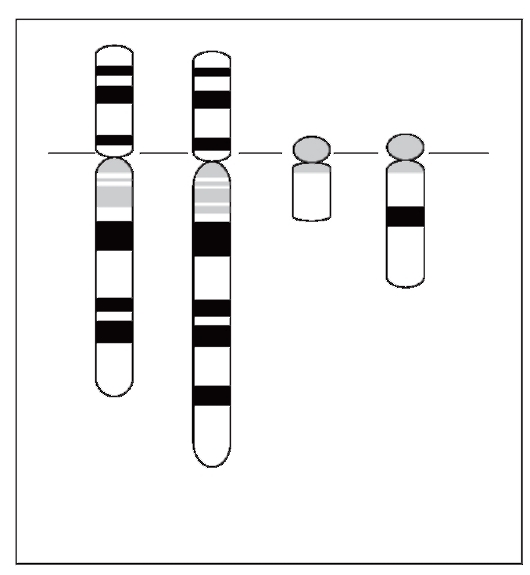
Рис. 6.6. Транслокация t(9;22) при хроническом миелолейкозе
образованию большого количества соответствующих онкопротеинов. Часто амплификация специфических протоонкогенов является признаком определенных опухолей (например, при мелкоклеточном раке легкого обнаруживается амплификация C-MYC, N-MYC и L-MYC протоонкогенов). - Точковые мутации в кодирующих последовательностях протоонкогенов приводят к синтезу онкогенных белков. Наряду с генными мутациями может наблюдаться его существенная амплификация в онкогене, что усиливает трансформирующие клетку свойства.
Вторая группа генов, мутации в которых приводят к развитию злокачественных новообразований, - опухолесупрессорные гены. Их функция заключается в ингибировании клеточного деления. В отличие от протоонкогенов, действие опухолесупрессоров рецессивно, т.е. для развития опухоли необходимо наличие мутации в обоих аллелях одного гена. С мутациями опухолесупрессорных генов связаны наследственные формы новообразований, обусловленные комбинацией герминативных и соматических мутаций. Некоторые опухолесупрессоры обладают свойством так называемых мутаторных генов - их инактивация обусловливает резкое возрастание частоты мутаций в других локусах генома. Как правило, продукты таких генов вовлечены в регуляцию процессов репликации и репарации ДНК, клеточного цикла и апоптоза.
Механизмы действия опухолесупрессорных генов были открыты при исследовании ретинобластомы - злокачественной опухоли сетчатки глаза у детей. Впервые ретинобластома как самостоятельное заболевание с аутосомно-доминантным типом наследования была описана еще в 1902 г. В 1971 г. А. Кнудсен предположил, что для возникновения болезни необходимы мутации в каждой копии гена, отвечающего за развитие опухоли.
Семейные случаи ретинобластомы (примерно 40% всех случаев заболевания) характеризуются более ранним возрастом начала болезни, предрасположенностью к другим типам злокачественных опухолей и являются в основном двусторонними или мультифокальными. Согласно предположению А. Кнудсена, семейные формы возникают в результате наследования одной мутации от родителей, а вторая мутация возникает уже в соматических клетках в нормальном аллеле. Шанс второй мутации достаточно высок, что и приводит к «доминантной предрасположенности» к развитию опухоли.
Спорадические случаи ретинобластомы возникают в результате двух соматических мутаций. Для них характерен поздний возраст начала заболевания, а также отсутствие предрасположенности к другим типам злокачественных новообразований. Как правило, спорадическая ретинобластома односторонняя (имеет один фокус возникновения).
Данные рассуждения А. Кнудсена легли в основу его «двухударной гипотезы» канцерогенеза, нашедшей хорошее подтверждение для большинства наследственных форм злокачественных новообразований (рис. 6.7). Индивид наследует от одного из родителей мутацию в опухолесупрессорном гене. Это может быть точковая мутация или микроделеция. Гетерозиготность индивида по данному локусу страхует его от возникновения опухоли. Однако в течение жизни в соматических клетках может возникнуть мутация и в нормальном аллеле опухолесупрессорного гена. Это приведет к потере гетерозиготности и, как результат, к полному отсутствию продукта опухолесупрессорного гена в клетке. Очевидно, что подобные события (две мутации) могут возникать и у индивидов, не несущих герминативных мутаций, однако вероятность их возникновения будет значительно ниже, поскольку
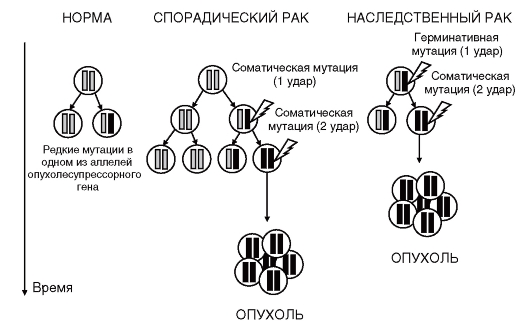
Рис. 6.7. «Двухударная гипотеза» канцерогенеза Кнудсена
требуется последовательное повреждение обоих нормальных аллелей конкретного опухолесупрессорного гена.
Ген ретинобластомы (RB1) стал первым открытым опухолесупрессорным геном у человека. Это открытие было сделано в 1986 г., т.е. спустя 15 лет после формулирования «двухударной гипотезы». Ген RB1 локализован в сегменте 13q14 и кодирует ретинобластомный белок (pRB) - негативный регулятор клеточного цикла. В гипофосфорилированном состоянии pRB связывает семейство транскрипционных факторов E2F, подавляя таким образом деление клетки. В нормальных условиях запуск клеточного деления осуществляется комплексом циклинов и циклинзависимых киназ, которые фосфорилируют pRB и освобождают тем самым факторы транскрипции. Точковые мутации в гене RB1 или аберрантное гиперметилирование его промоторного региона, а также микроделеции участка хромосомы 13q14 приводят к отсутствию опухолесупрессорного белка. В результате клетки, имеющие повреждения и в других локусах генома, не останавливаются в своем размножении, что увеличивает шансы развития опухолевого процесса.
У человека описан ряд злокачественных новообразований, возникающих за счет потери гетерозиготности (табл. 6.13). Для многих форм известны не только хромосомная локализация опухолесупрессорного гена, но и его структура, мутации и первичные продукты. Часто для возникновения одной и той же опухоли необходима потеря гетерозиготности не в одном, а в нескольких локусах. Кроме того, нужны еще мутации в онкогенах. Многокомпонентность генетических событий канцерогенеза очевидна.
Таблица 6.13. Опухоли, возникающие в связи с потерей гетерозиготности
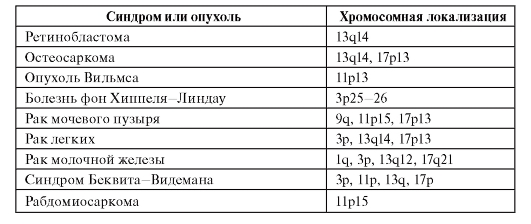
Окончание таблицы 6.13
Злокачественные новообразования, развивающиеся на основе унаследованных мутаций, иногда имеют характер семейных синдромов. Признаки таких синдромов:
• высокая частота опухолей у родственников I и II степени родства;
• наличие в родословной несколько близких родственников со сходными формами опухолей, например, молочной железы и яичника, кишечника, эндометрия;
• наличие двух членов семьи со сходными редкими формами опухолей;
• необычно ранний возраст начала заболевания;
• двусторонние опухоли парных органов;
• синхронность или непрерывность возникновения опухолей;
• опухоли органов разных систем у одного индивида.
Одним из примеров таких семейных форм злокачественных новообразований является синдром Ли-Фраумени, впервые описанный в 1988 г. как заболевание с аутосомно-доминантным типом наследования. Диагноз основывается на нахождении от 2 до 6 типов опухолей в одной родословной и более. Саркомы начинаются в возрасте до 5 лет, остеосаркомы - в юношеском возрасте, а опухоль мозга, молочной железы, аденокарцинома желудка или лейкемия проявляются до 30-летнего возраста. В 1990 г. было показано, что больные
имеют мутации в гене-супрессоре опухоли TP53 (17p13.1). В норме продукт данного гена белок p53 практически не обнаруживается в клетках вследствие чрезвычайно короткого времени полураспада. Функция p53 заключается в контроле целостности ДНК, регуляции клеточного цикла и апоптоза. Белок определяет, произошла ли репарация повреждений ДНК в клетке. Если повреждение не может быть репарировано, то запускается программа апоптоза. При повреждении ДНК p53 активируется и стимулирует транскрипцию гена p21, который кодирует ингибитор циклинзависимой киназы. Белок p21 присоединяется к комплексу циклина и циклинзависимой киназы и инактивирует его. В результате клетка останавливается в G1-фазе клеточного цикла. Очевидно, что мутации в гене TP53 делают невозможным прохождение такого каскада реакций и увеличивают шанс злокачественной трансформации клеток. Мутации данного гена найдены более чем в 50% разных форм злокачественных новообразований. Скрининг мутаций гена TP53 имеет важное клиническое значение для прогноза как в семьях с синдромом Ли-Фраумени, так и при спорадических формах опухолей.
Около 5-10% случаев рака молочной железы составляют наследственные формы, обусловленные передачей потомству мутантных вариантов высокопенетрантных генов репарации ДНК и апоптоза. Особое место среди них занимают гены BRCA1 (17q21) и BRCA2 (13q12). Продукты этих генов активируются в ответ на повреждение ДНК и вместе с белком RAD51, вовлеченным в осуществление гомологичной рекомбинации, формируют комплекс, участвующий в репарации двухцепочечных разрывов ДНК. Открытие генов BRCA1 и BRCA2 имеет большое значение для прогноза возникновения наследственных форм рака молочной железы. Женщины, несущие мутации этих генов, имеют высокий риск развития опухолей молочной железы и яичников. Этот риск особенно повышается в семьях, где есть два случая рака молочной железы и более, а также отмечается раннее начало заболевания - в возрасте до 40 лет.
Наиболее распространенной из наследственных форм опухолей кишечника является наследственный неполипозный колоректальный рак. В его развитии существенную роль играют мутации в генах мисс-матч репарации ДНК - MSH2 (2p11), MSH6 (2p22), MLH1 (3p21.3), MLH3 (14q24.3), PMS1 (2q31) и PMS2 (7p22). Инактивация обоих аллелей одного из этих генов приводит к резкому накоплению ошибок репликации ДНК (гены с мутаторным эффектом), что выявляется
по высокой изменчивости числа копий микросателлитных повторов. Подобный феномен микросателлитной нестабильности, помимо наследственного колоректального рака, наблюдается примерно в 10-20% случаев спорадических опухолей разных типов, свидетельствуя о том, что дефекты в генах репарации ДНК - один из общих механизмов канцерогенеза.
Рассмотренные выше события канцерогенеза не исчерпывают всей многокомпонентности генетической предрасположенности к раку и многофакторности причин развития опухолевого процесса. Можно определить еще целый ряд наследственных характеристик индивида, имеющих отношение к канцерогенезу. Так, например, метаболизм канцерогенов в клетках определяется биохимическими системами, многие из которых генетически полиморфны. Полиморфизм активирующих ксенобиотики ферментов (эстеразы, оксигеназы, изоформы цитохрома Р450) и детоксицирующих ферментов (различные трансферазы) определяе т индивидуальную чувствительность организма к канцерогенным воздействиям (см. главу 7).
Метаанализ данных литературы показывает, что наследственный полиморфизм генов, вовлеченных в биотрансформацию ксенобиотиков, демонстрирует наиболее значимые ассоциации с риском развития онкологических заболеваний. Некоторые примеры ассоциаций приведены в табл. 6.14.
Таблица 6.14. Ассоциации генетических полиморфизмов с различными типами опухолей
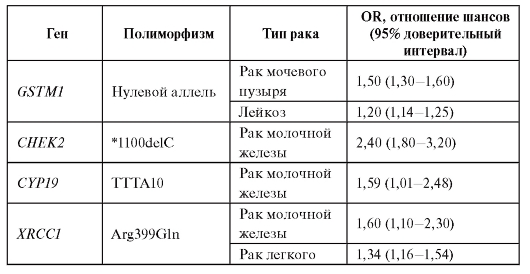
Окончание таблицы 6.14
Таким образом, медико-генетическое консультирование семьи с наследственной предрасположенностью к злокачественным новообразованиям - сложнейшая задача. Требуются специальная подготовка врачей-генетиков в вопросах генетических основ канцерогенеза и хорошая лабораторная база. Только такое сочетание позволяет правильно выявить предрасположенность членов семьи, определить необходимость их диспансерного наблюдения и характер профилактических мероприятий. С молекулярно-биологическими технологиями в современной онкологии можно ознакомиться в одноименной статье С.П. Коваленко на компакт-диске.
ЗНАЧЕНИЕ НАСЛЕДСТВЕННОЙ
ПРЕДРАСПОЛОЖЕННОСТИ В ОБЩЕЙ ПАТОЛОГИИ
ЧЕЛОВЕКА И КЛИНИЧЕСКОЙ ПРАКТИКЕ
Идентификация генетических вариантов подверженности широко распространенным заболеваниям, обозначаемая иногда как генетическое тестирование многофакторного заболевания, является активно развивающейся областью исследований, которая имеет важное теоретическое и практическое значение. Число публикаций по генетическим ассоциациям ежегодно в последнее десятилетие удваивается, и эта информация излагается в 1500 научных журналах на различных языках. Направления, по кото-
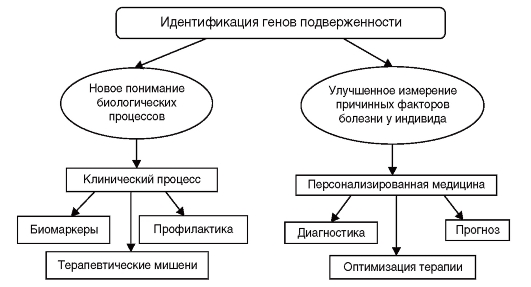
Рис. 6.8. Применение результатов исследования генетических ассоциаций
рым осуществляется систематизация накапливаемой информации, представлены на рис. 6.8.
Полногеномные ассоциативные исследования, анализирующие одновременно до 1 млн геномных вариантов, раскрывают биологические основы многофакторных заболеваний, открывая новые, до сих пор неизвестные метаболические пути формирования патологических фенотипов (болезней), обнаруживая терапевтические мишени, что способствует созданию новых лекарственных средств. Идентификация биомаркеров позволяет надеяться, что риск заболевания может быть снижен путем проведения оптимальных схем лечения. Даже умеренные ассоциации «генотип-фенотип» могут быть использованы для более широких возможностей перехода от теории к практике.
Однако следует заметить, что большинство идентифицированных к настоящему времени ассоциаций генетических полиморфизмов с многофакторными болезнями объясняют небольшой процент (2-10%) индивидуальной вариабельности в риске заболевания. В связи с этим, прежде чем «генетические профили» станут пригодны к широкому использованию в клинической практике, необходимы дополнительные уточняющие исследования и разработки. Они касаются совершенствования подходов к расчету рисков заболеваний, создания правового обоснования для применения генетических тестов, согла-
сованных действий исследователей, врачей и пациентов. Клиническая практика должна опираться на доказательную медицину. Результаты генетического тестирования многофакторного заболевания никогда не будут единственными ориентирами в принятии решений по лечебно-диагностическим и профилактическим мероприятиям - генетические тесты не вместо, а вместе с фенотипическими маркерами составляют основу в персонализированном прогнозе, всегда вероятностном.
КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯ
Аддитивное действие генов
Ассоциации болезней с маркерами (общая концепция) Ассоциация антигенов HLA с болезнями Ассоциация антигенов АВ0 с болезнями
Болезни с наследственной предрасположенностью (определение и классификация)
Генетико-эпидемиологический подход
Генетические основы индивидуальных различий метаболизма канцерогенов
Генетические основы предрасположенности Генетический маркер
Гетерозиготность как фактор предрасположенности Главные гены предрасположенности
Доказательства предрасположенности близнецовым методом Клинико-генетические доказательства предрасположенности Молекулярно-генетические механизмы канцерогенеза Наследственная предрасположенность и профилактика многофакторных заболеваний
Онкогены и гены-супрессоры опухолей
Основные группы болезней с наследственной предрасположенностью
Порог предрасположенности Потеря конституциональной гетерозиготности Предрасполагающие к раку наследственные синдромы Причины болезней с наследственной предрасположенностью Характеристика родословных при болезнях с наследственной предрасположенностью
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Аксенович Т.И. Статистические методы генетического анализа признаков человека: учеб. пос. - Новосибирск: Новосиб. гос. ун-т,
2001. - 128 с.
Генетический паспорт - основа индивидуальной предиктивной медицины / под ред. В.С. Баранова. - СПб.: Изд-во Н-Л, 2009. -
528 с.
Геномика - медицине. Научное издание / под ред. В.И. Иванова, Л.Л. Киселева. - М.: ИКЦ Академкнига, 2005. - 392 с.
Имянитов Е.Н., Хансон К.П. Молекулярная онкология: клинические аспекты. - СПб.: СПб МАПО, 2007. - 212 с.
Новик А.А., Камилова Т.А., Цыган В.Н. Введение в молекулярную биологию канцерогенеза: учеб. пос. / под ред. Ю.Л. Шевченко. - М.: ГЭОТАР-Медиа, 2004. - 224 с.
Пузырев В.П., Фрейдин М.Б., Кучер А.Н. Генетическое разнообразие народонаселения и болезни человека. - Томск: Печатная мануфактура, 2007. - 320 с.