Клиническая генетика : учебник / Н. П. Бочков, В. П. Пузырев, С. А. Смирнихина ; под ред. Н. П. Бочкова. - 4-е изд., доп. и перераб. - 2011. - 592 с. : ил.
|
|
|
|
Глава 3. СЕМИОТИКА И КЛИНИЧЕСКАЯ ДИАГНОСТИКА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ
ОБЩИЕ ЗАМЕЧАНИЯ
Общая врачебная подготовка предполагает знание клиникогенеалогического метода, синдромологического подхода к диагностике наследственных болезней, оценки результатов параклинических исследований, основных признаков и особенностей клинических проявлений наследственной патологии, общих принципов клинической диагностики, особенностей осмотра и физикального обследования пациентов и их родственников. Решающее слово в диагностике наследственных болезней имеют лабораторные анализы: цитогенетические, молекулярно-генетические, биохимические и др. Информация о них будет изложена в отдельной главе.
Термин «синдром» в клинической генетике употребляется не только для обозначения совокупности симптомов, объединенных единым патогенезом, но и для болезней, составляющих самостоятельные нозологические единицы. Нозологически идентифицированные наследственные болезни называют синдромами. Это обусловлено тем, что такие нозологические формы были первоначально описаны как симптомокомплексы без понимания их этиологии. Хотя в дальнейшем расшифровывалась наследственная природа (этиология) данного симптомокомплекса или синдрома вплоть до его полной генетической характеристики (хромосомные болезни, генные болезни, митохондриальные болезни), за наследственными болезнями, сначала описанными как синдромы, сохранился термин «синдром».
Например, после расшифровки этиологии синдрома Клайнфелтера была попытка называть его болезнью Клайнфелтера, но она оказалась безуспешной.
Термины «болезнь» и «синдром» для наследственной патологии равнозначны. Для обозначения некоторых нозологических форм употребляются оба термина (например, болезнь Дауна, синдром Дауна).
Однако люди, имеющие синдром Дауна, и их опекуны, по понятным причинам, чувствительны относительно используемых для описания этого хромосомного состояния терминов. В связи с этим после идентификации хромосомной основы синдрома Дауна в 1959 г. постепенно стали применять термин «трисомия 21».
ОСОБЕННОСТИ КЛИНИЧЕСКИХ ПРОЯВЛЕНИЙ
НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Любой вид патологии (инфекции, ожоги, травмы) имеет свои закономерности клинического проявления, в основе которого лежит взаимодействие повреждающего фактора с организмом. Знание этих закономерностей помогает врачу в диагностике заболеваний и лечении больных. Наследственная патология, несмотря на огромное нозологическое многообразие, имеет специфические черты, которые необходимо знать врачу в качестве ориентиров в диагностических поисках.
В основе клинических проявлений наследственной патологии лежат генетические закономерности действия и взаимодействия генов. Ниже изложены общие признаки наследственных болезней, позволяющие врачу заподозрить роль наследственных факторов в этиологии и патогенезе заболевания.
Семейный характер заболевания
Если врач при обследовании больного получает сведения о сходных случаях заболевания в семье, то это прямо указывает на их возможную наследственную природу. При семейных случаях заболевания необходим второй этап обследования больного, направленный на дифференциальную диагностику наследственной болезни. В то же время заболевание только у одного члена родословной не исключает наследственного характера болезни, поскольку заболевание может быть результатом новой доминантной мутации у одного из родителей или гетерозиготности обоих родителей по рецессивной болезни (сегрегация мутантного фенотипа).
Хроническое прогредиентное рецидивирующее течение
Наследственные болезни, начинающиеся в любом возрасте, имеют хроническое течение с прогредиентной клинической картиной.
Приведем несколько примеров. Хроническая пневмония с бронхоэктазами формируется у детей с легочной формой муковисцидоза. Длительные расстройства пищеварения возникают при целиакии (синоним: глютеновая энтеропатия), кишечной форме муковисцидоза, дисахаридазной недостаточности. Дети с миодистрофией Дюшенна постепенно теряют двигательную активность из-за атрофии мышц. В связи с прогредиентным течением эту болезнь называют прогрессирующей мышечной дистрофией.
Многие новые формы наследственных болезней были открыты при обследовании людей с хронической патологией.
Хронический процесс при наследственных болезнях развивается в результате постоянного действия мутантного гена. Хронизация и прогредиентность одного и того же заболевания по-разному выражены у разных больных, что объясняется взаимодействием генов (генотип каждого человека индивидуален). Рецидивирующее течение наследственных болезней обусловлено и генетическими, и средовыми факторами. К генетическим причинам относятся особенности функционирования генов у больного, т.е. регуляция их активности в установленных генотипом пределах. Средовые факторы - это и осложнения основного патологического процесса (активация микробного фактора, нарушение питания), и дополнительные повреждающие воздействия (охлаждение, инфекции, стрессы).
Специфические симптомы наследственных болезней
Редко встречающиеся специфические симптомы или их сочетания дают основание думать о наследственной природе заболевания. Например, вывих или подвывих хрусталика глаза характерен для синдромов Марфана, Вейля-Марчезани и гомоцистинурии. Голубые склеры бывают при несовершенном остеогенезе и некоторых других болезнях соединительной ткани.
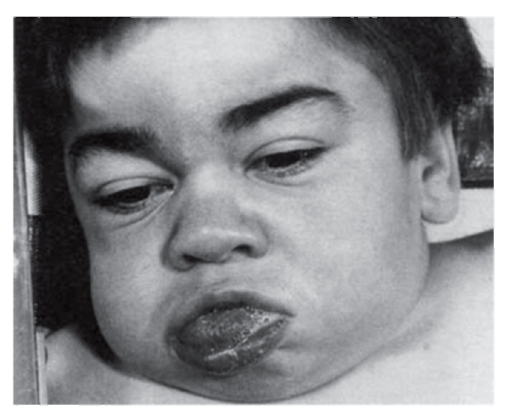
Рис. 3.1. Грубые черты лица у мальчика с мукополисахаридозом (синдром Хантера)
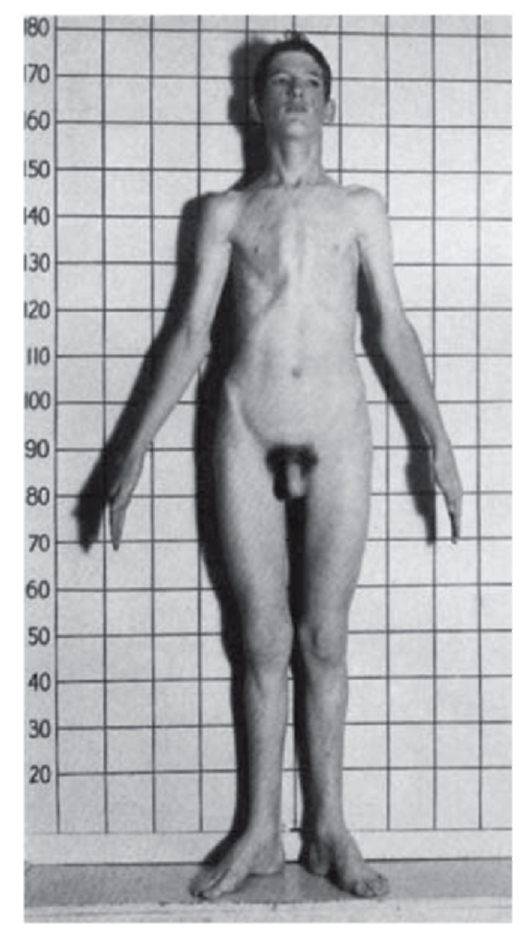
Рис. 3.2. Деформированная грудная клетка (килевидная или «куриная» грудь) при синдроме Марфана

Рис. 3.3. Женщина с ахондроплазией
При алкаптонурии моча на пеленках темнеет. От больных фенилкетонурией исходит мышиный запах. При кровоточивости можно думать о болезни фон Виллебранда или о гемофилии. Грубые черты лица имеют больные с мукополисахаридозами (рис. 3.1). Астеническое телосложение с деформированной грудной клеткой встречается при синдроме Марфана (рис. 3.2). Непропорциональные конечности и туловище, низкий рост, своеобразный лицевой череп говорят об ахондроплазии (рис. 3.3). Право-, левосторонняя асимметрия размеров лица и конечностей позволяет предполагать наследственную гемигипертрофию (рис. 3.4).
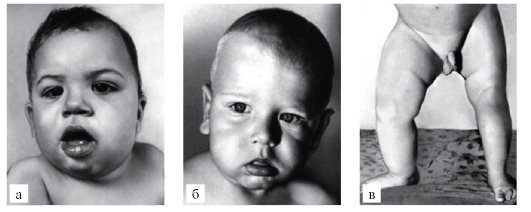
Рис. 3.4. Гемигипертрофия лица (а, б) и нижних конечностей (в) (нозологическая форма - гемигипертрофия)
Множественные патологические изменения органов и систем
Первичное вовлечение в патологический процесс многих органов или даже систем позволяет думать о наследственной причине заболевания. Большинство мутантных генов, вызывающих наследственные болезни, дают плейотропный эффект, в результате чего в процесс вовлекаются многие органы.
Плейотропное действие гена (плейотропия - влияние одного гена на формирование нескольких признаков) - универсальная генетическая закономерность, имеющая прямое отношение к клиническим проявлениям наследственной патологии. Хорошо известно, что любая моногенно детерминируемая наследственная болезнь всегда проявляется не отдельным симптомом, а специфическим сочетанием или комплексом нарушений разных органов и систем. С клиникогенетической точки зрения необходимо различать первичную и вторичную плейотропию.
Важность концепции плейотропии для медицинской генетики не раз пересматривалась. Первоначальное мнение, что все аспекты фенотипа и, следовательно, все проявления менделирующего синдрома зависят от одной функции (или дисфункции) мутантного аллеля, постоянно подкреплялось доказательствами. Однако восприятие важности плейотропии постепенно уменьшалось, особенно в 1940-х годах, когда была сформулирована гипотеза «один ген - один
фермент». Исследования млекопитающих и наблюдение за больными с наследственными нарушениями соединительной ткани поддерживают мнение, что генной плейотропии, вероятно, не существует. Однако отказ от понятия плейотропии был бы преждевременным. С медико-генетической точки зрения концепция о существовании плейотропии правомерна и в некоторых случаях помогает уяснить взаимосвязь клинических симптомов болезней.
Первичная плейотропия обусловлена биохимическими механизмами действия мутантного белка или фермента - первичных продуктов мутантных аллелей. Для иллюстрации этого положения приведем несколько примеров.
Мутантные аллели нескольких генов, контролирующих синтез коллагена и фибриллина, приводят к нарушению свойств волокнистой соединительной ткани. Поскольку соединительная ткань - основа всех органов и тканей, становятся понятными множественные влияния этих мутаций на клиническую картину (фенотип) при таких наследственных болезнях соединительной ткани, как, например, синдромы Элерса-Данло, Марфана: нарушения строения сосудистой стенки (особенно аорты), подвывих хрусталика, пролапс митрального клапана, гиперрастяжимость кожи, гиперподвижность суставов и т.д.
При фенилкетонурии нарушается обмен фенилаланина, в результате чего в организме не синтезируется тирозин. Вследствие этого уменьшается или прекращается образование меланина, что ведет к гипопигментации кожи, волос и радужки. С другой стороны, патологические метаболиты (фенилпировиноградная кислота и др.) нарушают процессы развития и функционирования нервной системы (повышенная возбудимость, тремор, судорожные припадки, умственная отсталость). В основе всех этих очень разнородных симптомов лежит первичный эффект недостаточности (или отсутствия) активности фенилаланингидроксилазы.
Вторичная плейотропия обусловлена осложнениями первичных патологических процессов. Например, при талассемии утолщение костей черепа и гепатолиенальный синдром - результат вторичных процессов, возникающих в связи с усиленным кроветворением и гемосидерозом паренхиматозных органов.
Муковисцидоз обусловлен ошибкой в синтезе трансмембранного белка, обеспечивающего ионный транспорт в клетках. Нарушение ионного транспорта натрия и хлора ведет к образованию густой слизи в бронхах и в экзокринной части поджелудочной железы. За этим следу-
ют вторичные легочные инфекции и нарушения переваривания пищи. И то и другое относится к вторичным плейотропным эффектам.
Таким образом, плейотропное действие генов обусловливает одну из особенностей клинического проявления наследственных болезней - вовлеченность в патологический процесс многих органов и систем. Этот важный обобщенный диагностический признак наследственной патологии должен служить ориентиром для врача.
Врожденный характер заболевания
И нормальные, и патологические аллели включаются в работу в разные периоды онтогенеза - от эмбрионального до старческого. Как подчеркивалось выше, врожденность патологических признаков не всегда свидетельствует о наследственной природе заболевания. Однако не менее 25% всех форм генных наследственных болезней и почти все хромосомные болезни начинают формироваться внутриутробно. Если ребенок рождается с комплексом патологических признаков, то болезнь считают врожденной. Примеры врожденных наследственных болезней - хромосомные синдромы, ахондроплазия, ихтиоз, Х-сцепленная гидроцефалия, аутосомно-рецессивная микроцефалия и др. Примеры врожденных, но ненаследственных болезней - краснушный, талидомидный, сифилитический, алкогольный, гидантоиновый и некоторые другие синдромы, этиология которых устанавливается при целенаправленном сборе анамнеза, относящегося к первым неделям беременности.
Врожденными нередко бывают наследственные болезни обмена веществ. Показания для биохимической и молекулярно-генетической диагностики таких болезней у младенцев - рвота, отказ от пищи, судороги, гипервентиляция, летаргия, кома, желтуха, гипертермия, измененный тонус мышц.
Резистентность к наиболее распространенным методам терапии
Одна из особенностей наследственных болезней - неэффективность лечения, хотя и не абсолютная. Это вполне понятно, потому что «исправить» первичные звенья, даже если известен первичный продукт мутантного гена, далеко не всегда удается (мукополисахаридозы, миодистрофия Дюшенна, нейрофиброматоз).
Естественно, что толерантность к лечению свойственна не всем болезням. Если расшифрованы ключевые звенья патогенеза, то раз-
рабатываются и успешные методы лечения. Некоторые заболевания из группы устойчивых к терапии переходят в группу поддающихся лечению (гепатолентикулярная дегенерация, целиакия, муковисцидоз).
ОБЩИЕ ПРИНЦИПЫ КЛИНИЧЕСКОЙ ДИАГНОСТИКИ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Как подчеркивалось выше, по мере развития медицины и здравоохранения наследственные болезни занимают все большую долю общей патологии человека. Большинство наследственных болезней имеет хроническое течение, вследствие чего высока повторная обращаемость таких больных. Особенно много больных с наследственными формами заболеваний поступает в специализированные клинические и диагностические отделения. В то же время наследственные формы диагностируются не всегда даже в клинических условиях. В определенной степени это понятно, поскольку диагностика наследственной патологии - сложный и трудоемкий процесс.
Трудности диагностики обусловлены прежде всего тем, что нозологический спектр наследственных болезней, каждая из которых имеет очень разнообразную клиническую картину, весьма широк (около 5000 форм). Так, известно более 500 наследственных форм нервных болезней; в дерматологии и офтальмологии таких форм более 250. Некоторые формы встречаются крайне редко, и врач может их никогда не видеть. В связи с таким разнообразием и сходством некоторых наследственных форм с ненаследственными болезнями (фенокопиями), а также в связи с редкостью наследственных болезней (1:200 000 и реже) врач не может активно владеть всем запасом знаний, необходимых для диагностики не только всех наследственных болезней, но даже редких форм по его специальности. Знание основных принципов клинической генетики поможет заподозрить нечасто встречающиеся наследственные болезни, а после дополнительных консультаций с врачом-генетиком, проведения параклинических и лабораторно-генетических обследований - установить точный диагноз.
Клиническая диагностика наследственных болезней основывается на данных клинического, генеалогического и параклинического обследования. Чтобы не пропустить наследственную болезнь, врач должен помнить о том, что наследственные болезни могут про-
текать под маской ненаследственных. В ряде случаев наследственная патология может сопутствовать основному ненаследственному заболеванию, по поводу которого больной обратился к врачу. Диагностика должна быть двухэтапной: общее клиническое обследование больного в соответствии с современными требованиями, описанными в соответствующих руководствах, и специализированное дифференциально-диагностическое обследование при подозрении на конкретную наследственную болезнь.
При общем клиническом обследовании любого больного диагностика должна завершиться четким диагнозом ненаследственного заболевания; четким диагнозом наследственной болезни; подозрением на наследственную этиологию основной или сопутствующей болезни. Первые две группы заключений составляют большинство, третья группа, как правило, требует применения специальных дополнительных методов обследования (параклинических, лабораторногенетических).
Для установления диагноза ненаследственного заболевания достаточно общего клинического и лабораторного обследования. Диагностика, например, конъюнктивита, острой пневмонии, дизентерии не требует генетического обследования.
Общие клинические методы также часто бывают основными в диагностике наиболее известных и распространенных наследственных болезней. Клиническая картина таких болезней была хорошо известна еще до установления их наследственной природы. Например, синдром Дауна (трисомия 21) с большой вероятностью можно диагностировать на основании данных клинического обследования больного. Однако известны случаи ошибочной диагностики синдрома Дауна, особенно на 1-м году жизни. Так, этот диагноз (без анализа кариотипа) на основании особенностей черт лица без учета других признаков синдрома Дауна иногда ставят больным с врожденным гипотиреозом.
Полного клинического обследования, включая использование параклинических методов, обычно достаточно для диагностики таких наследственных заболеваний, как ахондроплазия, нейрофиброматоз, хорея Гентингтона, ретинобластома, буллезный эпидермолиз и т.д. Классические случаи, как правило, затруднений у врача не вызывают, хотя возможны диагностические ошибки, особенно при неполном проявлении того или иного синдрома или при сходных по клиническим признакам других наследственных болезнях.
Казалось бы, вычленение наследственных форм заболевания - не такое уж трудное дело, но это не так. Кажущиеся на первый взгляд ненаследственными заболевания могут быть осложнением или проявлением скрытого наследственного патологического процесса. Вот несколько примеров.
Острая пневмония часто возникает у больных с хромосомными заболеваниями, с генерализованной патологией соединительной ткани, с наследственными болезнями обмена веществ, и она чаще, чем у «здоровых», принимает затяжное или хроническое течение. Пиелонефрит чаще возникает, а затем рецидивирует у больных с врожденными аномалиями мочевой системы. Нарушение ритма сердца может быть проявлением синдрома Элерса-Данло, наследственного удлиненного интервала Q-T.
ОСМОТР И ОБСЛЕДОВАНИЕ ПАЦИЕНТОВ
И ИХ РОДСТВЕННИКОВ
Выше изложены особенности клинических проявлений наследственной патологии. На их основе можно построить схему обследования больного, чтобы диагностировать наследственную болезнь или заподозрить ее.
Врожденные пороки развития. Генетические механизмы эмбрионального развития
Сначала рассмотрим наиболее очевидные признаки наследственной патологии - врожденные пороки развития.
Полный осмотр больного дает возможность выявить врожденный порок развития, который может быть наследственной болезнью, составной частью наследственной болезни либо следствием тератогенеза.
Под термином «врожденный порок развития» понимают морфологический дефект органа, части органа или большой области тела, ведущий к нарушению его/ее функции. Врожденные пороки развития - результат нарушенного органогенеза. В литературе можно встретить еще более общий термин - «врожденные аномалии» (или дефекты). Под этим термином подразумевается любая функциональная или структурная аномалия, которая есть у новорожденного или появляется позже, аномалия либо унаследованная, либо
вызванная средовым событием, предшествующим рождению. Это понятие охватывает не только пороки развития, но и наследственные болезни обмена.
Морфогенез - это реализация генетической программы в трехмерном пространстве и во времени, осуществляемая под влиянием многих факторов среды. В строго определенный период онтогенеза, в строго определенном месте начинается активация или репрессия строго определенных генов, ведущая к дифференцировке клеток и органогенезу. Вариации морфогенеза в конечном счете ведут к необозримому индивидуальному разнообразию людей. Выполнение морфогенетической программы начинается с оплодотворения, интенсивно продолжается во внутриутробном периоде, а затем в детстве и даже во взрослом состоянии.
Пренатальный период можно разделить на преэмбриональную, эмбриональную и плодную (фетальную) стадии. Временные характеристики основных событий эмбрио- и фетогенеза, которые помогают врачу в правильной интерпретации внутриутробного нарушения развития представлены в таблице 3.1.
Таблица 3.1. Основные события в пренатальном развитии человека
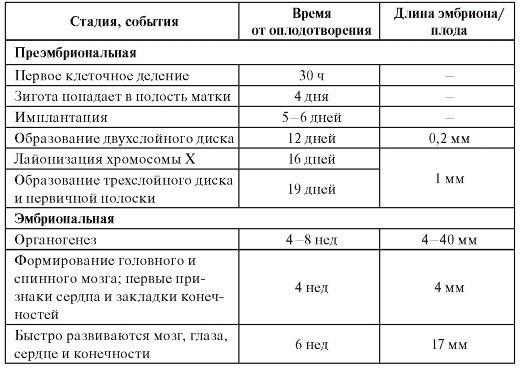
Окончание таблицы 3.1
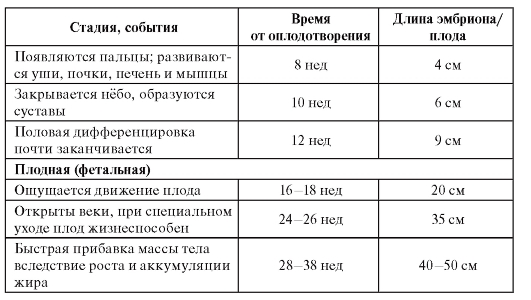
Морфогенез на эмбриональной стадии проявляется в установлении краниокаудальной и дорсовентральной осей. Клеточная агрегация и дифференциация ведут к образованию тканей и органов. На плодной стадии развития происходят быстрый рост и развитие органов.
Хотя сведения о генетических факторах, определяющих эмбриогенез, быстро пополняются, все же они недостаточны. Гены, ответственные за «оркестр» морфогенеза, особенно органогенеза, многообразны: транскрипционные факторы ДНК, ростовые факторы и сигнальные молекулы, лиганды, гены сигнальных путей трансдукции, белки внеклеточного матрикса, энзимы.
Многие вопросы генетического контроля морфогенеза первоначально были изучены на экспериментальных животных. Идентифицированы многие гены и генные семейства, играющие важную роль в раннем развитии. Гены эмбрионального развития человека гомологичны по нуклеотидным последовательностям генам дрозофилы и других животных. Большинство этих генов ответственны за выработку белков, называемых транскрипционными факторами. Они контролируют транскрипцию РНК с ДНК путем связывания специфических регуляторных последовательностей ДНК, образующих комплексы, которые начинают транскрипцию с помощью РНК-полимеразы.
Транскрипционные факторы могут активировать или подавлять экспрессию генов. Важнейшие транскрипционные факторы контролируют многие гены в координации последовательного каскада, включающего такие фундаментальные эмбриологические процессы, как сегментация, индукция, миграция и дифференциация клеток, апоптоз (программированная гибель клеток). Очевидно, эти процессы опосредуются факторами роста, клеточными рецепторами и др.
За 20 лет изучения генов эмбрионального развития установлена их большая роль в нормальном развитии и болезнях. Можно полагать, что дальнейший прогресс в этой области позволит определить генетически обоснованные подходы к лечению и профилактике многих болезней человека. Более подробная информация изложена в статье «Эмбриональные гены и транскрипционные факторы в эмбриогенезе человека» на компакт-диске.
Классификация и этиология врожденных пороков
Классификация врожденных пороков развития затруднена из-за многообразия их форм и сочетаний, исчисляющихся тысячами. Наиболее объективные критерии классификации - локализация и этиология пороков.
Врожденные пороки развития подразделяют на изолированные (в одном органе, например, стеноз привратника), системные (в пределах одной системы органов, например, хондродисплазии), множественные (в органах двух систем и более).
Синдромом множественных врожденных пороков развития называют такие сочетания пороков, при которых очевидна их этиологическая и патогенетическая связь, а также клинически очерчена морфологическая картина. Множественные аномалии, которые являются каскадом одного первичного нарушения, называют последовательностью (не причинная, а патогенетическая связь). Если множественные аномалии в определенных сочетаниях появились неслучайно у нескольких больных, то говорят об ассоциациях.
Этиология врожденных пороков развития может быть наследственной, экзогенной и многофакторной.
Наследственно обусловленные врожденные пороки развития возникают либо при генных мутациях, эффект которых проявляется в виде эмбрионального дисморфогенеза, либо при хромосомных и геномных мутациях (хромосомные болезни). Мутации в определен-
ных локусах могут нарушать процесс морфогенеза в эмбриональном и постэмбриональном периодах. Этому есть многочисленные доказательства, полученные в экспериментальной генетике и клиникогенетической практике. Нарушения морфогенеза (дисморфогенез) могут быть выражены в разной степени и различаться по специфичности. Мутации в определенных локусах ведут к наследственным синдромам врожденных пороков развития.
В результате интенсивного развития генетических технологий и применения их для изучения некоторых этапов эмбриогенеза человека установлена молекулярно-генетическая природа многих изолированных и синдромальных форм врожденных пороков. В табл. 3.2 приведены примеры таких генов.
Таблица 3.2. Фенотипы (синдромы), обусловленные генными мутациями
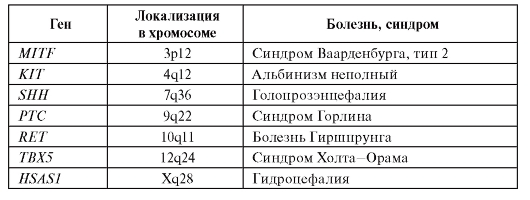
Как видно из табл. 3.2, генетические нарушения морфогенеза могут затрагивать любые системы. Большинство этих мутаций являются вновь возникшими.
Классификации генов, нарушающих морфогенез, еще нет.
Экзогенно обусловленные пороки развития становятся следствием действия тератогенных факторов в эмбриональном периоде, когда осуществляется органогенез. Механизм их действия во многом неясен. Тератогены могут оказывать цитоповреждающее действие, вызывать нарушение дифференцировки клеток в зачатках органов или мутации (генетические соматические повреждения). Хорошо доказано тератогенное действие ионизирующей радиации, лекарственных веществ (талидомид, стрептомицин, гидантоин, варфарин, вальпроевая кислота, аминоптерин, стероидные гормоны и др.), никотина и алкоголя, недостаточного питания (дефицита витаминов и
микроэлементов), биологических факторов (краснухи, цитомегалии). Синдромы, вызываемые тератогенами, имеют специфические наборы признаков дисморфогенеза, поэтому их выделяют в самостоятельные нозологические формы (гидантоиновый, краснушный синдромы, алкогольная эмбриофетопатия).
Чувствительность зародыша человека наибольшая в конце 1-й - начале 2-й недели гестации и между 3-й и 6-й неделями. Эти два срока называют критическими периодами развития.
Промежуток времени, в течение которого повреждающий фактор может вызвать в органе развитие порока, называют тератогенным терминационным периодом. Чувствительность закладок разных органов к действию экзогенных факторов в разные сроки пренатального онтогенеза сильно различается (рис. 3.5). На видно, что раньше других органов и систем нарушается развитие центральной нервной системы (ЦНС) и сердца. Выраженные врожденные пороки развития всех органов формируются в первые 7-8 недель пренатального развития.
В экспериментах на животных под влиянием тератогенов получены пороки развития, сходные с наследственными. Их называют фенокопиями. Однако в клинической практике достоверных случаев
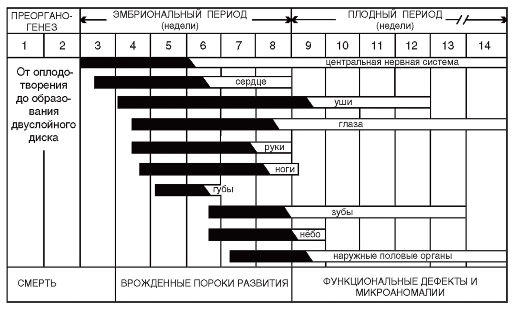
Рис. 3.5. Тератогенные терминационные периоды для разных органов
фенокопий врожденных пороков развития генной или хромосомной этиологии не описано.
Многофакторными врожденными пороками развития называют такие пороки, которые вызваны совместным действием наследственных и экзогенных факторов, причем ни один из них сам по себе не является причиной порока.
Относительный вклад разных этиологических факторов в возникновение врожденных пороков развития можно оценить лишь в общей форме. Согласно данным разных авторов, генетически обусловленные формы (генные и хромосомные) составляют примерно 20-30% всех врожденных пороков развития, многофакторные - 30-40%, экзогенные (тератогенные) - 2-5%, случаи неясной этиологии - 25-50%.
В зависимости от стадии онтогенеза, на которой действовал патогенный фактор, врожденные пороки развития бывают следствием гаметопатий, бластопатий, эмбриопатий и фетопатий.
Такие состояния, когда в гаметах есть мутации, нарушающие нормальное развитие организма, называются гаметопатиями. Этим термином обозначают также и аномалии гамет ненаследственной природы, которые приводят к нарушению оплодотворения или гибели зиготы. Все врожденные наследственные пороки развития - следствие гаметопатий.
Пороки, возникающие в результате поражения бластоцисты, называют бластопатиями. Следствием бластопатий являются такие пороки развития, как циклопия, сиреномелия, а также мозаичные формы хромосомных и реже генных болезней.
Эмбриопатии - нарушение развития зародыша (эмбриона). В строгом смысле этого слова все врожденные пороки развития независимо от этиологии являются эмбриопатиями, поскольку именно в эмбриональном периоде происходит формирование органов. Однако к эмбриопатиям целесообразно относить лишь пороки тератогенной природы, т.е. те пороки, которые возникают в результате действия повреждающего фактора в период от 15-го дня после оплодотворения до конца 8-й недели внутриутробного развития.
Пороки или аномалии, возникающие на плодной (фетальной) стадии развития, называются фетопатиями. Они возникают в результате воздействия повреждающих (тератогенных) факторов в период от 9-й недели внутриутробного развития до родов. К фетопатиям относятся нарушения развития плода, вызванные интоксикацией у матери
(диабетическая, алкогольная, инфекционная). В плодном периоде действие тератогенных факторов формирует в основном функциональные нарушения.
Антропометрия
Важным методом при обследовании больного с клиникогенетической точки зрения является антропометрия. Нарушения роста скелета (замедление или ускорение, избыточность или недоразвитие в целом), диспропорциональность отдельных частей скелета создают специфические антропометрические и визуальные характеристики наследственных болезней. Приведем несколько примеров. Высокий рост (более 180 см), определяемый в основном длиной нижних конечностей, длинные руки, длинные пальцы (арахнодактилия), долихостеномелия указывают на синдром Марфана. Укороченные конечности по сравнению с длиной туловища, запавшая переносица указывают на ахондроплазию. Уменьшение черепа (микроцефалия) - симптом многих наследственных болезней.
Для диагностики наследственных болезней полезны показатели роста, массы тела, телосложения, длины конечностей (иногда их отдельных частей), окружности груди и черепа, а также соотношение сагиттального и латерального размеров черепа. Все эти данные сравнивают с популяционными нормами. Антропометрические показатели у лиц с наследственной болезнью, имеющих нарушения роста и развития, выходят за пределы допустимых вариаций (перцентилей).
Признаки дисморфогенеза в диагностике наследственной и врожденной патологии
Многочисленные признаки дисморфогенеза или пороки развития являются составной частью многих наследственных и врожденных болезней. Они встречаются практически во всех системах и имеют весьма разнообразные проявления. Некоторые представления об их видах и числе можно найти в словаре признаков дисморфогенеза (см. «Приложение»). Большинство признаков дисморфогенеза нарушают функцию того органа, к которому они относятся (кожа, глаза, нёбо, конечность и т.д.), хотя несколько десятков признаков функцию не нарушают. Это микроаномалии развития, или врожденные морфогенетические варианты, они выходят за пределы нормальных вариаций, но не нарушают функцию органа (в отличие от врожденного порока развития). Они являются неспе-
цифическими признаками эмбрионального дисморфогенеза и отражают либо небольшие отклонения в гомеостазе развития, либо наследственную патологию, либо отклонения, вызванные тератогенными факторами. Врожденные морфогенетические варианты встречаются и у здоровых людей, но наличие нескольких признаков требует более внимательного обследования больного на предмет врожденной или наследственной патологии.
Поскольку любое нарушение морфогенеза имеет диагностическую значимость, необходимо внимательно осмотреть больного для выявления признаков дисморфогенеза.
Ниже приводится перечень наиболее распространенных признаков пре- и постнатального дисморфогенеза, оценка которых необходима для дифференциальной диагностики наследственных синдромов и болезней. Часть из них представлена на рис. 3.6-3.56. На каждом рисунке, как правило, можно видеть не один, а несколько признаков дисморфогенеза.
Признаки дисморфогенеза
- Кожа: ангиомы, телеангиэктазии, венозная сеть, пигментные пятна, депигментация, темно-коричневые веснушки (более 20), гипертрихоз, гирсутизм, липомы, фибромы, келоидные рубцы, повышенная растяжимость, складчатость, вялость (рис. 3.6), нарушение потоотделения, гиперкератоз (рис. 3.7).

Рис. 3.6. Складчатая вялая кожа (девочка 6 лет) (cutis laxa - кожа вялая)
- Подкожная жировая клетчатка: избыточное отложение, уменьшенное количество.
- Мышцы: гипертрофия, гипотрофия, аплазия.
- Волосы: сухие, редкие, шерстистые (рис. 3.8); алопеция (тотальная, гнездная), седая
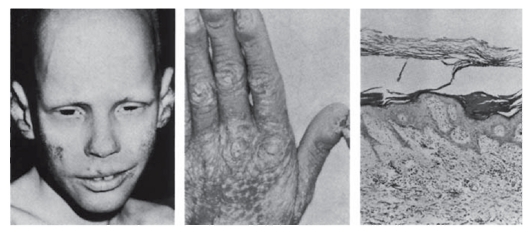
Рис. 3.7. Гиперкератоз (ихтиозоформная эритродермия)
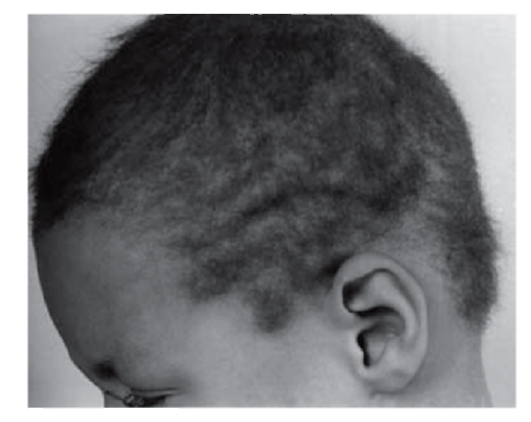
Рис. 3.8. Шерстистые волосы (синдром скрученных волос и глухоты)

Рис. 3.9. Седая прядь волос (синдром Ваарденбурга)
прядь над лбом (рис. 3.9), «мыс вдовы», низкий рост волос на лбу или на шее.
- Череп: гидроцефалия (рис. 3.10), микроцефалия, макроцефалия , брахицефалия, долихоцефалия, тригоноцефалия, акроцефалия (рис. 3.11), выступающий лоб, выступающий затылок (рис. 3.12), плоский затылок.
- Ушные раковины: анотия, макротия (рис. 3.13), микротия (рис. 3.14), деформированные, низко расположен-

Рис. 3.10. Гидроцефалия (синдром Х-сцепленной гидроцефалии)
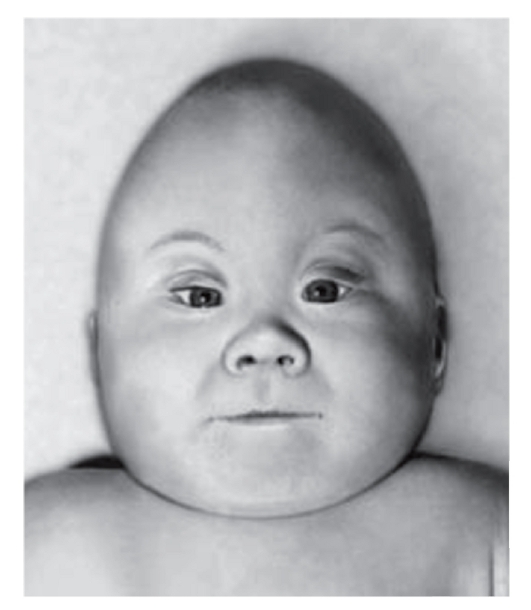
Рис. 3.11. Акроцефалия, широкая переносица, низко расположенные уши, большие щеки, короткие глазные щели, короткая шея (акроцефалополисиндактилия, или синдром Карпентера)
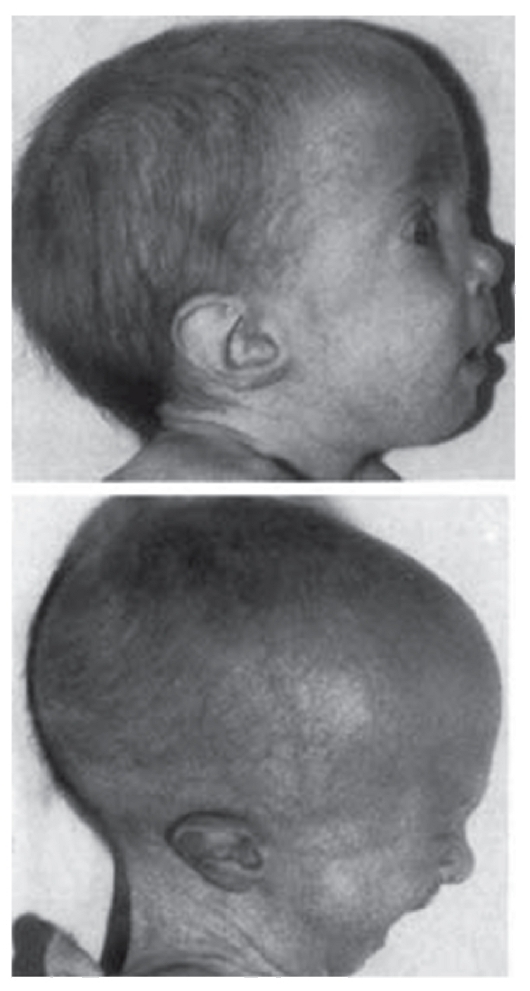
Рис. 3.12. Выступающий затылок, низко посаженные и отклоненные назад ушные раковины (трисомия 18)
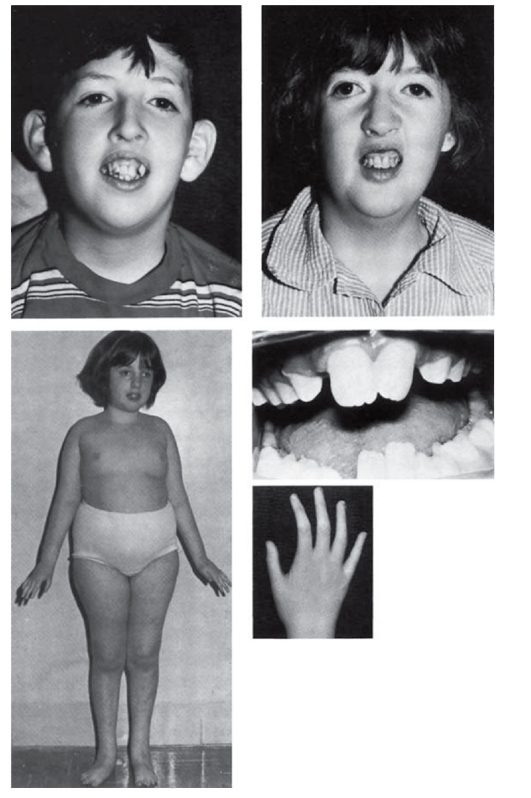
Рис. 3.13. Макротия и другие аномалии (синдром Коэна)
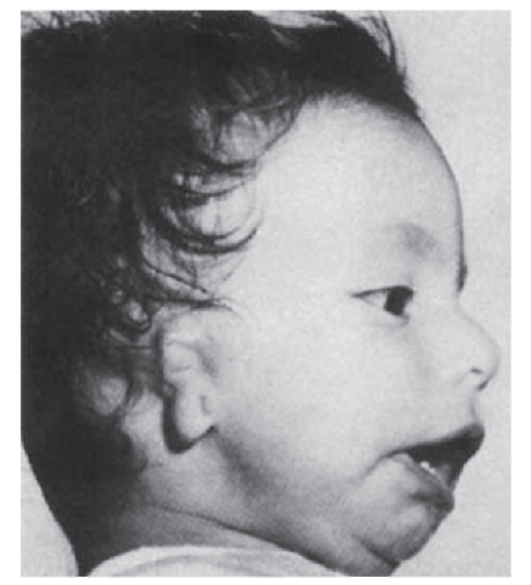
Рис. 3.14. Микротия, микрогнатия, макростомия (врожденная аномалия неясной этиологии)
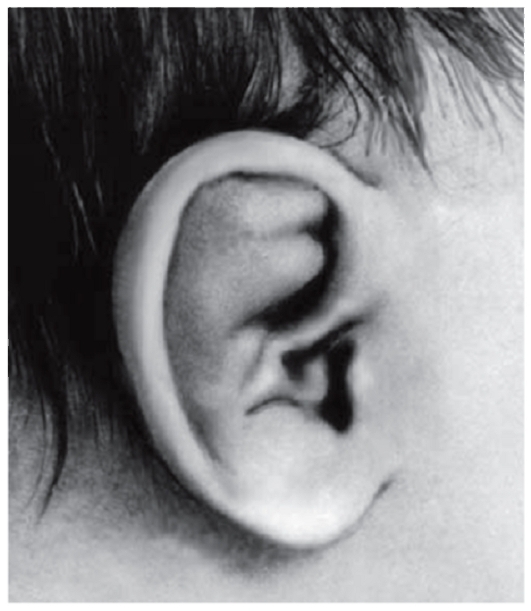
Рис. 3.15. Упрощенная форма завитка и утолщенный противозавиток (дистрофическая дисплазия)
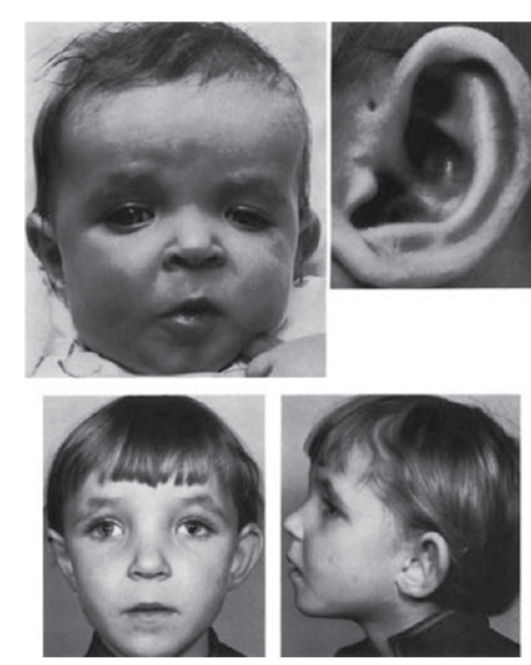
Рис. 3.16. Предушная фистула, отсутствие мочки уха, гипертелоризм (синдром «кошачьего глаза» - патология хромосомы 22)
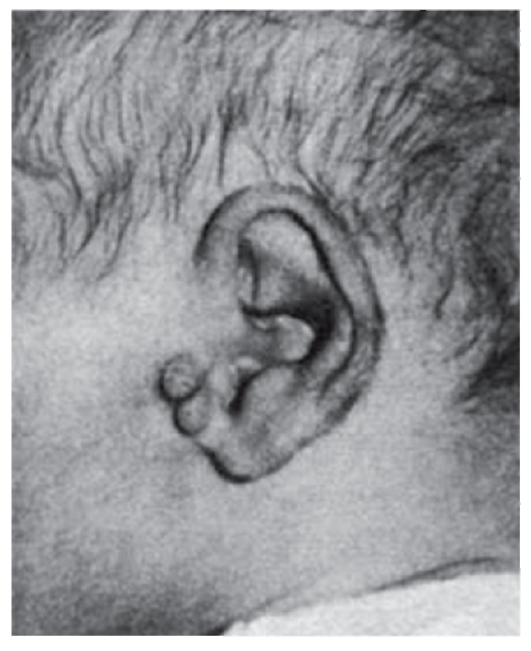
Рис. 3.17. Предушные папилломы (разные формы врожденных нарушений слуха)
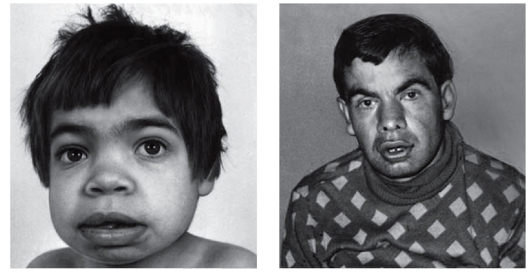
Рис. 3.18. Грубые черты лица (маннозидоз)
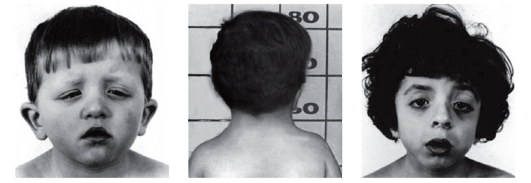
Рис. 3.19. Антимонголоидный разрез глаз, птоз, короткая шея (синдром Нунан)
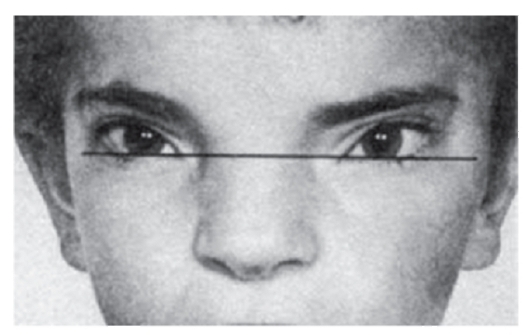
Рис. 3.20. Монголоидный разрез глаз (синдром Грейга)
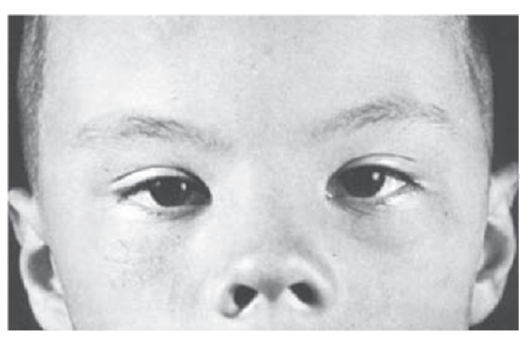
Рис. 3.21. Телекант, эпикант, плоская переносица; открытые вперед ноздри (синдром Элерса-Данло)
ные (рис. 3.11), оттопыренные, отклоненные назад (рис. 3.12), завитки со сглаженным упрощенным рисунком (рис. 3.15), предушные фистулы (рис. 3.16), предушные папилломы (рис. 3.17).
- Лицо: плоское, круглое, треугольное, вытянутое, грубые черты (рис. 3.18).
- Область глаз и глаза: антимонголоидный (рис. 3.19) и монголоидный (рис. 3.20) разрез глаз, эпикант (рис. 3.21), телекант (рис. 3.21), гипертелоризм (рис. 3.22), гипотелоризм, птоз (рис. 3.23), блефарофимоз, косоглазие (рис. 3.24), микрофтальм, экзофтальм, короткая глазная
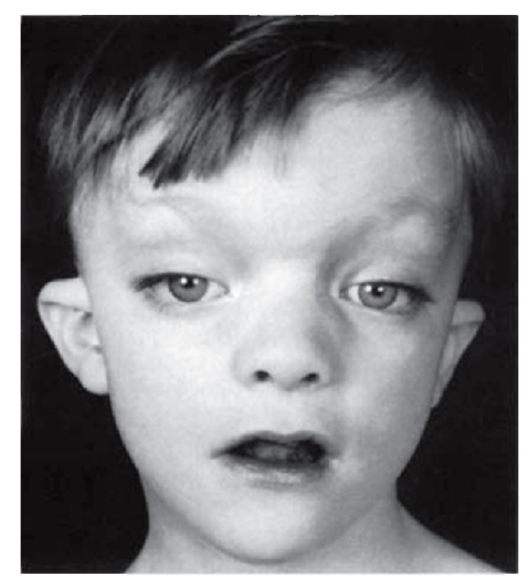
Рис. 3.22. Гипертелоризм, птоз, низко посаженные деформированные ушные раковины (синдром Аарскога)
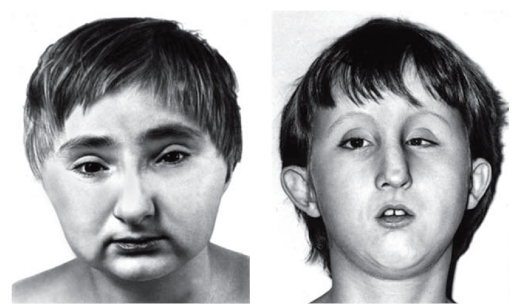
Рис. 3.23. Птоз, косоглазие, оттопыренные, низко посаженные ушные раковины с упрощенным рисунком, маленький рот (синдром Халлермана- Штрайфа)
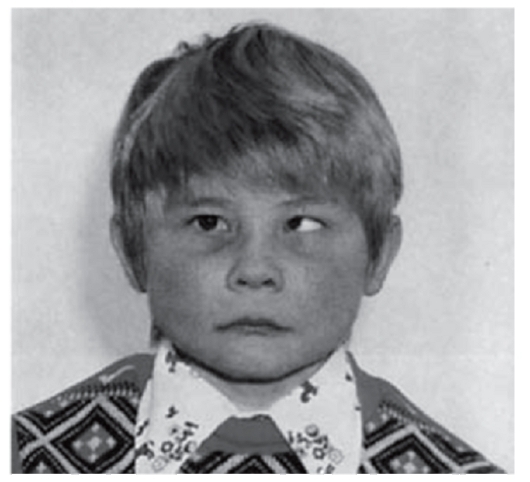
Рис. 3.24. Косоглазие
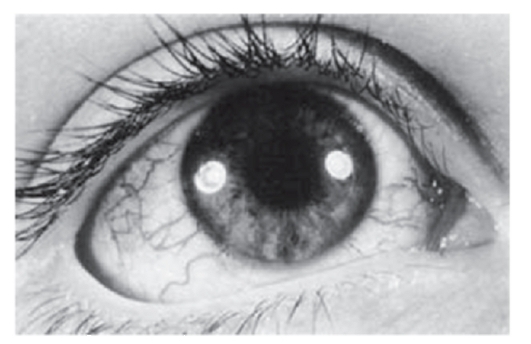
Рис. 3.25. Телеангиэктазии склеры (атаксия-телеангиэктазия)
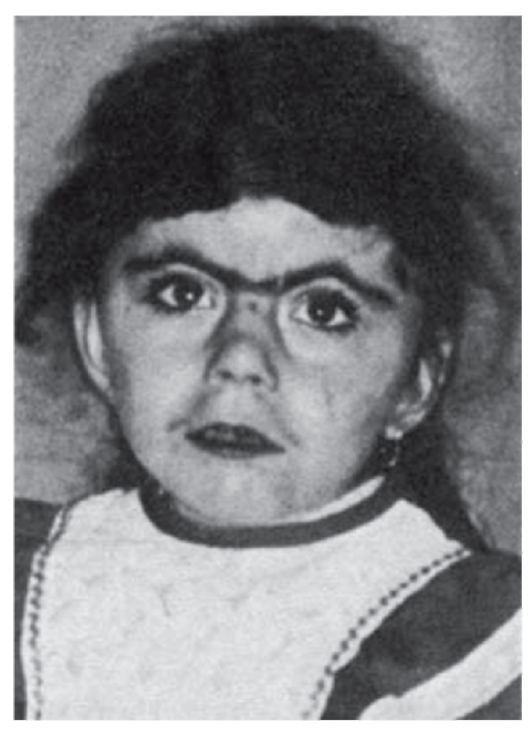
Рис. 3.26. Синофриз (синдром Корнелии де Ланге)
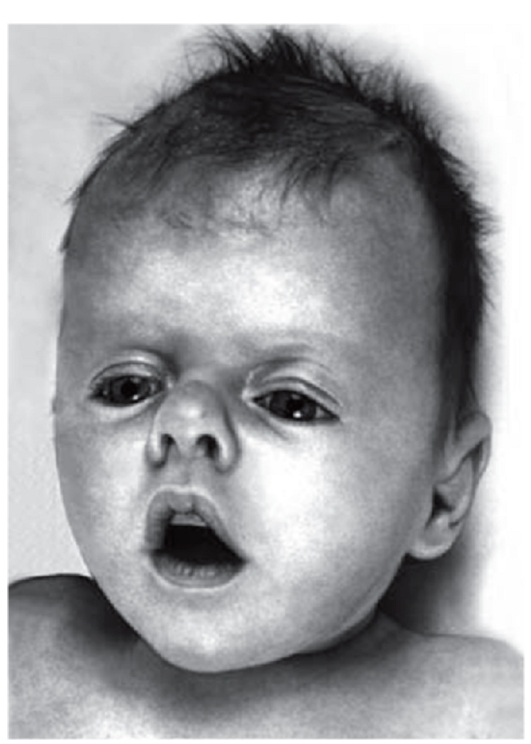
Рис. 3.27. Седловидная переносица, антимонголоидный разрез глаз (синдром Пфайфера)
щель, двойной или тройной ряд ресниц, колобома радужки, гетерохромия радужек, голубые склеры, телеангиэктазии (рис. 3.25), миопия, гиперметропия, синофриз (рис. 3.26).
- Hoc: короткий, клювовидный, седловидная переносица (рис. 3.27), широкая плоская переносица, плоские крылья носа, открытые вперед ноздри
(рис. 3.28).
- Фильтр: длинный (рис. 3.29), короткий, плоский, глубокий.
- Челюсти: прогения, ретрогения, макрогения (рис. 3.30) и
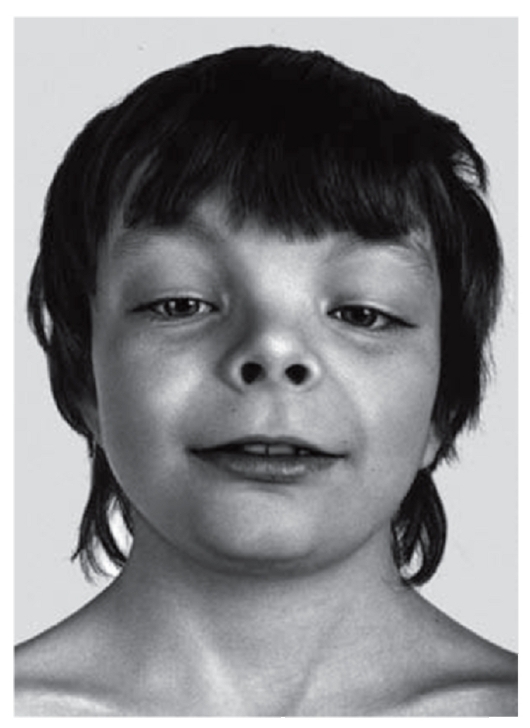
Рис. 3.28. Открытые вперед ноздри, короткий нос, антимонголоидный разрез глаз, гипертелоризм, широкая переносица (синдром Аарскога)

Рис. 3.29. Длинный фильтр, короткий нос с недоразвитыми крыльями, микрогения, низко посаженные уши (диабетическая эмбриопатия)

Рис. 3.30. Макрогения (синдром Х-сцепленной умственной отсталости)
микрогения (рис. 3.31), микрогнатия и макрогнатия.
- Губы и полость рта: макростомия и микростомия; губы тонкие, толстые; нёбо плоское, высокое, арковидное, готическое, расщелина нёба (рис. 3.32); раздвоение язычка; макроглоссия (рис. 3.33) и микроглоссия, короткая уздечка языка, множественные уздечки губ.
- Зубы: неправильное расположение, неправильная форма, врожденный избыток или врожденное отсутствие одного или нескольких зубов, гипоплазия эмали, диастема (верхняя, нижняя) (рис. 3.34), тремы (рис. 3.35).
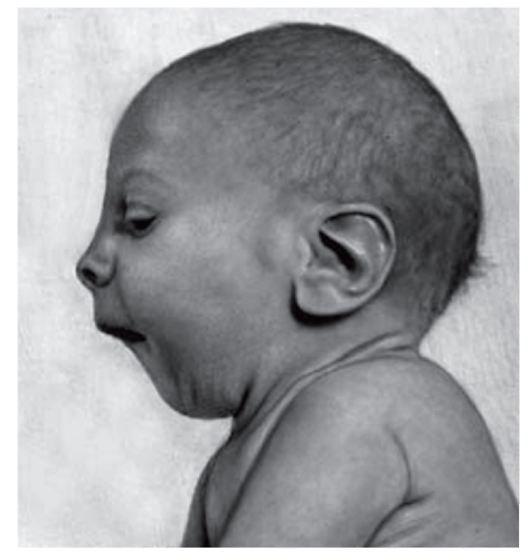
Рис. 3.31. Микрогения, крупные ушные раковины (синдром Смита- Лемли-Опитца)
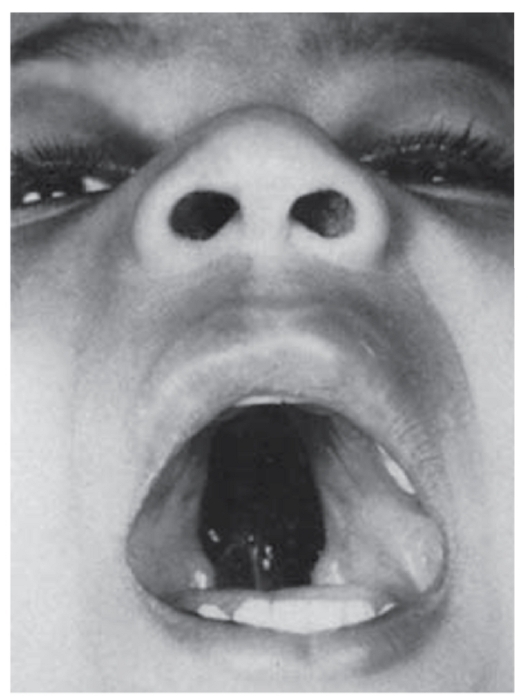
Рис. 3.32. Расщелина нёба (врожденный порок как следствие противосудорожной терапии беременной)
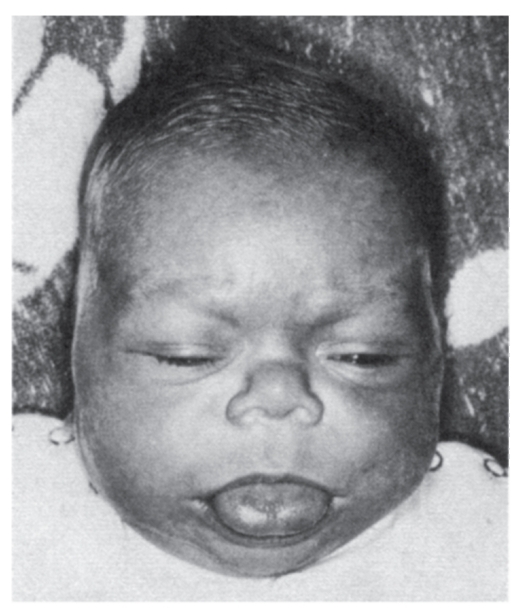
Рис. 3.33. Макроглоссия (синдром Беквита-Видемана)

Рис. 3.34. Верхняя и нижняя диастема (распространена среди нубийцев Египта)

Рис. 3.35. Тремы - широкие промежутки между зубами (изолированный надклапанный стеноз аорты)
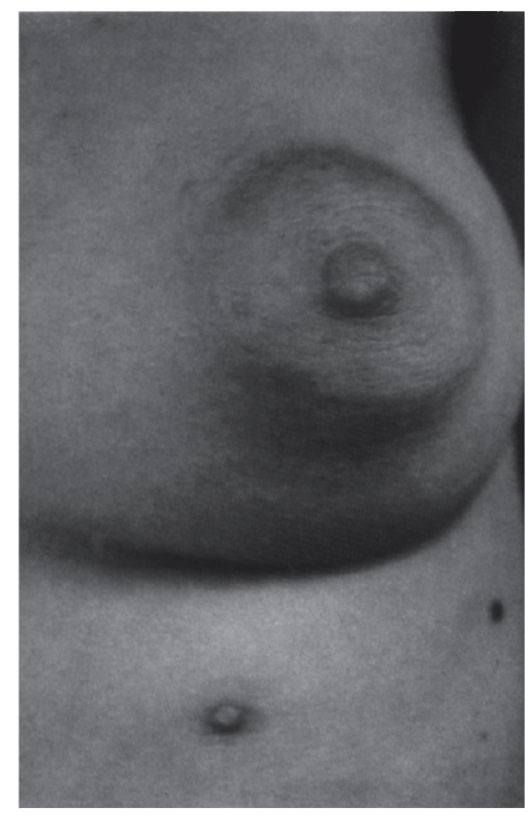
Рис. 3.36. Полителия (добавочный сосок) (встречается при частичной трисомии 12)
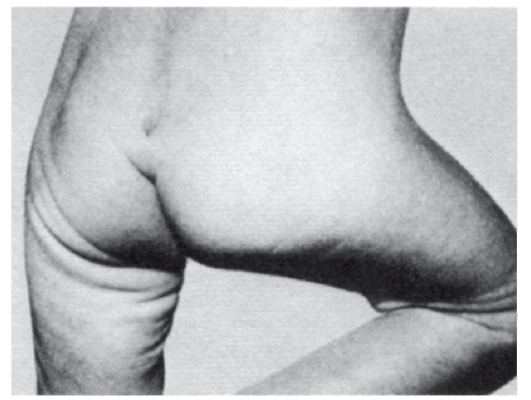
Рис. 3.37. Пилонидальная ямка (встречается при разных хромосомных и генных синдромах, а также при диабете у матери)
- Шея: короткая, длинная, кривошея, крыловидные складки, низкая линия роста волос.
- Грудная клетка и туловище: долихостеномелия, воронкообразная, килевидная, добавочные соски (полителия) (рис. 3.36), гипертелоризм сосков, сколиоз, лордоз, кифоз, пилонидальная ямка
(рис. 3.37).
- Конечности: укороченные, удлиненные, вальгусная дефор-
мация(Х-образные)иливарусная деформация (О-образные) (рис. 3.38), полидактилия (преаксиальная и постаксиальная) (рис. 3.39, рис. 3.40), олигодактилия (рис. 3.41), брахидактилия (рис. 3.42), укорочение отдельных пальцев (рис. 3.43, 3.44), арахнодактилия, синдактилия (рис. 3.45, рис. 3.46), клинодактилия (рис. 3.47), камптодактилия (рис. 3.48), широкий I палец, гипоплазия I пальца, трехфаланговый I палец кисти (рис. 3.49), конусовидная форма пальцев (рис. 3.50), поперечная ладонная складка (рис. 3.51), сиднеевская складка, одна складка на V пальце кисти, глубокая складка на стопе (рис. 3.52), сандалевидная щель на стопе, полая стопа, конская стопа, косолапость, плоскостопие,
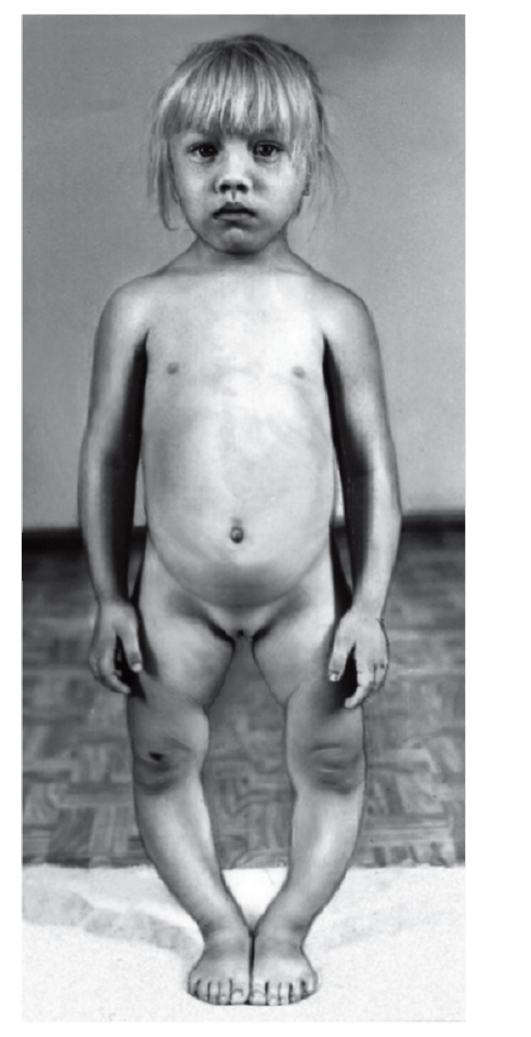
Рис. 3.38. Варусная деформация нижних конечностей; гипертелоризм сосков (гипофосфатемия или витамин D-резистентный рахит)
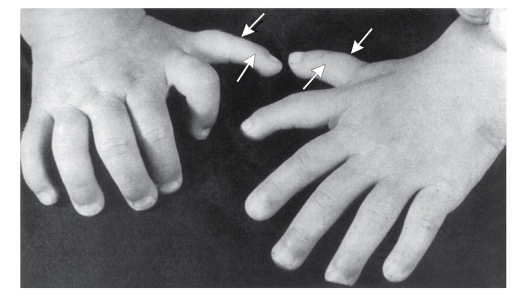
Рис. 3.39. Полидактилия преаксиальная (обозначено стрелкой)
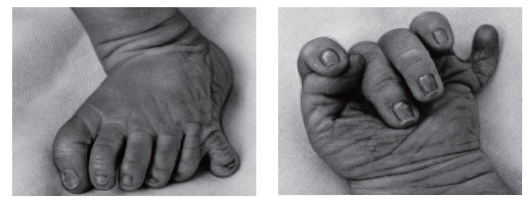
Рис. 3.40. Полидактилия кисти и стопы постаксиальная (синдром Смита- Лемли-Опитца)
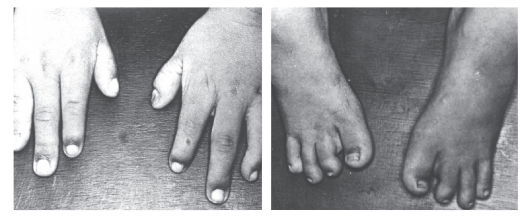
Рис. 3.41. Олигодактилия кистей и стоп, гипоплазия отдельных пальцев и ногтей (постаксиальный акрофациальный дизостоз)
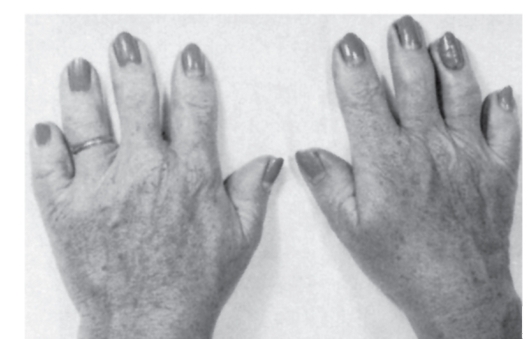
Рис. 3.42. Брахидактилия (тип А1)
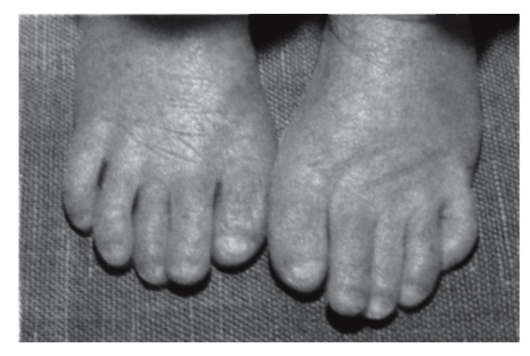
Рис. 3.43. Укороченные I пальцы стоп, гипоплазия ногтей (синдром Вольфа-Хиршхорна - делеция хромосомы 4р)
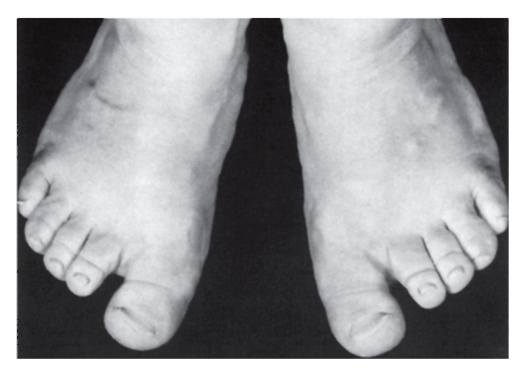
Рис. 3.44. Короткие пальцы стопы, короткие ногти, широкий I палец, сандалевидная щель (периферический дизостоз)
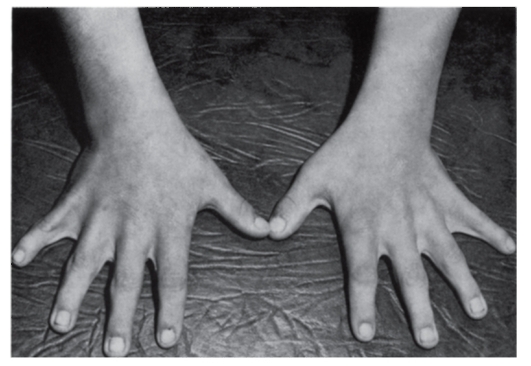
Рис. 3.45. Синдактилия кожная (синдром Аарскога)
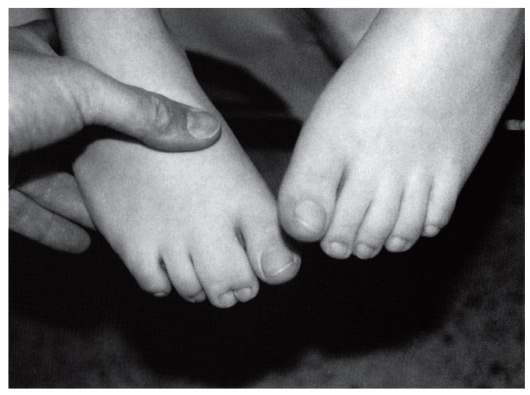
Рис. 3.46. Синдактилия II-III пальцев стоп разной выраженности (аминоптериновый синдром)
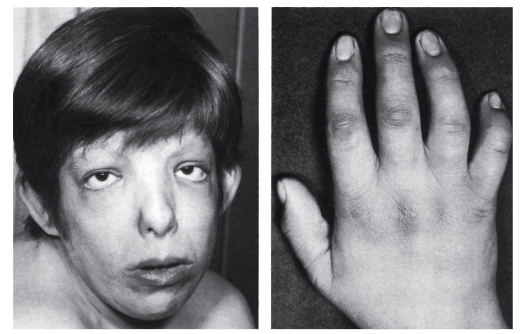
Рис. 3.47. Клинодактилия (синдром трисомии 9)
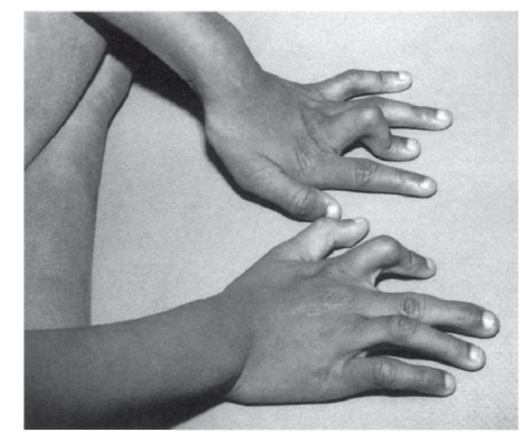
Рис. 3.48. Камптодактилия II пальца правой кисти и III пальца левой кисти (вальпроевый синдром)
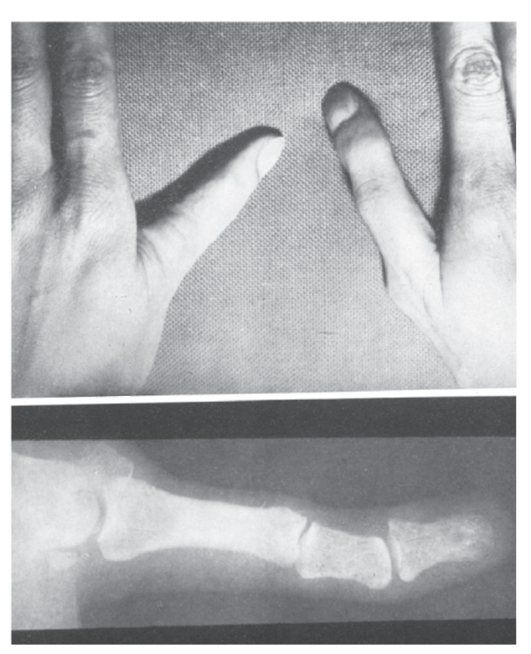
Рис. 3.49. Трехфаланговый I палец кисти (анемия и трехфаланговые I пальцы)
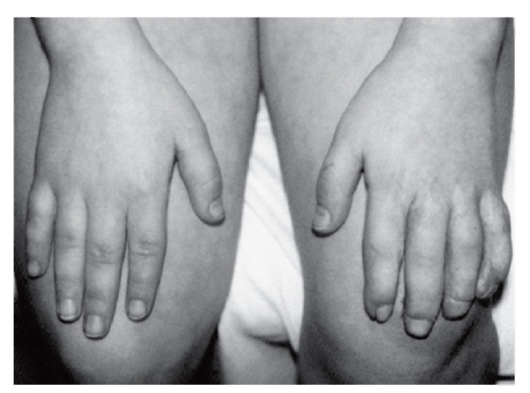
Рис. 3.50. Конусовидные пальцы (аминоптериновый синдром)
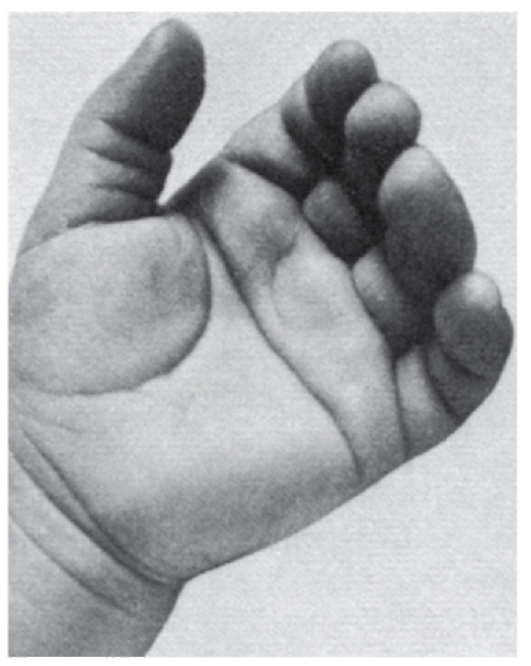
Рис. 3.51. Четырехпальцевая поперечная («обезьянья») ладонная складка (часто встречается при хромосомных и генных болезнях, а также у 2-4% здоровых людей)
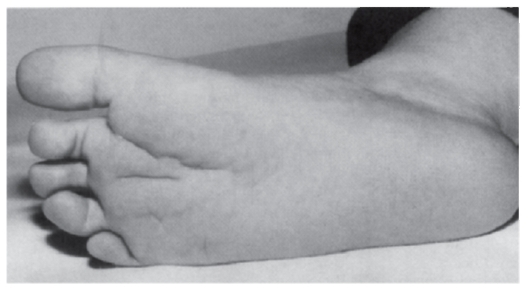
Рис. 3.52. Глубокая складка на стопе (трисомия 8, мозаичная форма)
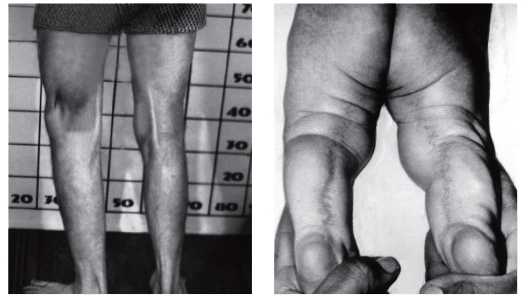
Рис. 3.53. Подколенная складка (синдром подколенного птеригиума)
переразгибание суставов, гемигипертрофия, подколенная складка (рис. 3.53).
- Ногти: широкие, короткие (рис. 3.54), вогнутые, аплазия, гипоплазия (рис. 3.55), дистрофия, «часовые стекла».
- Мочеполовая система: крипторхизм, гипоспадия, шалевидная мошонка (рис. 3.56), увеличенный клитор.

Рис. 3.54. Гипоплазия концевых фаланг (брахидактилия, тип Д)
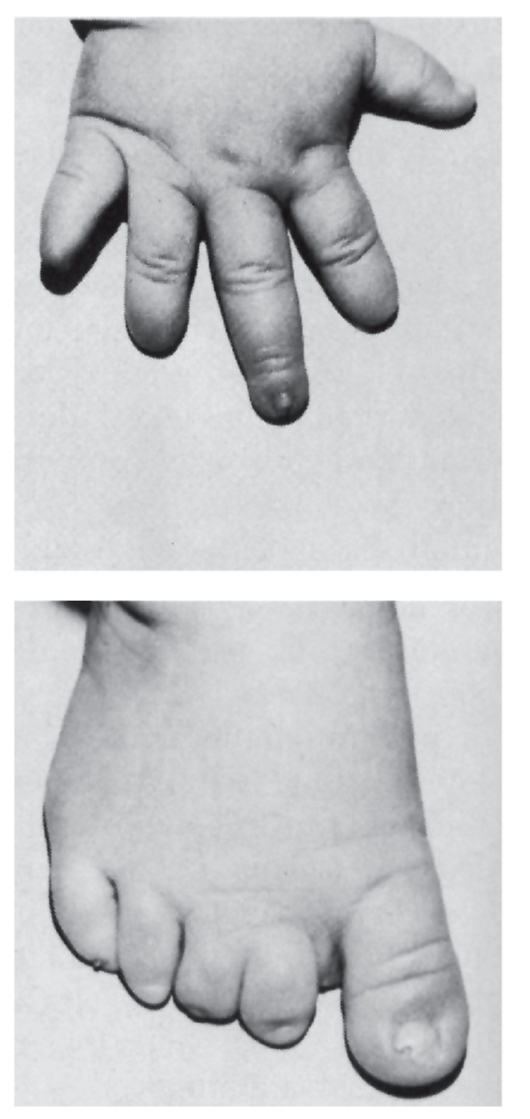
Рис. 3.55. Гипоплазия концевых фаланг и ногтей (гидантоиновый синдром)
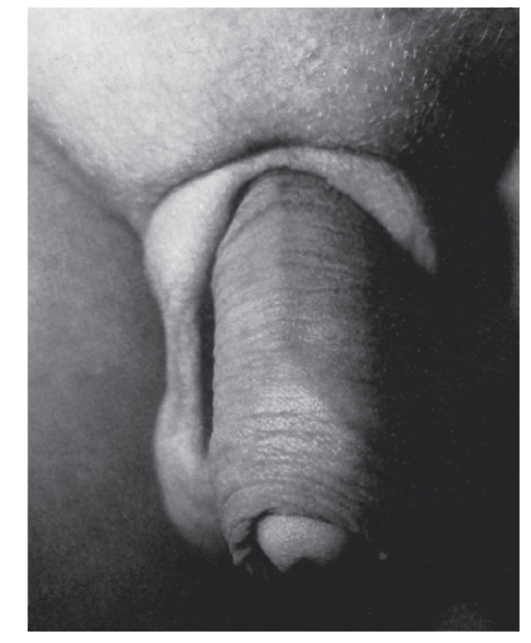
Рис. 3.56. Шалевидная мошонка (синдром Аарскога)
Течение беременности
На наследственную или врожденную патологию тератогенной природы может указывать нарушение течения беременности и пренатального развития плода.
Угроза прерывания беременности, мало- и многоводие, малая подвижность плода могут быть признаками врожденных и наследственных болезней плода.
Например, ограничение движений плода в матке бывает при артрогрипозах гетерогенной генетической этиологии.
При задержке внутриутробного развития или пренатальной гипоплазии размеры и масса плода или новорожденного не соответствуют гестационному сроку. Это состояние обобщенно отражает неблагополучие пренатального периода развития и требует дифференциальной диагностики наследственной патологии.
Возможные причины задержки внутриутробного развития: хромосомные аномалии, генные мутации (например, семейная дизавтономия, синдромы Корнели де Ланге, Дубовица и др.), хронические инфекционные заболевания плода (цитомегалия, врожденная краснуха, сифилис), радиационное поражение, многоплодная беременность, аплазия поджелудочной железы у плода. Развитие плода может задерживаться также под влиянием некоторых факторов материнского организма (токсикоз, курение, гемоглобинопатия и др.).
Внутриутробную задержку развития необходимо отличать от генетически обусловленных малых размеров плода (врожденная гипофункция щитовидной железы, различные формы наследственной карликовости).
При некоторых наследственных болезнях происходит избыточное развитие в пренатальном периоде (внутриутробная макросомия). Дети с синдромами Сотоса, Беквита-Видемана, диабетической фетопатией рождаются с повышенной массой тела.
КЛИНИКО-ГЕНЕАЛОГИЧЕСКИЙ МЕТОД
Генеалогия в широком смысле слова - это родословная. Генеалогический метод - метод родословных, т.е. прослеживание болезни (или признака) в семье или роду с указанием типа родственных связей между членами родословной. В медицинской генетике этот метод называется клинико-генеалогическим, поскольку речь идет
о наблюдении патологических признаков с помощью клинического обследования.
Генеалогический метод относится к наиболее универсальным методам в медицинской генетике. Его широко применяют в целях медико-генетического консультирования для установления наследственного характера признака, при определении типа наследования и пенетрантности гена, анализе сцепления генов и картировании хромосом, изучении интенсивности мутационного процесса, расшифровке механизмов взаимодействия генов.
Эмпирические наблюдения родословных с наследованием патологических признаков известны давно. Например, в Талмуде отражено понимание сцепленного с Х-хромосомой наследования гемофилии. В середине XVIII в. П. Мопертюи описал наследование доминантного признака полидактилии и правильно проанализировал расщепление признака в потомстве. В начале XIX в. Дж. Адамс на основе эмпирического анализа родословных описал доминантный и рецессивный типы наследования. Несколько врачей подробно разобрались в наследовании гемофилии и цветовой слепоты. Эти и некоторые другие попытки анализа родословных можно рассматривать как предпосылки формирования генеалогического метода, которое закончилось в начале XX в., вскоре после рождения генетики как науки. С этого времени генеалогический метод широко использовали в генетике человека и медицинской генетике. Его дальнейшее усовершенствование шло по линии как составления родословных, так и (особенно) разработки методов статистического анализа данных. Метод находил все более широкое применение в клинической генетике и в генетике человека (изучение мутационного процесса, сцепления генов и др.).
Суть генеалогического метода сводится к выявлению родословных связей и прослеживанию признака или болезни среди близких и дальних прямых и непрямых родственников. Технически он складывается из двух этапов: составления родословной и генеалогического анализа.
Составление родословной
Сбор сведений о семье начинается с консультирующегося или с пробанда. Консультирующимся называется лицо, обратившееся к врачу или первым попавшее в поле зрения исследователя. Пробанд - это больной или носитель изучаемого признака. Во многих случаях консультирующийся и пробанд - один и тот же человек. Дети одной
родительской пары называются сибсами (братья и сестры). Название «сибс» происходит от английской аббревиатуры SIBS: Sisters-BrotherS. Семьей в узком смысле называют родительскую пару и их детей, но иногда и более широкий круг кровных родственников, хотя в последнем случае лучше использовать термин «род».
Обычно родословную собирают по одному или по нескольким признакам. Чисто технически ее нельзя составить по всем известным
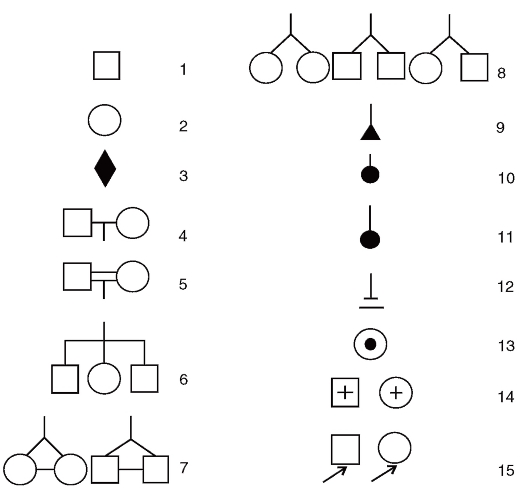
Рис. 3.57. Символы, используемые при составлении родословных: 1- лицо мужского пола; 2 - лицо женского пола; 3 - пол неизвестен; 4 - брак; 5 - родственный брак; 6 - сибсы; 7 - монозиготные близнецы; 8 - дизиготные близнецы; 9 - выкидыш; 10 - аборт; 11 - мертворожденный; 12 - бездетный брак; 13 - гетерозиготная носительница мутантного гена в Х-хромосоме; 14 - умершие; 15 - пробанд
признакам, да в этом и нет надобности. Врач всегда интересуется каким-то конкретным заболеванием или признаком либо несколькими дополнительными признаками, сопутствующими основному.
В зависимости от цели исследования родословная может быть полной или ограниченной. Желательно, конечно, стремиться к наиболее полному составлению родословных по восходящему, нисходящему и боковым направлениям. Эта задача не такая легкая, как может показаться на первый взгляд. Чем больше поколений вовлекается в родословную, тем она обширнее. Это влечет за собой неточность получаемых сведений и, следовательно, неточность родословной в целом.
Составление родословной сопровождают краткой записью о каждом ее члене с точной характеристикой его родства с пробандом (легенда родословной). В дальнейшем для наглядности (или при публикации) родословную изображают графически. Для этого обычно пользуются стандартными символами (рис. 3.57). Перечислить все обозначения невозможно. Если рассматриваемых признаков в родословной много, то можно прибегать к буквенным или штриховым различиям внутри символов. Изображение родословной обязательно сопровождается описанием обозначений. Пример составления родословных приведен на рис. 3.58.
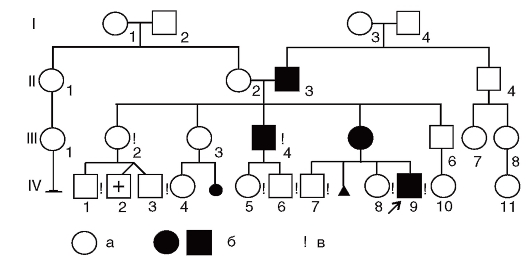
Рис. 3.58. Пример родословной. Обозначения стандартные (см. рис. 3.57):
а - больные диабетом; б - больные нейрофиброматозом; в - лично обследованные. I, II, III, IV - поколения
Поколения обозначают римскими цифрами сверху вниз, обычно слева от родословной. Арабскими цифрами нумеруют потомство одного поколения (весь ряд) последовательно слева направо. Братья и сестры располагаются в родословной в порядке рождений. Таким образом, каждый член родословной имеет свой шифр, например II-2, III-8. Супругов членов родословной можно обозначать тем же номе-
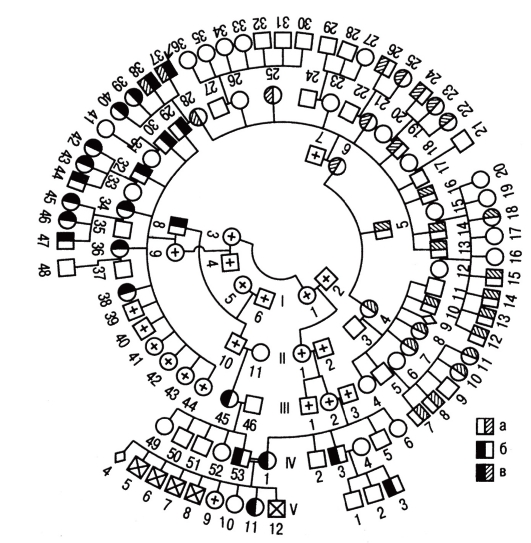
Рис. 3.59. Большая родословная с концентрическим расположением поколений: а - больные гемоглобинозом Е; б - больные талассемией; в - больные гемоглобинозом Е и талассемией
ром, но со строчной буквой вслед за цифрой, если супруги кровно не связаны с членами родословной. Если супруг не обследован на наличие рассматриваемого признака и его родословная не приводится, желательно его вообще не изображать. Внесение такого значка в родословную не дает никакой информации, но затрудняет восприятие основной части родословной. Все индивиды должны располагаться строго по поколениям в один горизонтальный ряд. «Подвешивание» символов между рядами поколений - довольно грубая ошибка.
Если родословная очень обширная, то разные поколения располагают не горизонтальными рядами, а концентрически (рис. 3.59).
В настоящее время все шире используются вспомогательные репродуктивные технологии. В связи с этим предложены новые символы в записях родословных для таких семей (рис. 3.60).
При применении генеалогического метода в родословной важно отмечать обследованных на наличие признака (можно использовать
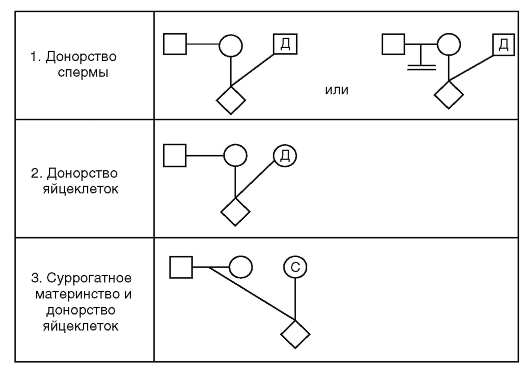
Рис.3.60. Символы вспомогательных репродуктивных технологий в родословных: Д - доноры спермы и яйцеклеток; С - суррогатная мать (вынашивающая беременность)
сведения из объективного источника, например из истории болезни) и необследованных, сведения о которых почерпнуты из ответов пробанда или родственников, а также из анкет. Грубая ошибка - искусственное укорочение звеньев родословной из-за трудностей обследования лиц II и III степеней родства, особенно если не указывается, у кого из членов родословной действительно не было родственников, а у кого сведения не собраны.
Получить сведения о родственниках непросто. Во-первых, не все пациенты знают о болезнях родственников, во-вторых, они нередко скрывают семейные случаи из-за ложного стыда или, наоборот, «открывают» их у родственников супруга, стараясь свалить на них вину за болезнь ребенка.
Для получения семейных сведений можно применять анкетирование. При правильном перечне вопросов и доступности формулировок для понимания членами семьи, не имеющими медицинского образования, анкетирование дает достаточно полную информацию. Очень важно провести личный осмотр и дополнительное обследование родственников больного, если это необходимо. При сборе семейного анамнеза желательно использовать и другие источники медицинской и генеалогической информации (выписки из истории болезни, домовые книги, церковные записи и т.д.).
Подробное клинико-генеалогическое исследование проводится в случае подозрения на наследственную болезнь при первичном клиническом осмотре. Обследование членов семьи должно быть подробным, в отличие от первичных элементов семейного анализа, которые применяются при любом первичном осмотре больного.
Одна из распространенных ошибок в применении генеалогического метода - ограничение анализа только опросом родственников (или о родственниках). Даже подробного опроса, как правило, недостаточно. Некоторые члены родословной часто нуждаются в полном клиническом, параклиническом или лабораторно-генетическом обследовании (цитогенетическом, биохимическом и т.п.), что требует дополнительных расходов. План такого обследования необходимо тщательно рассмотреть с генетической точки зрения в соответствии с принципом «меньше нельзя, а больше не нужно».
Помощь клинико-генеалогического метода в диагностике наследственной патологии очевидна. Так, если в родословной обнаружена наследственная болезнь и анализ показывает возможность ее передачи пробанду, то даже при стертой клинической симптоматике у про-
банда (что и стало причиной подробного генеалогического обследования) можно установить диагноз данной наследственной болезни.
При тематическом целенаправленном применении клиникогенеалогического метода данные лучше регистрировать в таблице (табл. 3.3). Из таблицы 3.3 видно, что у пробанда имеется отягощенность по злокачественным новообразованиям мочевого пузыря.
Таблица 3.3. Табличный способ регистрации генеалогических сведений
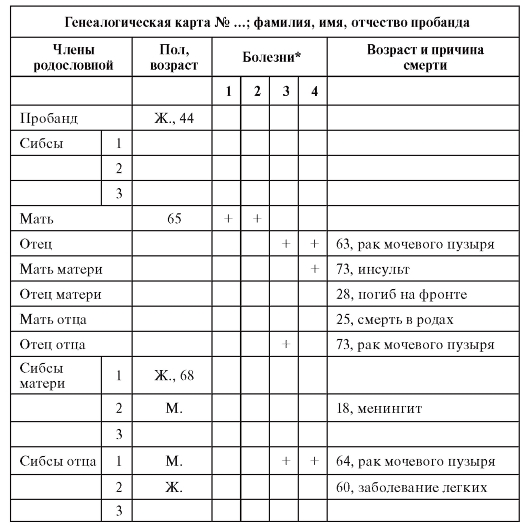
*В данной родословной отмечены такие болезни как: сахарный диабет (1), ишемическая болезнь сердца (2), злокачественные новообразования (3), гипертоническая болезнь (4).
Генеалогический анализ
Первая задача при анализе родословной - установление наследственного характера признака. Если в родословной встречается один и тот же признак (или болезнь) несколько раз, то можно думать о его наследственной природе, но, прежде всего, надо исключить возможность фенокопии. Например, если патогенный фактор действовал на женщину во время всех беременностей, то могут родиться несколько детей с врожденными пороками. Другой пример: одни и те же профессиональные вредности или внешние факторы могут вызывать сходные заболевания у членов одной семьи.
Если исключается действие сходных внешних факторов (для разных поколений оно исключается с большей вероятностью), то говорят о наследственном характере болезни. С помощью генеалогического метода были открыты многие наследственные болезни.
Вторая задача - установление типа наследования, если будет обнаружен наследственный характер признака (болезни). Для этого используют принципы генетического анализа и различные статистические методы обработки данных не из одной, а из многих родословных, что является уже исследовательской задачей.
Нетрудно понять, что в большинстве случаев простое отношение числа больных детей к числу здоровых даст неправильное представление о типе наследования, потому что, например, при рецессивном заболевании в поле зрения врача не попадают семьи-носители, в которых родились только здоровые дети.
Теоретически можно представить полное выявление супружеских пар, гетерозигот по патологическому гену, в том числе имеющих здоровых детей. Практически регистрация всегда начинается от больного потомка. В таком случае невыявленные семьи составляют, например, при одном ребенке и доминантном типе наследования 1/2, а при рецессивном - 3/4 Долю невыявленных гетерозиготных семей можно определить для любого числа детей при различных типах наследования. Следовательно, в расчеты отношения числа больных и здоровых детей нужно вводить поправки на долю невыявленных семей.
Освоение методов количественной оценки сегрегации (расщепления) болезней или дискретных менделирующих признаков в потомстве требует специальной подготовки. Здесь они излагаться не будут.
Определение типа наследования в конкретной родословной - всегда серьезная генетическая задача, хотя на первый взгляд она может показаться довольно легкой. Для решения генетических задач
по анализу родословных врач должен иметь специальную подготовку. Если необходим углубленный клинико-генеалогический анализ, то врач общей практики направляет семью в медико-генетическую консультацию к врачу-генетику. Вместе с тем врачу общей практики надо знать основные критерии разных типов наследования, которые приводятся ниже.
Болезни с аутосомно-доминантным типом наследования
При таком типе наследования для развития болезни достаточно унаследовать мутантный аллель от одного из родителей. Большинство болезней этого типа вызывают такие патологические состояния, которые не наносят серьезного ущерба здоровью и в большинстве случаев не влияют на способность иметь потомство. Родословные таких лиц, особенно описанные в прошлом, когда в браках было много детей, дали возможность установить типичные признаки аутосомнодоминантных форм наследственной патологии (рис. 3.61).
- Болезнь встречается в каждом поколении родословной, что называют передачей болезни по вертикали.
- Соотношение больных и здоровых приближается к 1:1.
- У нормальных детей больных родителей рождаются нормальные дети.
- Число больных мальчиков и девочек равное.
- Больные мужчины и женщины с равной вероятностью передают болезнь своим дочерям и сыновьям.
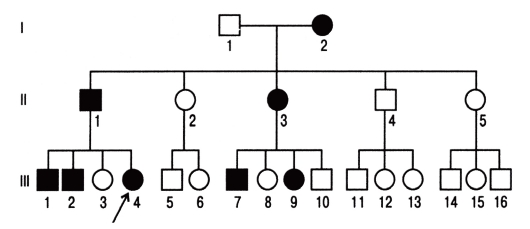
Рис. 3.61. Родословная с аутосомно-доминантным типом наследования болезни (синдром Марфана)
- Чем больше болезнь отражается на репродукции, тем больше доля спорадических случаев (новые мутации).
- Гомозиготы могут рождаться от двух больных родителей. Болезнь у них протекает обычно тяжелее, чем у гетерозигот (так называемое полудоминантное наследование). Доминантно наследуемые состояния имеют полиморфные клинические проявления не только в разных семьях, но и у членов одной семьи. Например, при нейрофиброматозе у одних больных в семье могут быть множественные нейрофибромы, а у других - лишь единичные кожные проявления. Особенность ряда доминантных болезней - высокая вариабельность сроков начала болезни даже в пределах одной семьи. Наглядным примером служит хорея Гентингтона. Распределение больных по возрасту начала болезни описывается нормальным распределением со средним значением 38-40 лет. Объясняется это различным числом унаследованных триплетов (см. ниже).
При тяжелых заболеваниях, когда у больных снижена возможность иметь потомство (сниженная фертильность), родословные не типичны. Если мутация возникает впервые в зародышевых клетках, то родословная показывает спорадический случай.
Наиболее часто встречаются следующие генные болезни с аутосомно-доминантным типом наследования: нейрофиброматоз I типа (болезнь Реклингхаузена), синдромы Марфана, Элерса-Данло, ахондроплазия, несовершенный остеогенез, миотоническая дистрофия, хорея Гентингтона.
Болезни с аутосомно-рецессивным типом наследования
Заболевания с данным типом наследования проявляются только у гомозигот. Гетерозиготы фенотипически (клинически) не отличаются от здоровых людей с двумя нормальными аллелями (рис. 3.62).
При редких аутосомно-рецессивных заболеваниях обычно отмечают следующее.
- Родители обычно клинически здоровы.
- Чем больше детей в семье, тем больше вероятность иметь более одного больного ребенка.
- Чем реже встречается мутантный ген в популяции, тем чаще родители больного ребенка являются кровными родственниками.
- Если больны оба супруга, то все дети будут больными.

Рис. 3.62. Родословная с аутосомно-рецессивным типом наследования болезни (синдром Тея-Сакса -GM2-ганглиозидоз)
- В браке больного со здоровым рождаются нормальные дети (если здоровый не гетерозиготен).
- В браке больного с носителем мутантного аллеля рождается 50% больных детей, что имитирует доминантный тип наследования (псевдодоминирование).
- Оба пола поражаются одинаково.
Браки, в которых оба родителя гетерозиготны, встречаются наиболее часто. Сегрегация потомства соответствует менделевскому соотношению 1 (здоровый): 2 (гетерозиготы): 1 (больной). Риск появления больного ребенка в таком браке составляет 25%. Малодетность современных семей затрудняет установление рецессивного типа наследования болезни. Однако если родители ребенка являются родственниками, то имеется высокая вероятность аутосомно-рецессивного заболевания.
На рис. 3.63 показаны примеры кровнородственных браков.
Пропорция общих генов и коэффициент инбридинга - очень высокие. При оценке генетической ситуации в таких семьях надо принимать во внимание высокую вероятность встречи редких аллелей и, следовательно, гомозиготность, т.е. рецессивное заболевание.
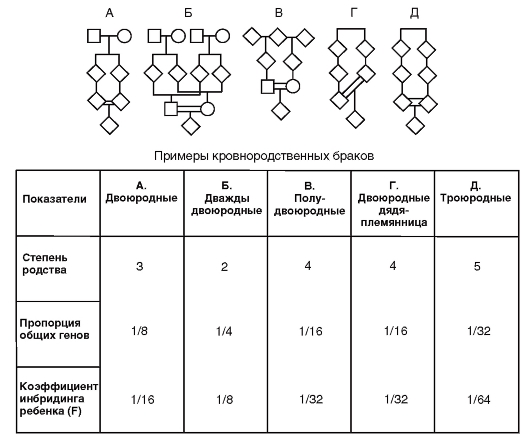
Рис. 3.63. Примеры кровнородственных браков
Браки двух гомозиготных людей очень редки. Естественно, что все дети в этих семьях будут гомозиготами, а потому больными. В тех семьях, в которых у больных родителей (например, альбиносов) рождались здоровые дети, такое несоответствие объясняется мутациями в разных генах. Такие дети являются двойными гетерозиготами.
Браки гетерозигот (здоровых) с гомозиготами (больными) в основном кровнородственные. Соотношение числа больных и здоровых равно 1:1.
Наиболее типичными болезнями с аутосомно-рецессивным типом наследования являются муковисцидоз, фенилкетонурия, галактоземия, гепатолентикулярная дегенерация (болезнь Вильсона-Коновалова), адреногенитальный синдром, мукополисахаридозы .
Болезни с Х-сцепленным доминантным типом наследования
Особенности наследования этих болезней обусловлены тем, что у женщин две Х-хромосомы, а у мужчин одна. Унаследовав от одного из родителей патологический аллель, женщина является гетерозиготой, а мужчина - гемизиготой. Основные характеристики родословных при этом типе наследования следующие (рис. 3.64).
- Поражаются и мужчины, и женщины, но больных женщин в 2 раза больше, чем мужчин.
- Больные женщины передают патологический аллель в среднем 50% сыновей и 50% дочерей.
- Больной мужчина передает патологический аллель всем дочерям и не передает сыновьям, поскольку они получают от отца Y-хромосому.
- В среднем женщины (если они гетерозиготны) болеют менее тяжело, чем мужчины (если они гемизиготны). Болезнь более вариабельна по клиническим проявлениям у гетерозиготных женщин.
По Х-сцепленному доминантному типу наследуется витамин D-резистентный рахит (наследственная гипофосфатемия). Если болезнь тяжелая и летальна у гемизигот (синдром недержания пигмента, ротолицепальцевой синдром, синдром Гольтца-Горлина), то все мальчики погибают. Больными бывают только девочки.
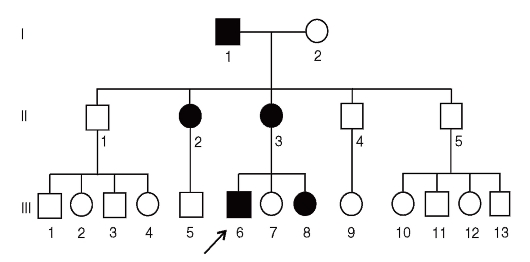
Рис. 3.64. Родословная с Х-сцепленным доминантным типом наследования болезни (витамин D-резистентный рахит)
Болезни с Х-сцепленным рецессивным типом наследования
При редко встречающихся болезнях с этим типом наследования женщины практически всегда гетерозиготны, т.е. они фенотипически нормальны (здоровы) и являются носителями. Больными бывают только мужчины. Характеристики болезней этого типа различаются в зависимости от репродуктивного статуса. Если репродуктивная функция у больных нарушена (мышечная дистрофия Дюшенна-Беккера), то родословные имеют следующие характеристики (рис. 3.65).
- Больные - только мальчики.
- Около 2/3 случаев передается от матерей-носительниц, 1/3 возникает в результате новых мутаций в Х-хромосоме матери.
- В унаследованных случаях у больных мальчиков могут быть больные братья и дяди по матери.
- Новые мутации являются спорадическими случаями.
- Сестры больных братьев при унаследованных случаях имеют 50% вероятность быть носителями патологического аллеля.
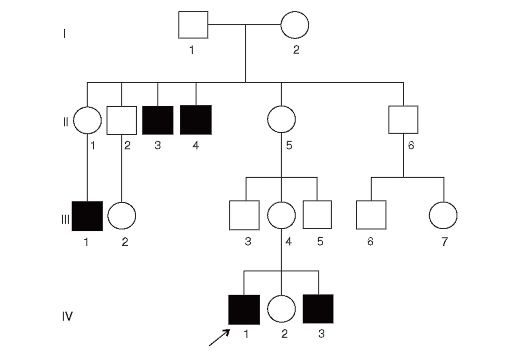
Рис. 3.65. Родословная с Х-сцепленным рецессивным типом наследования болезни (патология нарушает репродуктивную функцию - миодистрофия Дюшенна)
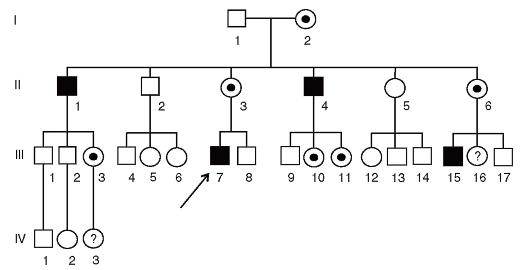
Рис. 3.66. Родословная с Х-сцепленным рецессивным типом наследования болезни (репродукция не нарушена - гемофилия)
- Сестры-носительницы передают ген 50% сыновей (они больны) и 50% дочерей (они носительницы).
- Здоровые мужчины не передают болезни.
Если репродукция при конкретной болезни не нарушена (гемофилия, недостаточность Г6ФДГ), то наследование будет следующим (рис. 3.66).
- Доля унаследованных случаев - более 2/3.
- Больные мужчины передают патологический аллель всем своим дочерям и никому из сыновей.
- Все фенотипически нормальные дочери больных мужчин являются носительницами.
- В браке женщины-носительницы с больным мужчиной среди дочерей 50% - больные, 50% - носительницы, среди сыновей 50% - больные, 50% - здоровые.
- Иногда гетерозиготные женщины могут быть больными в связи со случайной гетерохроматинизацией хромосомы с нормальным аллелем во всех или почти во всех клетках.
К Х-сцепленным рецессивным болезням относятся гемофилия, мышечная дистрофия Дюшенна-Беккера, синдром Хантера (мукополисахаридоз II типа), синдром Леша-Найхана.
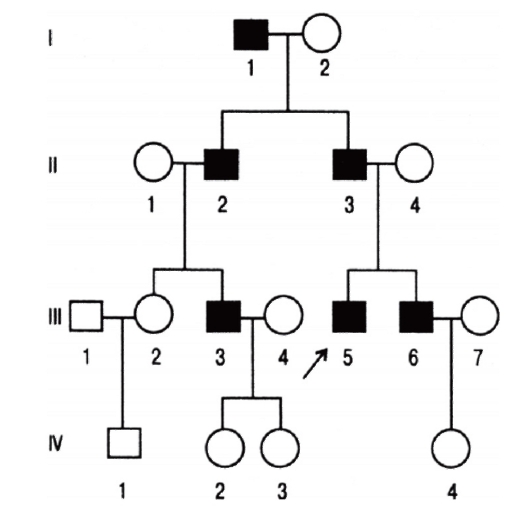
Рис. 3.67. Родословная с Y-сцепленным типом наследования признака (оволосение ушной раковины)
Y-сцепленный тип наследования
Долго полагали, что Y-хромосома содержит только гетерохроматиновые участки (без генов). Новейшие исследования позволили обнаружить и локализовать в Y-хромосоме ряд генов: детерминирующий развитие семенников, отвечающий за сперматогенез (фактор азооспермии), контролирующий интенсивность роста тела, конечностей и зубов, определяющий оволосение ушной раковины. На примере этого признака можно видеть характерные черты Y-сцепленного типа передачи (рис. 3.67).
Признак передается всем мальчикам. Естественно, что патологические мутации, затрагивающие формирование яичек или сперматогенез, наследоваться не могут, потому что такие индивиды стерильны.
Митохондриальная наследственность
Митохондрии передаются с цитоплазмой яйцеклеток. Спермии не имеют митохондрий, поскольку цитоплазма элиминируется при созревании мужских половых клеток. В каждой яйцеклетке содержится около 25 000 митохондрий. Каждая митохондрия имеет кольцевую хромосому. Описаны мутации различных генов митохондрий. Генные мутации в мтДНК обнаружены при атрофии зрительного
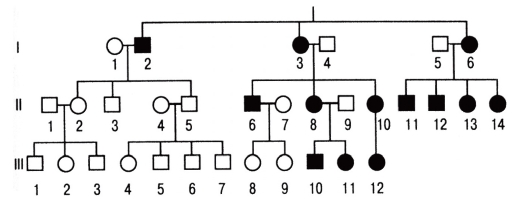
Рис. 3.68. Родословная, иллюстрирующая передачу нейтрального признака через митохондрии (фрагмент ДНК)
нерва Лебера, митохондриальных миопатиях, доброкачественной опухоли (онкоцитоме), прогрессирующих офтальмоплегиях.
Митохондриальная наследственность имеет следующие признаки (рис. 3.68).
- Болезнь передается только от матери.
- Больны и девочки, и мальчики.
- Больные отцы не передают болезнь ни дочерям, ни сыновьям.
СИНДРОМОЛОГИЧЕСКИЙ ПОДХОД К ДИАГНОСТИКЕ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
В наследственной патологии не существует патогномоничных признаков. Один и тот же признак встречается при нескольких или даже при многих формах. Например, деформация грудной клетки в виде воронки или киля бывает не менее чем при 30 наследственных болезнях, искривление позвоночника - более чем при 50 наследственных синдромах. Аномалии почек известны при 30 синдромах. Гипоплазия или дисплазия ногтей может наблюдаться при 25 различных наследственных болезнях. Люди с этими синдромами часто бывают пациентами отделений сердечно-сосудистой хирургии.
Если внимательно осматривать больного, то можно выявить признаки, существенно облегчающие дифференциальную диагностику. Например, у больного с врожденным пороком сердца нужно тщательно осмотреть руки: укорочение I пальца кисти или 3 фаланги
на нем вместо 2 сразу наводит на мысль о доминантно наследуемом синдроме Холта-Орама (или, как его еще называют, синдром «рука-сердце»).
Важное место в диагностике наследственных синдромов или болезней занимает анализ строения лицевой области. Так, резко выступающие надбровные дуги могут быть признаком синдрома фронтометафизарной дисплазии (Х-сцепленная форма остеодисплазии Мелника-Нидлза), а запавшая переносица - мукополисахаридоза или ахондроплазии. Гипертелоризм позволяет заподозрить один из 50-60 наследственных синдромов.
Искривление нижних конечностей - результат не только рахита, как полагали ранее. Оно может быть следствием нарушенного обмена в костях при 25 различных наследственных болезнях.
Большое диагностическое значение имеет осмотр зубов, особенно у молодых людей. Более чем при 20 синдромах наблюдаются изменения зубов: неправильная форма, раннее выпадение, множественный кариес, сверхкомплектность или срастание, врожденное отсутствие резца или клыка. Изменения зубов нередко отмечаются при мукополисахаридозах, синдроме Элерса-Данло, пахионихии и многих других наследственных болезнях.
Умственная отсталость сопровождает многочисленные моногенные болезни (более 700 форм) и большинство хромосомных.
Можно перечислить около 200 внешних симптомов и признаков, которые выявляют при наследственных болезнях без применения специальных дополнительных методов обследования. Однако чтобы выявить эти признаки, их нужно прицельно искать.
Большинство наследственных болезней встречаются редко (1:100 000 и реже). Однако среди больных какого-либо профиля вероятность обнаружения конкретного вида наследственной патологии существенно увеличивается. Так, больных с синдромом Марфана и гомоцистинурией можно встретить в глазных (высокая миопия) и хирургических (деформация грудной клетки) клиниках. Больные низкого роста чаще наблюдаются у эндокринолога; к ортопедам обращаются пациенты с наследственными деформациями костей и суставов и т.д.
Наследственные формы часто встречаются в практике офтальмологов. Атрофия зрительных нервов наблюдается, по крайней мере, при 15, катаракты и помутнения хрусталика - более чем при 30 наследственных болезнях. Птоз, косоглазие, нистагм, помутнение
роговицы, отслойка сетчатки и т.д. - симптомы многих наследственных болезней, распознавание которых улучшается, если врач знает синдромологию наследственных и врожденных болезней.
Хотя наследственных болезней очень много, их представительство в определенных клиниках ограничено небольшим числом форм, как правило, с более легким течением. Освоение этих форм не представляет трудностей для врача любой специальности; нетрудно изучить и симптомы, на основании которых можно заподозрить синдром.
ПАРАКЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ В КЛИНИЧЕСКОЙ ГЕНЕТИКЕ
Проявления наследственных болезней весьма разнообразны по направленности и глубине изменений многих органов и систем, что обусловлено большим числом и тяжестью нозологических форм. В связи с этим параклинические исследования занимают существенное место в диагностике наследственных болезней.
Уже в самом начале XX в., когда генетика человека еще только получила основы для своего развития, английский врач А. Гаррод применил биохимический анализ мочи для диагностики наследственной болезни обмена веществ - алкаптонурии. В 30-х годах ХХ в. норвежский врач И.А. Феллинг открыл метод диагностики фенилкетонурии на основе реакции мочи с хлоридом железа (если в моче есть фенилпировиноградная кислота, появляется сине-зеленая окраска). Однако широкое применение параклинических методов для диагностики наследственных болезней началось с периода интенсивного развития клинической генетики (50 годы XX в.).
В настоящее время для диагностики наследственных болезней и оценки состояния больного используют клинико-биохимические, гематологические, иммунологические, эндокринологические, электрофизиологические, рентгенорадиологические методы. Более углубленные варианты составили отдельную группу лабораторногенетических методов (см. главу 9).
Значение параклинических методов для клинической генетики трудно переоценить, но подробно описать их невозможно. Приведем лишь отдельные примеры их использования в диагностических целях. Клинико-биохимические исследования проводят при муковисцидозе, семейной гиперхолестеринемии, фенил-
кетонурии, болезни Вильсона-Коновалова. Гематологические исследования проводят для подтверждения гемоглобинопатий, наследственного гемохроматоза. Эндокринологические исследования назначают при врожденном гипотиреозе, адреногенитальном синдроме, нанизме. Иммунологические исследования необходимы при первичных иммунодефицитах, атаксии-телеангиэктазии (синдром Луи-Бар). Электрофизиологические исследования проводят при нервно-мышечных заболеваниях, многих наследственных болезнях нервной системы. УЗИ обязательно назначают при врожденных пороках внутренних органов, аномалиях половой дифференцировки. Рентгенорадиологические исследования необходимы при диагностике хондродистрофии, нейрофиброматоза.
КОМПЬЮТЕРНЫЕ ПРОГРАММЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
«Охота» за генами и практическая потребность в диагностике многочисленных форм наследственных болезней и врожденных пороков развития невозможны без сравнительного анализа данных литературы. В связи с этим на протяжении последних 20 лет стали создаваться компьютерные информационные базы данных и диагностические программы. Их назначение - ускорить и объективизировать выбор из множества генетически разнородных, но клинически сходных синдромов и болезней.
Генетическая консультация и помощь при наследственных болезнях возможны при точном диагнозе. Поскольку генетические нарушения редки, большинство врачей и даже медицинских генетиков имеют собственный опыт только по нескольким случаям данной болезни. Следовательно, крайне важно знакомство с описанными в литературе случаями. Из-за генетической гетерогенности надо точно определить нозологическую форму, чтобы родители знали величину риска при повторных деторождениях. Базы данных особенно полезны для дифференциальной диагностики.
Каталоги необходимы для практической работы клинических генетиков. Они обеспечивают информацию или помогают ориентироваться в информации по диагностике, лечению и генетическому консультированию. Поиски по отдельным словам или комбинациям слов дают перечень форм, в которых появляется слово или комбина-
ция слов. Это хорошее начало в поисках диагноза неясной болезни для правильного лечения и консультирования конкретной семьи.
На рисунке 3.69 представлен принцип диагностики с использованием компьютерных программ. Симптомы, выявленные врачом, вводят в компьютер. На их основе осуществляется компьютерный поиск наиболее вероятных диагнозов. После этого врач может обратиться за справкой в базу данных по выбранным диагнозам и получить описание синдрома (или болезни) и даже фотографии больных. Таким образом, врач принимает решение о диагнозе и выбирает способ его верификации, если в этом есть необходимость. В случае, представленном на рис. 3.69, была нужна цитогенетическая верификация. Более подробная информация по интерактивным базам данных представлена на компакт-диске в разделе «Компьютерные базы данных для постановки диагноза наследственного заболевания».

Рис. 3.69. Компьютерная диагностика наследственных болезней
КЛЮЧЕВЫЕ СЛОВА И ПОНЯТИЯ
Врожденные морфогенетические варианты Гамето-, бласто-, эмбрио- и фетопатии Генеалогический анализ Генетические основы морфогенеза
Значение антропометрии в диагностике
Классификация врожденных пороков развития
Клиническая диагностика наследственных болезней
Критические периоды развития
Области применения генеалогического метода
Особенности клиники наследственных болезней
Параклинические методы в диагностике
Плейотропия (первичная и вторичная)
Последовательности и ассоциации
Пренатальная гипоплазия
Пробанд, сибсы, семья, род
Семиотика наследственных болезней
Синдромологический подход
Тератогенез, тератогены
Тератогенный терминационный период
Термин «синдром»
Типы наследования (критерии)
Фенокопии
Характеристика генеалогического метода Эмбриональный дисморфогенез Этиология врожденных пороков развития
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Акуленко Л.В., Богомазов Е.А., Захарова О.М. и др. Медицинская и клиническая генетика для стоматологов. - М.: ГЭОТАР-Медиа, 2008. - 398 с.
Баранов В.С., Кузнецова Т.В. Цитогенетика эмбрионального развития человека: научно-практические аспекты. - СПб.: Изд-во Н-Л,
2007. - 640 с.: ил.
Беляков Ю.А. Наследственные болезни и синдромы в стоматологической практике. - М.: Медицина, 2008. - 238 с.
Иллариошкин С.Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНКдиагностика и медико-генетическое консультирование в неврологии. - М.: МИА, 2002. - 591 с.
Кадурина Т.И., Горбунова В.Н. Дисплазия соединительной ткани. Руководство для врачей. - СПб.: Элби-СПб, 2009. - 704 с.
Козлова С.И., Демикова Н.С. Наследственные синдромы и медикогенетическое консультирование: атлас-справочник. - 3-е изд., доп.
и перераб. - М.: Т-во научных изданий КМК; Авторская академия,
2007. - 448 с.
Лазюк Г.И. Тератология человека. - М.: Медицина, 1979. - 440 с. Мордовцев В.Н., Мордовцева В.В., Мордовцева В.В. Наследственные болезни и пороки развития кожи. Клиника. Морфология. Лечение:
Атлас. - М.: Наука, 2004. - 174 с.
Наследственные болезни нервной системы: руководство / под ред. Ю.Е. Вельтищева, П.А. Темина. - М.: Медицина, 1998. - 496 с.