Клиническая генетика: Учебник. - 3-е изд., испр. и доп. -Н.П. Бочков, 2004. - 480 с.: ил.
|
|
|
|
ГЛАВА 5. ХРОМОСОМНЫЕ БОЛЕЗНИ
Общие вопросы
Хромосомные болезни - это большая группа врожденных наследственных болезней с множественными врожденными пороками развития. В их основе лежат хромосомные или геномные мутации. Эти два разных типа мутаций для краткости объединяют термином «хромосомные аномалии».
Нозологическое выделение по меньшей мере трех хромосомных болезней как клинических синдромов врожденных нарушений развития сделано до установления их хромосомной природы.
Наиболее часто встречающаяся болезнь, трисомия 21, клинически была описана в 1866 г. английским педиатром Л. Дауном и получила название «синдром Дауна». В дальнейшем причина синдрома не раз подвергалась генетическому анализу. Высказывались предположения о доминантной мутации, о врожденной инфекции, о хромосомной природе.
Первое клиническое описание синдрома моносомии по Х-хромосоме как отдельной формы болезни было сделано русским клиницистом Н.А. Шерешевским в 1925 г., а в 1938 г. Г. Тернер также описал этот синдром. По фамилии этих ученых моносомию по Х-хромосоме называют синдромом Шерешевского-Тернера. В зарубежной литературе в основном используют название «синдром Тернера», хотя никто не оспаривает заслугу Н.А. Шерешевского.
Аномалии в системе половых хромосом у мужчин (трисомия XXY) как клинический синдром впервые описал Г. Клайнфелтер в 1942 г.
Перечисленные заболевания стали объектом первых клиникоцитогенетических исследований, проведенных в 1959 г. Расшифровка этиологии синдромов Дауна, Шерешевского-Тернера и Клайнфелтера открыла новую главу в медицине - хромосомные болезни.
В 60-х годах XX века благодаря широкому развертыванию цитогенетических исследований в клинике полностью сложилась клиническая цитогенетика. Была показана роль хромосомных и геномных мутаций в патологии человека, расшифрована хромосомная этиология многих синдромов врожденных пороков разви-
тия, определена частота хромосомных болезней среди новорожденных и при спонтанных абортах.
Наряду с изучением хромосомных болезней как врожденных состояний начались интенсивные цитогенетические исследования в онкологии, особенно при лейкозах. Роль хромосомных изменений в опухолевом росте оказалась очень значимой.
По мере совершенствования цитогенетических методов, особенно таких, как дифференциальная окраска и молекулярная цитогенетика, открывались новые возможности для обнаружения ранее не описанных хромосомных синдромов и установления связи между кариотипом и фенотипом при небольших изменениях хромосом.
В результате интенсивного изучения хромосом человека и хромосомных болезней на протяжении 35-40 лет сложилось учение о хромосомной патологии, которая имеет большое значение в современной медицине. Данное направление в медицине включает не только хромосомные болезни, но и патологию внутриутробного периода (спонтанные аборты, выкидыши), а также соматическую патологию (лейкозы, лучевая болезнь). Число описанных типов хромосомных аномалий приближается к 1000, из них более 100 форм имеют клинически очерченную картину и называются синдромами. Диагностика хромосомных аномалий необходима в практике врачей разных специальностей (генетик, акушер-гинеколог, педиатр, невропатолог, эндокринолог и др.). Во всех многопрофильных современных больницах (более 1000 коек) в развитых странах имеются цитогенетические лаборатории.
Этиология и классификация
Этиологическими факторами хромосомной патологии являются все виды хромосомных мутаций и некоторые геномные мутации. Хотя геномные мутации в животном и растительном мире многообразны, у человека обнаружены только 3 типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Из всех вариантов анеуплоидий встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий встречается только моносомия Х.
Что касается хромосомных мутаций, то у человека обнаружены все их типы (делеции, дупликации, инверсии, транслокации). С
клинико-цитогенетической точки зрения делеция в одной из гомологичных хромосом означает нехватку участка или частичную моносомию по этому участку, а дупликация - избыток или частичную трисомию. Современные методы молекулярной цитогенетики позволяют выявлять мелкие делеции на уровне гена. Таким образом, стирается грань между генной и хромосомной патологией.
Если транслокация реципрокная (взаимная) без потери участков вовлеченных в нее хромосом, то она называется сбалансированной.
Как и инверсия, она не дает патологических эффектов у носителя. Однако в результате сложных механизмов кроссинговера и редукции числа хромосом при образовании гамет у носителей сбалансированных транслокаций и инверсий могут образовываться несбалансированные гаметы, т.е. гаметы с частичной дисомией или с частичной нуллисомией либо с той и другой аномалией из разных участков (в норме каждая гамета моносомна).
Транслокация между двумя акроцентрическими хромосомами с потерей их коротких плеч приводит к образованию одной метаили субметацентрической хромосомы вместо двух акроцентрических. Такие транслокации называются робертсоновскими. Формально их носители имеют моносомию по коротким плечам двух акроцентрических хромосом. Однако такие носители здоровы, потому что потеря коротких плеч двух акроцентрических хромосом компенсируется работой таких же генов в остальных 8 акроцентрических хромосомах. У носителей робертсоновских транслокаций может образовываться 6 типов гамет (рис. 5.1), но нуллисомные гаметы должны приводить к моносомии по аутосомам в зиготе, а такие зиготы не развиваются.
Клиническая картина простых и транслокационных форм трисомии по акроцентрическим хромосомам одинаковая.
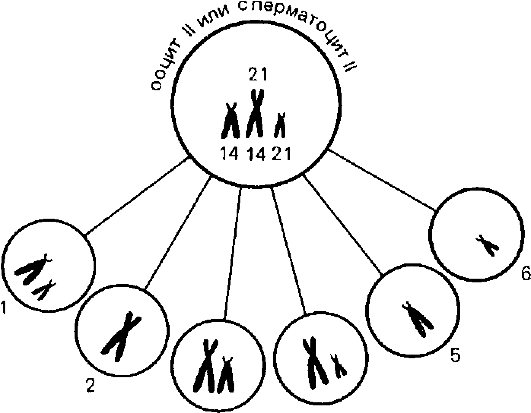 Рис. 5.1. Типы гамет у носителей робертсоновской транслокации 21/14.
Рис. 5.1. Типы гамет у носителей робертсоновской транслокации 21/14.
1 - моносомия 14 и 21 (норма); 2 - моносомия 14 и 21 с робертсоновской транслокацией; 3 - дисомия 14 и моносомия 21; 4 - дисомия 21, моносомия 14; 5 - нуллисомия 21; 6 - нуллисомия 14.
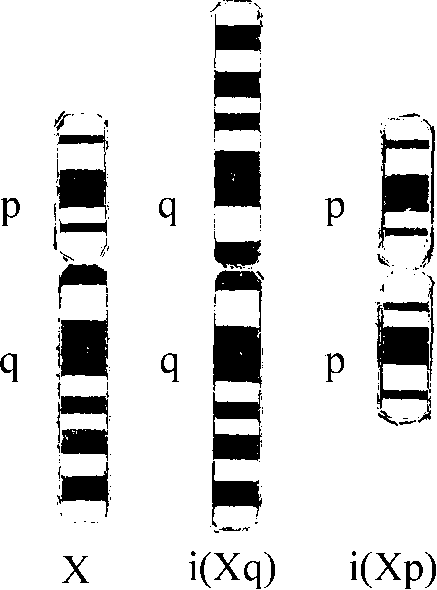 Рис. 5.2. Изохромосомы Х по длинному [i(Xq)] и короткому [i(Xp)] плечу.
Рис. 5.2. Изохромосомы Х по длинному [i(Xq)] и короткому [i(Xp)] плечу.
В случае концевых делеций в обоих плечах хромосомы возникает кольцевая хромосома. У индивида, унаследовавшего кольцевую хромосому от одного из родителей, будет частичная моносомия по двум концевым участкам хромосомы.
Иногда разрыв хромосомы проходит через центромеру. Каждое плечо, разъединенное после репликации, имеет две сестринские хроматиды, соединенные оставшейся частью центромеры. Сестринские хроматиды одного и того же плеча становятся плечами одной хромосомы (рис. 5.2). Со следующего митоза эта хромосома начинает реплицироваться и передаваться из клетки в клетку как само-
стоятельная единица наряду с остальным набором хромосом. Такие хромосомы называют изохромосомами. У них одинаковые по набору генов плечи. Каков бы ни был механизм образования изохромосом (он еще полностью не выяснен), их наличие вызывает хромосомную патологию, потому что это одновременно и частичная моносомия (по отсутствующему плечу), и частичная трисомия (по присутствующему плечу).
Недавно у человека обнаружено явление однородительских дисомий. У таких индивидов число хромосом по всем парам нормальное, но одна пара представлена хромосомами от одного родителя. В основе этого явления могут лежать несколько механизмов: нуллисомная по определенной хромосоме набора гамета сливается с дисомной по этой же хромосоме другой гаметой; у первоначально трисомного по какой-либо хромосоме зародыша (или даже в зиготе) теряется единственная хромосома, происходящая от одного из родителей, а две хромосомы от другого родителя сохраняются (редукция трисомии); у моносомной по какой-либо хромосоме зиготы при митозах в процессе дробления данная хромосома дублируется и воспроизводится в последующих делениях в двойном от одного родителя наборе (постзиготическая дупликация моносомии).
В основе классификации хромосомной патологии лежат 3 принципа, позволяющие точно охарактеризовать форму хромосомной патологии и ее варианты у обследуемого.
Первый принцип - характеристика хромосомной или геномной мутации (триплоидия, простая трисомия по хромосоме 21, частичная моносомия и т.д.) с учетом конкретной хромосомы. Этот принцип можно назвать этиологическим.
Клиническая картина хромосомной патологии определяется типом геномной или хромосомной мутации, с одной стороны, и индивидуальной хромосомой - с другой. Нозологическое подразделение хромосомной патологии основывается, таким образом, на этиологическом и патогенетическом принципе: для каждой формы хромосомной патологии устанавливается, какая структура вовлечена в патологический процесс (хромосома, сегмент) и в чем состоит генетическое нарушение (недостаток или избыток хромосомного материала). Дифференциация хромосомной патологии на основании клинической картины не имеет существенного значения, поскольку разным хромосомным аномалиям свойственна большая общность нарушений развития.
Второй принцип - определение типа клеток, в которых возникла мутация (в гаметах или зиготе). Гаметические мутации ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию.
Если хромосомная аномалия возникает в зиготе или на ранних стадиях дробления (такие мутации называют соматическими, в отличие от гаметических), то развивается организм с клетками разной хромосомной конституции (два типа и более). Такие формы хромосомных болезней называют мозаичными.
Для возникновения мозаичных форм, по клинической картине совпадающих с полными формами, нужно не менее 10% клеток с аномальным набором.
Третий принцип - это выявление поколения, в котором возникла мутация: возникла она заново в гаметах здоровых родителей (спорадические случаи) или родители уже имели такую аномалию (наследуемые, или семейные, формы).
О наследуемых хромосомных болезнях говорят тогда, когда мутация имеется в клетках родителя, в том числе и в гонадах. Это могут быть и случаи трисомии. Например, у индивидов с синдромами Дауна и трипло-Х образуются нормальные и дисомные гаметы. Такое происхождение дисомных гамет - следствие вторичного нерасхождения, т.е. нерасхождения хромосом у индивида с трисомией. Большая часть наследуемых случаев хромосомных болезней связана с робертсоновскими транслокациями, сбалансированными реципрокными транслокациями между двумя (реже более)
хромосомами и инверсиями у здоровых родителей. Клинически значимые хромосомные аномалии в этих случаях возникли в связи со сложными перестройками хромосом в процессе мейоза (конъюгация, кроссинговер).
Таким образом, для точной диагностики хромосомной болезни необходимо определить: 1) тип мутации; 2) вовлеченную в процесс хромосому; 3) форму (полная или мозаичная); 4) встречаемость в родословной - спорадический или наследуемый случай. Такая диагностика возможна только при цитогенетическом обследовании пациента, а иногда и его родителей и сибсов.
Эффекты хромосомных аномалий в онтогенезе
Хромосомные аномалии вызывают нарушение общего генетического баланса, той скоординированности в работе генов и системности регуляции, которые сложились в процессе эволюции каждого вида. Неудивительно, что патологические эффекты хромосомных и геномных мутаций проявляются на всех стадиях онтогенеза и, возможно, даже на уровне гамет, влияя на их формирование (особенно у мужчин).
Изучение первичных эффектов хромосомных аномалий началось в начале 60-х годов вскоре после открытия хромосомных болезней и продолжается до сих пор. Главные эффекты хромосомных аномалий проявляются в двух связанных между собой вариантах: летальности и врожденных пороках развития.
Летальность. Имеются убедительные свидетельства того, что патологические эффекты хромосомных аномалий начинают проявляться уже со стадии зиготы. Летальность - один из главных факторов внутриутробной гибели, достаточно высокой у человека.
Выявить количественный вклад хромосомных аномалий в гибель зигот и бластоцист (первые 2 нед после оплодотворения) в полной мере трудно, поскольку в этот период беременность ни клинически, ни лабораторно еще не диагностируется. Однако некоторые прямые исследования бластоцист и результаты экстраполяций позволяют предположить, что 30-40% оплодотворенных яйцеклеток погибает на стадии зиготы - бластоцисты, т.е. до имплантации и, следовательно, до лабораторного или клинического установления беременности. В этих случаях происходит резкое нарушение ранних морфогенетических процессов (до гаструляции и
формирования зародышевых листков). Такие случаи ранней остановки развития можно объяснить тем, что нарушение геномного баланса вследствие развития какой-то определенной формы хромосомной аномалии приводит к дискоординации включения и выключения генов на соответствующей стадии развития (временной фактор) или в соответствующем месте бластоцисты (пространственный фактор). Это вполне понятно: поскольку в процессах развития на ранних стадиях участвуют примерно 1000 генов, локализованных во всех хромосомах, хромосомная аномалия нарушает взаимодействие генов и инактивирует какие-то конкретные процессы развития (межклеточные взаимодействия, дифференцировка клеток и др.).
Многочисленные цитогенетические исследования материала спонтанных абортов, выкидышей и мертворожденных позволяют объективно судить об эффектах разных типов хромосомных аномалий во внутриутробном периоде индивидуального развития. Летальный или дизморфогенетический эффект хромосомных аномалий обнаруживается на всех стадиях внутриутробного онтогенеза (имплантация, эмбриогенез, органогенез, рост и развитие плода). Суммарный вклад хромосомных аномалий во внутриутробную гибель (после имплантации) у человека составляет 45%. При этом чем раньше прерывается беременность, тем вероятнее, что это обусловлено аномалиями развития эмбриона, вызванными хромосомным дисбалансом. У 2-4-недельных абортусов (эмбрион и его оболочки) хромосомные аномалии обнаруживают в 60-70% случаев. В I триместре гестации хромосомные аномалии встречаются у 50% абортусов. У плодов-вькидышей II триместра такие аномалии находят в 25-30% случаев, а у плодов, погибших после 20 нед гестации,- в 7% случаев.
Среди перинатально погибших плодов частота хромосомных аномалий составляет 6%.
Наиболее тяжелые формы по дисбалансу хромосомного набора встречаются у ранних абортусов. Это полиплоидии (25%), полные трисомии по аутосомам (50%). Трисомии по некоторым аутосомам (1; 5; 6; 11; 19) встречаются крайне редко даже у элиминированных эмбрионов и плодов, что свидетельствует о большой морфогенетической значимости этих аутосом. Данные аномалии прерывают развитие в доимплантационном периоде или нарушают гаметогенез.
Высокая морфогенетическая значимость аутосом еще более отчетлива при полных аутосомных моносомиях. Последние редко
обнаруживаются даже в материале ранних спонтанных абортов из-за летального эффекта такого дисбаланса.
Врожденные пороки развития. Если хромосомная аномалия не дает летального эффекта на ранних стадиях развития, то ее последствия проявляются в виде врожденных пороков развития. Практически все хромосомные аномалии (кроме сбалансированных) приводят к врожденным порокам развития, сочетания которых известны как нозологические формы хромосомных болезней и синдромов.
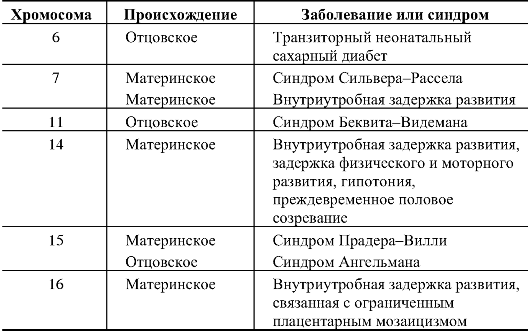
Эффекты однородительских дисомий. Явление однородительских дисомий небезразлично для индивида. Во-первых, может присходить гомозиготизация по рецессивным патологическим генам, т.е. болезнь будет получена от одного родителя. Во-вторых, по некоторым хромосомам однородительские дисомии приводят к синдромам или внутриутробной задержке роста плода в связи с импринтингом локуса в отцовской или материнской хромосоме (моноаллельная, а не биаллельная экспрессия). Примеры влияния однородительских дисомий на развитие индивида приведены в табл. 5.1.
Таблица 5.1. Примеры однородительских дисомий человека, приводящих к аномалиям фенотипа
 Эффекты хромосомных аномалий в соматических клетках. Роль
хромосомных и геномных мутаций не ограничивается их влиянием на
развитие патологических процессов в ранних периодах онтогенеза
(незачатие, спонтанный аборт, мертворождение, хромосомная болезнь). Их
эффекты прослеживаются в течение всей жизни.
Эффекты хромосомных аномалий в соматических клетках. Роль
хромосомных и геномных мутаций не ограничивается их влиянием на
развитие патологических процессов в ранних периодах онтогенеза
(незачатие, спонтанный аборт, мертворождение, хромосомная болезнь). Их
эффекты прослеживаются в течение всей жизни.
Хромосомные аномалии, возникающие в соматических клетках в постнатальном периоде, могут вызывать различные последствия: остаться нейтральными для клетки, обусловить гибель клетки, активировать деление клетки, изменить функцию. Хромосомные аномалии возникают в соматических клетках постоянно с невысокой частотой (около 2%). В норме такие клетки элиминируются иммунной системой, если они проявляют себя чужеродно. Однако в некоторых случаях (активация онкогенов при транслокациях, делециях) хромосомные аномалии становятся причиной злокачественного роста. Например, транслокация между хромосомами 9 и 22 вызывает миелолейкоз. Облучение и химические мутагены индуцируют хромосомные аберрации. Такие клетки гибнут, что наряду с действием других факторов способствует развитию лучевой болезни, аплазии костного мозга. Имеются экспериментальные доказательства накопления клеток с хромосомными аберрациями в процессе старения.
Патогенез
Несмотря на хорошую изученность клиники и цитогенетики хромосомных болезней, их патогенез даже в общих чертах еще неясен. Не разработана общая схема развития сложных патологических процессов, обусловленных хромосомными аномалиями и приводящих к появлению сложнейших фенотипов хромосомных болезней. Ключевое звено в развитии хромосомной болезни ни при одной форме не выявлено. Некоторые авторы предполагают, что это звено - несбалансированность генотипа или нарушение общего генного баланса. Однако такое определение ничего конструктивного не дает. Несбалансированность генотипа - условие, а не звено патогенеза, она должна реализовываться через какие-то специфические биохимические или клеточные механизмы в фенотип (клиническую картину) болезни.
Систематизация данных о механизмах нарушений при хромосомных болезнях показывает, что при любых трисомиях и частич-
ных моносомиях можно выделить 3 типа генетических эффектов: специфические, полуспецифические и неспецифические.
Специфические эффекты должны быть связаны с изменением числа структурных генов, кодирующих синтез белка (при трисомии их число увеличивается, при моносомии уменьшается). Многочисленные попытки найти специфические биохимические эффекты подтвердили это положение лишь для немногих генов или их продуктов. При трисомии 21 обнаружено 50% повышение активности супероксиддисмутазы (ген локализован в 21-й хромосоме). Подобный «эффект дозы гена» выявлен для нескольких десятков генов при трисомиях по разным хромосомам.
Однако биохимическое изучение фенотипа хромосомных болезней пока не привело к пониманию путей патогенеза, возникающих вследствие хромосомных аномалий врожденных нарушений морфогенеза в широком смысле слова. Обнаруженные биохимические отклонения пока трудно связать с фенотипическими характеристиками болезней на органном и системном уровнях. Изменение числа аллелей гена не всегда вызывает пропорциональное изменение продукции соответствующего белка. При хромосомной болезни всегда существенно меняется активность других ферментов или количество белков, гены которых локализованы на не вовлеченной в дисбаланс хромосоме. Ни в одном случае не обнаружено белка-маркера при хромосомных болезнях.
Полуспецифические эффекты при хромосомных болезнях могут быть обусловлены изменением числа генов, и в норме представленных в виде многочисленных копий. К таким генам относятся гены рибосомных и транспортных РНК, гистоновых и рибосомных белков, сократительных белков актина и тубулина. Эти белки в норме контролируют ключевые этапы метаболизма клетки, процессов ее деления, межклеточных взаимодействий. Каковы фенотипические эффекты дисбаланса этой группы генов, как компенсируется их недостаток или избыток, пока неизвестно.
Неспецифические эффекты хромосомных аномалий связывают с измененным содержанием гетерохроматина в клетке. Важная роль гетерохроматина в клеточных делениях, клеточном росте и других биологических функциях не вызывает сомнений. Таким образом, неспецифические и частично полуспецифические эффекты приближают нас к клеточным механизмам патогенеза,
безусловно, играющим важнейшую роль при врожденных пороках развития.
Большой фактический материал позволяет провести сопоставление клинического фенотипа болезни с цитогенетическими изменениями (фенокариотипические корреляции).
Общее для всех форм хромосомных болезней - множественность поражения. Это черепно-лицевые дизморфии, врожденные пороки развития внутренних и наружных органов, замедленные внутриутробные и постнатальные рост и развитие, отставание психического развития, нарушения функций нервной, эндокринной и иммунной систем. При каждой форме хромосомных болезней наблюдается 30-80 различных отклонений, перекрывающих формы. Лишь небольшое число хромосомных болезней проявляется только определенным сочетанием отклонений в развитии, но не специфическими пороками, что и используют в клинической и патологоанатомической диагностике.
Патогенез хромосомных болезней развертывается в раннем внутриутробном и продолжается в постнатальном периоде. Множественные врожденные пороки развития как главное фенотипическое проявление хромосомных болезней формируются в раннем эмбриогенезе, поэтому к периоду постнатального онтогенеза все основные пороки развития уже налицо (кроме пороков развития половых органов). Раннее и множественное поражение систем организма объясняет некоторую общность клинической картины разных хромосомных болезней.
Фенотипическое проявление хромосомных аномалий, т.е. формирование клинической картины, зависит от следующих главных факторов: 1) индивидуальности вовлеченной в аномалию хромосомы или ее участка (специфический набор генов); 2) типа аномалии (трисомия, моносомия; полная, частичная); 3) размера недостающего (при делеции) или избыточного (при частичной трисомии) материала; 4) степени мозаичности организма по аберрантным клеткам; 5) генотипа организма; 6) условий среды (внутриутробная или постнатальная).
Степень отклонений в развитии организма зависит от качественной и количественной характеристики унаследованной хромосомной аномалии. При исследовании клинических данных у человека полностью подтверждается доказанная у других видов относительно невысокая биологическая ценность гетерохроматиновых районов хромосом. Полные трисомии у живорожденных наблюдаются только по аутосомам, богатым
гетерохроматином (8; 9; 13; 18; 21). Так же объясняется полисомия (до пентасомии) по половым хромосомам, в которой Y-хромосома имеет мало генов, а добавочные Х-хромосомы бывают гетерохроматинизированы.
Клиническое сопоставление полных и мозаичных форм болезни показывает, что мозаичные формы протекают в среднем легче. По-видимому, это объясняется присутствием нормальных клеток, частично компенсирующих генетический дисбаланс. В индивидуальном прогнозе прямой связи тяжести течения заболевания и соотношения аномальных и нормальных клонов не обнаруживается.
По мере изучения фено- и кариотипических корреляций при разных протяженностях хромосомной мутации выясняется, что наиболее специфичные для того или иного синдрома проявления обусловлены отклонениями в содержании сравнительно небольших сегментов хромосом. Дисбаланс по значительному объему хромосомного материала делает клиническую картину более неспецифичной. Так, специфические клинические симптомы синдрома Дауна проявляются при трисомии по сегменту длинного плеча хромосомы 21q22.1. Для развития синдрома «кошачьего крика» при делециях короткого плеча аутосомы 5 наиболее важна средняя часть сегмента (5р15). Характерные черты синдрома Эдвардса связаны с трисомией сегмента хромосомы 18q11.
Каждой хромосомной болезни свойствен клинический полиморфизм, в общей форме обусловленный генотипом организма и условиями среды. Вариации в проявлениях патологии могут быть очень широкими: от летального эффекта до незначительных отклонений в развитии. Так, 60-70% случаев трисомии 21 заканчиваются гибелью во внутриутробном периоде, в 30% случаев рождаются дети с синдромом Дауна, имеющим очень различные клинические проявления. Моносомия по Х-хромосоме среди новорожденных (синдром Шерешевского-Тернера) - это 10% всех моносомных по Х-хромосоме зародышей (остальные погибают), а если учитывать еще доимплантационную гибель зигот Х0, то живорожденные с синдромом Шерешевского-Тернера составляют только 1%.
Несмотря на недостаточное понимание закономерностей патогенеза хромосомных болезней в целом, некоторые звенья общей цепи событий в развитии отдельных форм уже известны и их количество постоянно увеличивается.
Клинико-цитогенетические характеристики наиболее распространенных хромосомных болезней
Синдром Дауна
Синдром Дауна, трисомия 21, - наиболее изученная хромосомная болезнь. Частота синдрома Дауна среди новорожденных равна 1:700-1:800, не имеет какой-либо временной, этнической или географической разницы при одинаковом возрасте родителей. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца (рис. 5.3).
С возрастом существенно увеличивается вероятность рождения детей с синдромом Дауна. Так, у женщин в возрасте 45 лет она составляет около 3%. Высокая частота детей с синдромом Дауна (около 2%) наблюдается у рано рожающих женщин (до 18 лет). Следовательно, для популяционных сравнений частоты рождения детей с синдромом Дауна надо принимать во внимание распреде-
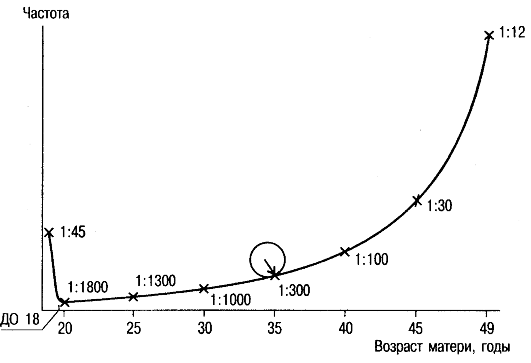 Рис. 5.3. Зависимость частоты рождения детей с синдромом Дауна от возраста матери.
Рис. 5.3. Зависимость частоты рождения детей с синдромом Дауна от возраста матери.
ление рожающих женщин по возрасту (доля женщин, рожающих после 30-35 лет, в общем числе рожающих). Это распределение иногда меняется в течение 2-3 лет для одного и того же населения (например, при резком изменении экономической ситуации в стране). Рост частоты синдрома Дауна с увеличением материнского возраста известен, но большинство детей с синдромом Дауна все-таки рождены матерями моложе 30 лет. Это связано с большим числом беременностей в этой возрастной группе по сравнению с женщинами более старшего возраста.
В литературе описана «пучковость» рождения детей с синдромом Дауна в определенные промежутки времени в некоторых странах (городах, провинциях). Эти случаи можно объяснить скорее стохастическими колебаниями спонтанного уровня нерасхождения хромосом, чем воздействием предполагаемых этиологических факторов (вирусная инфекция, низкие дозы радиации, хлорофос).
Цитогенетические варианты синдрома Дауна разнообразны. Однако основную долю (94-95%) составляют случаи простой полной трисомии 21 вследствие нерасхождения хромосом в мейозе. Вклад материнского нерасхождения в эти гаметические формы болезни составляет 80-90%, а отцовского - только 10-20%. Причины такой разницы неясны. Около 2% детей с синдромом Дауна имеют мозаичные формы (47+21/46). Примерно 3-4% больных с синдромом Дауна имеют транслокационную форму трисомии по типу робертсоновских транслокаций между акроцентриками (D/21 и G/21). Почти 50% транслокационных форм наследуются от родителей-носителей, а 50% транслокаций возникают de novo.
Соотношение мальчиков и девочек с синдромом Дауна составляет 1:1.
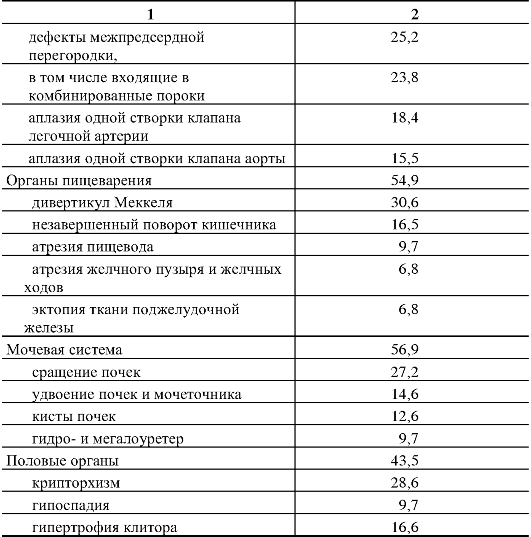
Клиническая симптоматика синдрома Дауна разнообразна: это и врожденные пороки развития, и нарушения постнатального развития нервной системы, и вторичный иммунодефицит и т.п. Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8-10% ниже средних величин). Многие симптомы синдрома Дауна заметны уже при рождении и в последующем проявляются более четко. Квалифицированный педиатр устанавливает правильный диагноз синдрома Дауна в родильном доме не менее чем в 90% случаев. Из черепно-лицевых дизморфий отмечаются монголоидный разрез глаз (по этой причине синдром Дауна долго называли монголоидизмом), брахицефалия, круглое уплощенное лицо, плоская спинка носа, эпикант, крупный (обычно высунутый) язык,
деформированные ушные раковины (рис. 5.4). Мышечная гипотония сочетается с разболтанностью суставов (рис. 5.5). Часто встречаются врожденный порок сердца, клинодактилия, типичные изменения дерматоглифики [четырехпальцевая, или «обезьянья», складка на ладони (рис. 5.6), две кожные складки вместо трех на мизинце, высокое положение трирадиуса и др.]. Пороки ЖКТ наблюдаются редко.
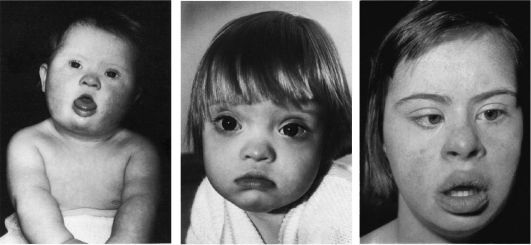 Рис. 5.4. Дети
разного возраста с характерными чертами синдрома Дауна (брахицефалия,
круглое лицо, макроглоссия и открытый рот, эпикант, гипертелоризм,
широкая переносица, «карпий рот», косоглазие).
Рис. 5.4. Дети
разного возраста с характерными чертами синдрома Дауна (брахицефалия,
круглое лицо, макроглоссия и открытый рот, эпикант, гипертелоризм,
широкая переносица, «карпий рот», косоглазие).
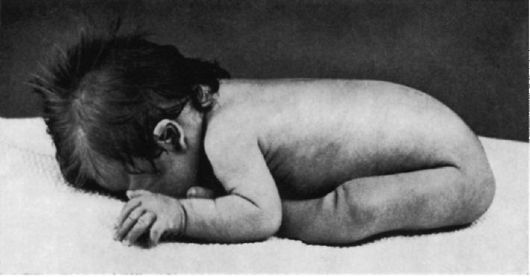 Рис. 5.5. Резкая гипотония у пациента с синдромом Дауна.
Рис. 5.5. Резкая гипотония у пациента с синдромом Дауна.
 Рис. 5.6. Ладони взрослого мужчины с синдромом Дауна (усиленная морщинистость, на левой руке четырехпальцевая, или «обезьянья», складка).
Рис. 5.6. Ладони взрослого мужчины с синдромом Дауна (усиленная морщинистость, на левой руке четырехпальцевая, или «обезьянья», складка).
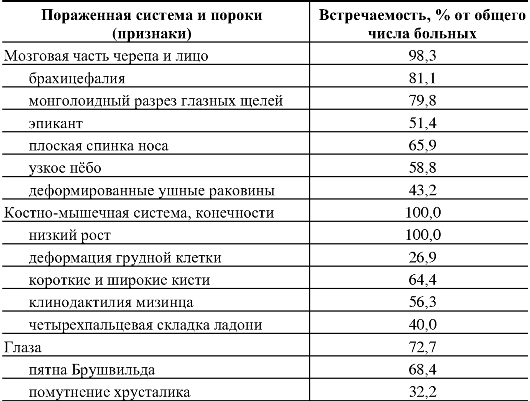
Какие-либо симптомы, кроме низкого роста, выявляются не у всех больных. В табл. 5.2 и 5.3 представлена частота внешних признаков и основных врожденных пороков внутренних органов при синдроме Дауна.
Таблица 5.2. Наиболее частые внешние признаки синдрома Дауна (по Г.И. Лазюку, с дополнениями)
 Диагноз
синдрома Дауна устанавливают на основании сочетания нескольких
симптомов. Следующие 10 признаков наиболее важны для установления
диагноза, наличие 4-5 из них достоверно указывает на синдром Дауна: 1)
уплощение профиля лица (90%); 2) отсутствие сосательного рефлекса (85%);
3) мышечная гипотония (80%); 4) монголоидный разрез глазных щелей
(80%); 5) избыток кожи на шее (80%); 6) разболтанность суставов (80%);
7) диспластичный таз (70%); 8) диспластичные (деформированные) ушные
раковины (60%); 9) клинодактилия мизинца (60%); 10) четырехпальцевая
сгибательная складка (поперечная линия) ладони (45%). Большое значение
для диагностики имеет динамика физического и умственного разви-
Диагноз
синдрома Дауна устанавливают на основании сочетания нескольких
симптомов. Следующие 10 признаков наиболее важны для установления
диагноза, наличие 4-5 из них достоверно указывает на синдром Дауна: 1)
уплощение профиля лица (90%); 2) отсутствие сосательного рефлекса (85%);
3) мышечная гипотония (80%); 4) монголоидный разрез глазных щелей
(80%); 5) избыток кожи на шее (80%); 6) разболтанность суставов (80%);
7) диспластичный таз (70%); 8) диспластичные (деформированные) ушные
раковины (60%); 9) клинодактилия мизинца (60%); 10) четырехпальцевая
сгибательная складка (поперечная линия) ладони (45%). Большое значение
для диагностики имеет динамика физического и умственного разви-
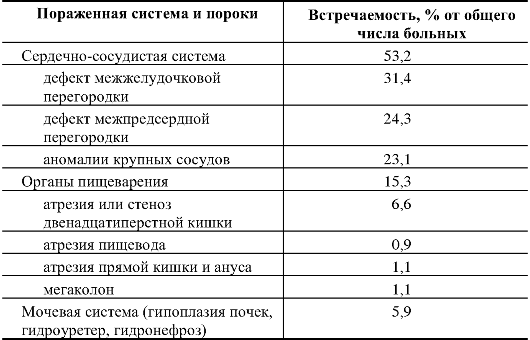
Таблица 5.3. Основные врожденные пороки внутренних органов при синдроме Дауна (по Г.И. Лазюку, с дополнениями)
 тия
ребенка - при синдроме Дауна оно задерживается. Рост взрослых больных
на 20 см ниже среднего. Задержка умственного развития может достигать
уровня имбецильности без специальных методов обучения. Дети с синдромом
Дауна ласковые, внимательные, послушные, терпеливые при обучении.
Коэффициент умственного развития (англ. IQ) у разных детей может
составлять от 25 до 75.
тия
ребенка - при синдроме Дауна оно задерживается. Рост взрослых больных
на 20 см ниже среднего. Задержка умственного развития может достигать
уровня имбецильности без специальных методов обучения. Дети с синдромом
Дауна ласковые, внимательные, послушные, терпеливые при обучении.
Коэффициент умственного развития (англ. IQ) у разных детей может
составлять от 25 до 75.
Реакция детей с синдромом Дауна на воздействия окружающей среды часто патологическая в связи со слабым клеточным и гуморальным иммунитетом, снижением репарации ДНК, недостаточной выработкой пищеварительных ферментов, ограниченными компенсаторными возможностями всех систем. По этой причине дети с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела, выражен гиповитаминоз.
Врожденные пороки внутренних органов, сниженная приспособляемость детей с синдромом Дауна часто приводят к смерти в первые 5 лет. Следствием измененного иммунитета и недостаточности репарационных систем (для поврежденной ДНК) являются лейкозы, часто возникающие у больных с синдромом Дауна.
Дифференциальная диагностика проводится с врожденным гипотиреозом, другими формами хромосомных аномалий. Цитогенетическое обследование детей показано не только при подозрении на синдром Дауна, но и при клинически установленном диагнозе, поскольку цитогенетическая характеристика пациента необходима для прогноза здоровья будущих детей у родителей и их родственников.
Этические проблемы при синдроме Дауна многоплановы. Несмотря на повышение риска рождения ребенка с синдромом Дауна и другими хромосомными синдромами, врач должен избегать прямых рекомендаций по ограничению деторождения у женщин старшей возрастной группы, так как риск по возрасту остается достаточно низким, особенно с учетом возможностей пренатальной диагностики.
Неудовлетворенность у родителей часто вызывает форма сообщения врачом о диагнозе синдрома Дауна у ребенка. Диагностировать синдром Дауна по фенотипическим признакам обычно можно сразу после родоразрешения. Врач, пытающийся отказаться от установления диагноза до исследования кариотипа, может потерять уважение родственников ребенка. Важно сообщить родителям как можно скорее после рождения ребенка, по крайней мере о ваших подозрениях, но не следует полностью информировать родителей ребенка о диагнозе. Нужно дать достаточно сведений, отвечая на непосредственные вопросы, и поддерживать контакт с родителями до того дня, когда станет возможным более детальное обсуждение. Немедленная информация должна включать объяснение этиологии синдрома для исключения взаимных обвинений супругов и описание исследований и процедур, необходимых для того, чтобы полностью оценить здоровье ребенка.
Полное обсуждение диагноза нужно провести, как только родильница более или менее оправится от стресса родоразрешения, обычно в 1-е сутки после родов. К этому времени у матерей возникает множество вопросов, на которые необходимо отвечать точно и определенно. Важно приложить все усилия, чтобы на этой встрече присутствовали оба родителя. Ребенок становится предметом непосредственного обсуждения. В этот период еще рано нагружать родителей всей информацией о заболевании, так как новые и сложные понятия требуют времени для осмысления.
Не пытайтесь давать прогнозы. Бесполезно пытаться точно предвидеть будущее любого ребенка. Древние мифы вроде: «По крайней мере он будет всегда любить и наслаждаться музыкой» - непростительны. Нужно представить картину, написанную широкими мазка-
ми, и отметить, что способности каждого ребенка развиваются индивидуально.
90% детей с синдромом Дауна, рожденных в России, родители оставляют на попечение государства. Родители (а часто и педиатры) не знают, что при правильном обучении такие дети могут стать полноценными членами общества.
Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врожденные пороки сердца устраняются оперативно. Постоянно проводится общеукрепляющее лечение. Питание должно быть полноценным. Необходимы внимательный уход за больным ребенком, защита от действия вредных факторов окружающей среды (простуда, инфекции). Большие успехи в сохранении жизни детей с синдромом Дауна и их развитии обеспечивают специальные методы обучения, укрепления физического здоровья с раннего детства, некоторые формы лекарственной терапии, направленные на улучшение функций ЦНС. Многие больные с трисомией 21 теперь способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семьи.
Синдром Патау - трисомия 13
Синдром Патау выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования у детей с врожденными пороками развития. Частота синдрома Патау среди новорожденных равна 1:5000-1:7000. Цитогенетические варианты этого синдрома следующие. Простая полная трисомия 13 как следствие нерасхождения хромосом в мейозе у одного из родителей (главным образом у матери) встречается у 80-85% больных. Остальные случаи обусловлены, в основном, передачей дополнительной хромосомы (точнее, ее длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Обнаружены и другие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации), но они встречаются крайне редко. Клиническая и патологоанатомическая картина простых трисомных форм и транслокационных форм не различается.
Соотношение полов при синдроме Патау близко к 1:1. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией (на 25-30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок гестации 38,3 нед). Ха-
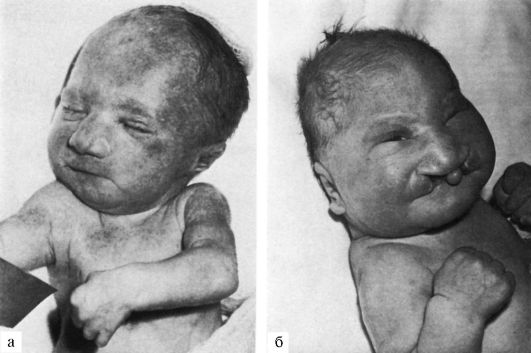 Рис. 5.7. Новорожденные
с синдромом Патау [тригоноцефалия (б); двусторонняя расщелина верхней
губы и нёба (б); узкие глазные щели (б); низко расположенные (б) и
деформированные (а) ушные раковины; микрогения (а); флексорное положение
кистей].
Рис. 5.7. Новорожденные
с синдромом Патау [тригоноцефалия (б); двусторонняя расщелина верхней
губы и нёба (б); узкие глазные щели (б); низко расположенные (б) и
деформированные (а) ушные раковины; микрогения (а); флексорное положение
кистей].
рактерное осложнение беременности при вынашивании плода с синдромом Патау - многоводие: оно встречается почти в 50% случаев.
Синдром Патау сопровождается множественными врожденными пороками развития головного мозга и лица (рис. 5.7). Это патогенетически единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак синдрома Патау - расщелины верхней губы и нёба (обычно двусторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разных комбинациях: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдаются полидактилия (чаще двусторонняя и на руках) и флексорное положение кистей. Частота разных симптомов у детей с синдромом Патау представлена в табл. 5.4.
Таблица 5.4. Основные врожденные пороки при синдроме Патау (по Г.И. Лазюку)
 Окончание табл. 5.4
Окончание табл. 5.4
 Клиническая
диагностика синдрома Патау основывается на сочетании характерных
пороков развития. При подозрении на синдром Патау показано УЗИ всех
внутренних органов.
Клиническая
диагностика синдрома Патау основывается на сочетании характерных
пороков развития. При подозрении на синдром Патау показано УЗИ всех
внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни (95% умирают до 1 года). Однако некоторые больные живут несколько лет. Более того, в развитых странах отмечается тенденция
увеличения продолжительности жизни больных с синдромом Патау до 5 лет (около 15% больных ) и даже до 10 лет (2-3% больных).
Другие синдромы врожденных пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опица) по отдельным признакам совпадают с синдромом Патау. Решающий фактор в диагностике - исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей в семье.
Лечебная помощь детям с синдромом Патау неспецифическая: операции по поводу врожденных пороков развития (по жизненным показаниям), общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. Дети с синдромом Патау практически всегда глубокие идиоты.
Синдром Эдвардса - трисомия 18
Почти во всех случаях синдром Эдвардса обусловлен простой трисомной формой (гаметическая мутация у одного из родителей). Встречаются и мозаичные формы (нерасхождение на ранних стадиях дробления). Транслокационные формы крайне редки, и, как правило, это частичные, а не полные трисомии. Клинических различий между цитогенетически различающимися формами трисомии нет.
Частота синдрома Эдвардса среди новорожденных составляет 1:5000-1:7000. Соотношение мальчиков и девочек 1:3. Причины преобладания девочек среди больных пока неясны.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности (роды в срок). На рис. 5.8-5.11 показаны пороки при синдроме Эдвардса. Это множественные врожденные пороки развития лицевой части черепа, сердца, костной системы, половых органов. Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие; глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномальная стопа (пятка выступает, свод провисает), I палец стоп короче II пальца. Спинномозговая грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).
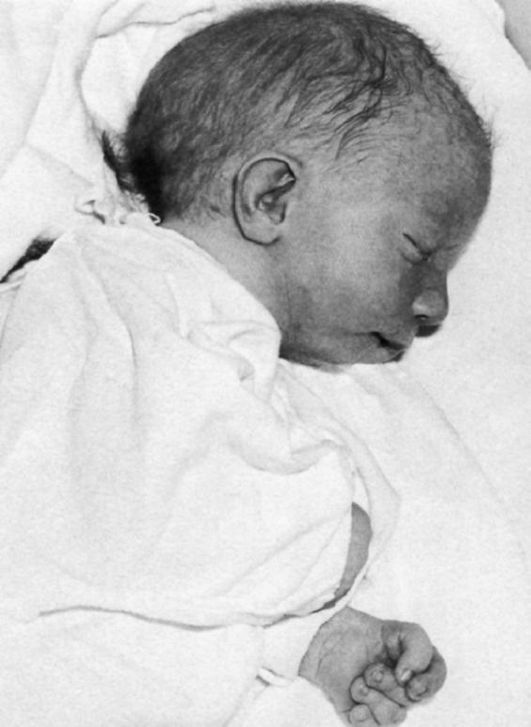 Рис. 5.8. Новорожденный с синдромом Эдвардса (выступающий затылок, микрогения, флексорное положение кисти).
Рис. 5.8. Новорожденный с синдромом Эдвардса (выступающий затылок, микрогения, флексорное положение кисти).
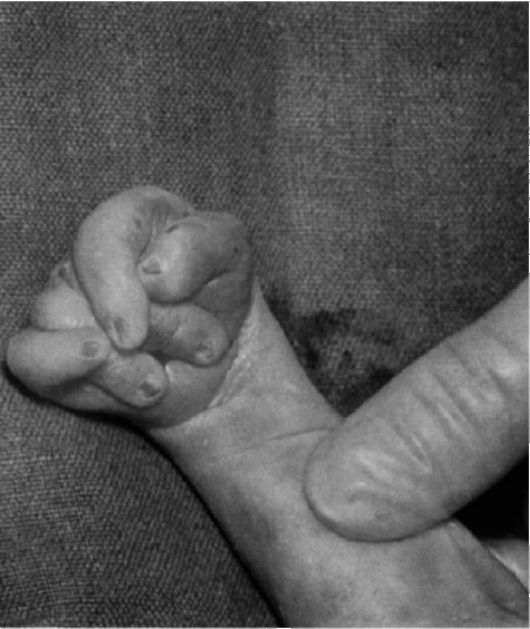 Рис. 5.9. Характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 мес).
Рис. 5.9. Характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 мес).
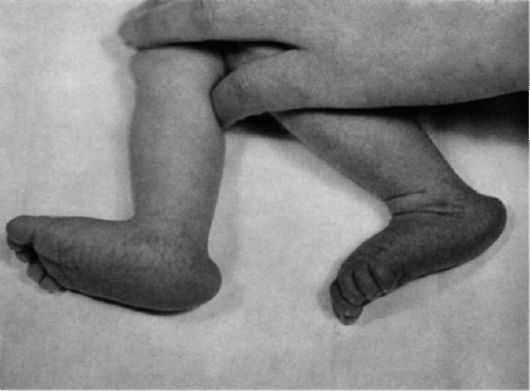 Рис. 5.10. Стопа-качалка (пятка выступает, свод провисает).
Рис. 5.10. Стопа-качалка (пятка выступает, свод провисает).
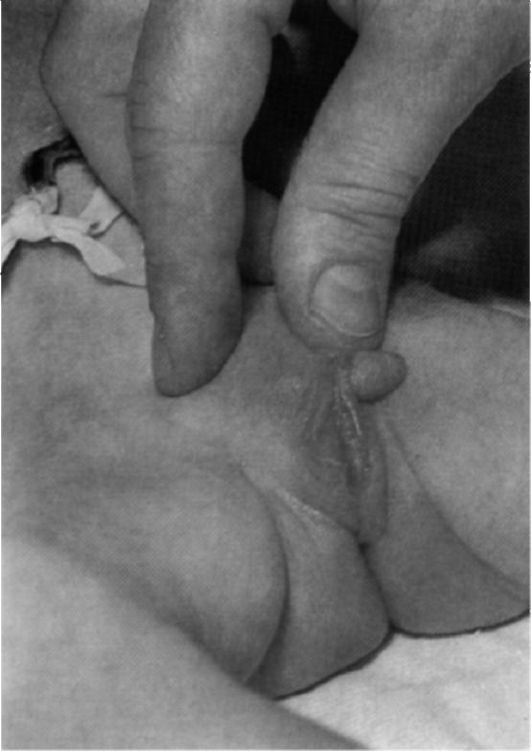 Рис. 5.11. Гипогенитализм у мальчика (крипторхизм, гипоспадия).
Рис. 5.11. Гипогенитализм у мальчика (крипторхизм, гипоспадия).
Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично. Частота отдельных врожденных пороков приведена в табл. 5.5.
Таблица 5.5. Основные врожденные пороки при синдроме Эдвардса (по Г.И. Лазюку)
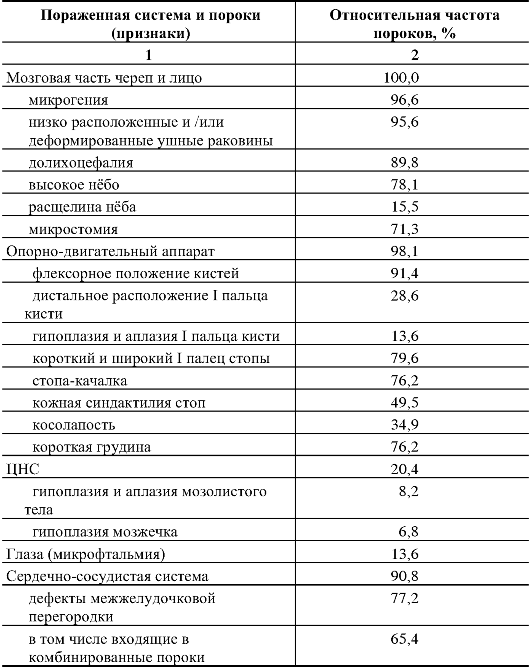 Окончание табл. 5.5
Окончание табл. 5.5
 Как
видно из табл. 5.5, наиболее значимы в диагностике синдрома Эдвардса
изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки
развития сердечно-сосудистой системы.
Как
видно из табл. 5.5, наиболее значимы в диагностике синдрома Эдвардса
изменения мозгового черепа и лица, опорно-двигательного аппарата, пороки
развития сердечно-сосудистой системы.
Дети с синдромом Эдвардса умирают в раннем возрасте (90% до 1 года) от осложнений, обусловленных врожденными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечнососудистая недостаточность). Клиническая и даже патологоанатомическая дифференциальная диагностика синдрома Эдвардса сложна, поэтому во всех случаях показано цитогенетическое исследование. Показания для него те же, что и при трисомии 13 (см. выше).
Трисомия 8
Клиническая картина синдрома трисомии 8 впервые описана разными авторами в 1962 и 1963 гг. у детей с отставанием в умственном развитии, отсутствием надколенника и другими врожденными пороками развития. Цитогенетически был констатирован мозаицизм по хромосоме из группы С или D, поскольку индивидуальной идентификации хромосом в тот период еще не было. Полная трисомия 8, как правило, летальна. Ее часто обнаруживают у пренатально погибших эмбрионов и плодов. Среди новорожденных трисомия 8 встречается с частотой не более чем 1:5000, преобладают мальчики (соотношение мальчиков и девочек 5:2). Большинство описанных случаев (около 90%) относится к мозаичным формам. Заключение о полной трисомии у 10% больных основывалось на исследовании одной ткани, чего в строгом смысле недостаточно для исключения мозаицизма.
Трисомия 8 - результат вновь возникшей мутации (нерасхождение хромосом) на ранних стадиях бластулы, за исключением редких случаев новой мутации в гаметогенезе.
Различий в клинической картине полных и мозаичных форм не выявлено. Тяжесть клинической картины широко варьирует. Причины таких вариаций неизвестны. Корреляций между тяжестью заболевания и долей трисомных клеток не обнаружено.
Дети с трисомией 8 рождаются доношенными. Возраст родителей из общей выборки не выделяется.
Для болезни наиболее характерны отклонения в строении лица, пороки опорно-двигательного аппарата и мочевой системы (рис. 5.12- 5.14). Это выступающий лоб, косоглазие, эпикант, глубоко посаженные глаза, гипертелоризм глаз___
и сосков, высокое нёбо (иногда расщелина), толстые губы, вывернутая нижняя губа, большие ушные раковины с толстой мочкой, контрактуры суставов, камптодактилия, аплазия надколенника, глубокие борозды между межпальцевыми подушечками, четырехпальцевая складка, аномалии ануса. При УЗИ выявляются аномалии позвоночника (добавочные позвонки, неполное закрытие по-
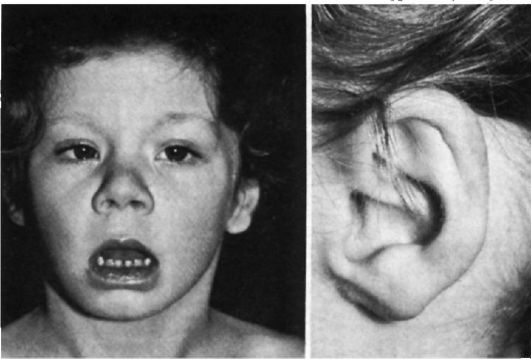 Рис. 5.12. Трисомия 8 (мозаицизм) (вывернутая нижняя губа, эпикант, аномальная ушная раковина).
Рис. 5.12. Трисомия 8 (мозаицизм) (вывернутая нижняя губа, эпикант, аномальная ушная раковина).
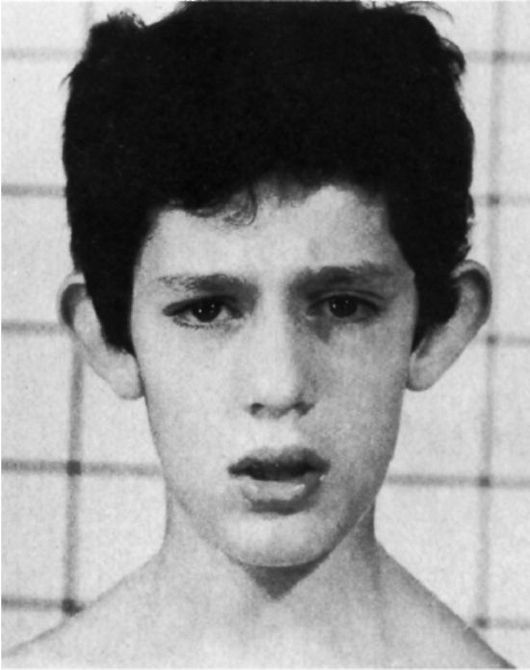 Рис. 5.13. 10-летний мальчик с трисомией 8 (умственная недостаточность, большие оттопыренные ушные раковины с упрощенным рисунком).
Рис. 5.13. 10-летний мальчик с трисомией 8 (умственная недостаточность, большие оттопыренные ушные раковины с упрощенным рисунком).
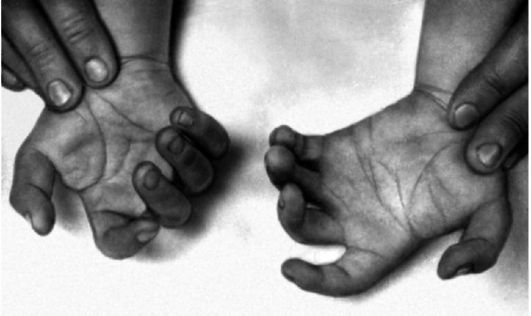 Рис. 5.14. Контрактуры межфаланговых суставов при трисомии 8.
Рис. 5.14. Контрактуры межфаланговых суставов при трисомии 8.
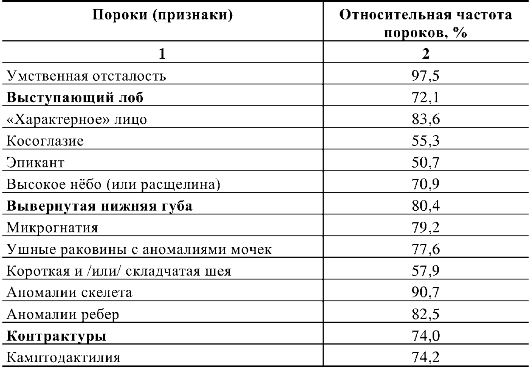
звоночного канала), аномалии формы и положения ребер или добавочные ребра. В табл. 5.6 приведены обобщенные данные о встречаемости отдельных симптомов (или пороков) при трисомии 8.
Таблица 5.6. Основные признаки трисомии 8 (по Г.И. Лазюку)
 Окончание табл. 5.6.
Окончание табл. 5.6.
 Примечание. Полужирным шрифтом выделены наиболее значимые для диагностики признаки.
Примечание. Полужирным шрифтом выделены наиболее значимые для диагностики признаки.
Число симптомов у новорожденных составляет от 5 до 15 и более.
При трисомии 8 прогноз физического, психического развития и жизни неблагоприятный, хотя описаны пациенты в возрасте 17 лет. Со временем у больных проявляются умственная отсталость, гидроцефалия, паховая грыжа, новые контрактуры, аплазия мозолистого тела, кифоз, сколиоз, аномалии тазобедренного сустава, узкий таз, узкие плечи.
Методов специфического лечения нет. Оперативные вмешательства проводят по жизненным показаниям.
Полисомии по половым хромосомам
Это большая группа хромосомных болезней, представленная различными комбинациями дополнительных Х- или Y-хромосом, а в случаях мозаицизма - комбинациями разных клонов. Общая
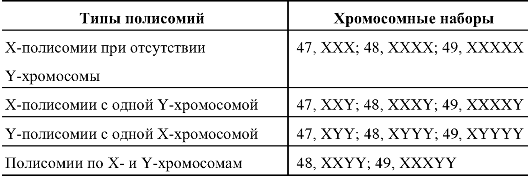
частота полисомии по Х- или Y-хромосомам среди новорожденных составляет 1,5:1000-2:1000. В основном это полисомии XXX, XXY и XYY. Мозаичные формы составляют примерно 25%. В табл. 5.7 представлены типы полисомий по половым хромосомам.
Таблица 5.7. Типы полисомий по половым хромосомам у человека
 Синдром трипло-Х (47,ХХХ). Среди
новорожденных девочек частота синдрома составляет 1:1000. Женщины с
кариотипом XXX в полном или мозаичном варианте имеют в основном
нормальное физическое и психическое развитие, обычно выявляются случайно
при обследовании. Это объясняется тем, что в клетках две X-хромо- сомы
гетерохроматинизированы (два тельца полового хроматина), а функционирует
лишь одна, как и у нормальной женщины. Как правило, у женщины с
кариотипом XXX нет отклонений в половом развитии, она имеет нормальную
плодовитость, хотя риск хромосомных нарушений у потомства и
возникновения спонтанных абортов повышен. Интеллектуальное развитие
нормальное или на нижней границе нормы. Лишь у некоторых женщин с
трипло-Х есть нарушения репродуктивной функции (вторичная аменорея,
дисменорея, ранняя менопауза и др.). Аномалии развития наружных половых
органов (признаки дизэмбриогенеза) обнаруживаются лишь при тщательном
обследовании, выражены незначительно и не служат поводом для обращения к
врачу.
Синдром трипло-Х (47,ХХХ). Среди
новорожденных девочек частота синдрома составляет 1:1000. Женщины с
кариотипом XXX в полном или мозаичном варианте имеют в основном
нормальное физическое и психическое развитие, обычно выявляются случайно
при обследовании. Это объясняется тем, что в клетках две X-хромо- сомы
гетерохроматинизированы (два тельца полового хроматина), а функционирует
лишь одна, как и у нормальной женщины. Как правило, у женщины с
кариотипом XXX нет отклонений в половом развитии, она имеет нормальную
плодовитость, хотя риск хромосомных нарушений у потомства и
возникновения спонтанных абортов повышен. Интеллектуальное развитие
нормальное или на нижней границе нормы. Лишь у некоторых женщин с
трипло-Х есть нарушения репродуктивной функции (вторичная аменорея,
дисменорея, ранняя менопауза и др.). Аномалии развития наружных половых
органов (признаки дизэмбриогенеза) обнаруживаются лишь при тщательном
обследовании, выражены незначительно и не служат поводом для обращения к
врачу.
Варианты синдрома Х-полисомии без Y-хромосомы с числом Х-хромосом более 3 встречаются редко. С увеличением числа дополнительных Х-хромосом нарастают отклонения от нормы. У женщин с тетра- и пентасомией описаны отклонения в умственном развитии, черепно-лицевые дизморфии, аномалии зубов, скелета и половых органов. Однако женщины даже с тетрасомией по Х-хромосоме имеют потомство.
Синдром Клайнфелтера включает случаи полисомии по половым хромосомам, при которых имеется не менее двух Х-хромосом и не менее одной Y-хромосомы. Наиболее часто встречающийся и типичный по клинической картине синдром - это синдром Клайнфелтера с набором 47,ХХY Этот синдром (в полном и мозаичном вариантах) встречается с частотой 1:500-1:750 новорожденных мальчиков. Варианты полисомии с большим числом Х- и Y-хромосом (см. табл. 5.7) встречаются редко. Клинически они также относятся к синдрому Клайнфелтера.
Присутствие Y-хромосомы определяет формирование мужского пола. До периода полового созревания мальчики развиваются почти нормально, лишь с небольшим отставанием в психическом развитии. Генетический дисбаланс в связи с добавочной Х-хромосомой клинически проявляется в период полового созревания в виде недоразвития яичек и вторичных мужских половых признаков.
Больные имеют высокий рост, женский тип телосложения, гинекомастию, слабое оволосение лица, подмышечных впадин и лобка (рис. 5.15). Яички уменьшены, гистологически обнаруживаются дегенерация герминативного эпителия и гиалиноз семенных канатиков. Больные бесплодны (азооспермия, олигоспермия).
Синдром дисомии по Y-хромосоме (47,XYY) встречается с частотой 1:1000 новорожденных мальчиков.
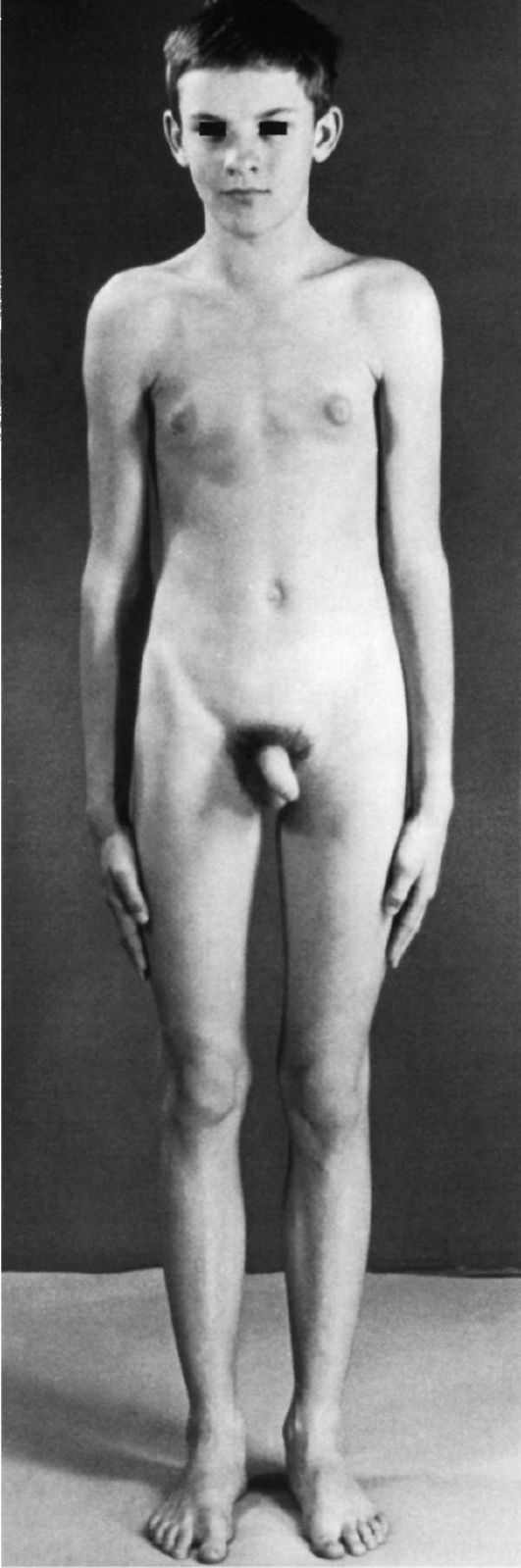 Рис. 5.15. Синдром Клайнфелтера. Высокий рост, гинекомастия, оволосение на лобке по женскому типу.
Рис. 5.15. Синдром Клайнфелтера. Высокий рост, гинекомастия, оволосение на лобке по женскому типу.
Большинство мужчин с таким набором хромосом не отличаются от нормы по физическому и умственному развитию, имеют рост немного выше среднего. Заметных отклонений ни в половом развитии, ни в гормональном статусе, ни в плодовитости у большинства XYY-индивидов нет. Не исключены некоторые особенности поведения: склонность к агрессивным и даже криминальным поступкам.
Синдром Шерешевского-Тернера (45,Х) - единственная форма моносомии у живорожденных. Не менее 90% зачатий с кариотипом 45,Х абортируется спонтанно. Моносомия Х составляет 15- 20% среди всех аномальных кариотипов абортусов.
Частота синдрома Шерешевского-Тернера равна 1:2000-1:5000 новорожденных девочек. Цитогенетика синдрома многообразна. Наряду с истинной моносомией во всех клетках (45,X) встречаются другие формы хромосомных аномалий по половым хромосомам. Это делеции короткого или длинного плеча Х-хромосомы [46,Х,Хр-; 46,X,Xq-], изохромосомы [46,X,i(Xq); 46,Х,i(Xp)], кольцевые хромосомы [46,X,R(X)], а также различные варианты мозаицизма. Лишь 50-60% пациенток с синдромом Шерешевского- Тернера имеют простую полную моносомию (45,Х). Единственная Х-хромосома в 80-85% случаев имеет материнское происхождение и лишь в 15-20% - отцовское.
В остальных случаях синдром обусловлен разнообразным мозаицизмом (в целом 30-40%) и более редкими вариантами делеций, изохромосом, кольцевых хромосом.
Клинически синдром Шерешевского-Тернера проявляется в 3 направлениях: 1) гипогонадизм, недоразвитие половых органов и вторичных половых признаков; 2) врожденные пороки развития; 3) низкий рост.
Со стороны половой системы отмечаются отсутствие гонад (агенезия гонад), гипоплазия матки и маточных труб, первичная аменорея, скудное оволосение лобка и подмышечных впадин, недоразвитие молочных желез, недостаточность эстрогенов, избыток гипофизарных гонадотропинов. У детей с синдромом Шерешевс- кого-Тернера часто (до 25% случаев) встречаются разные врожденные пороки сердца и почек.
Внешний вид больных достаточно своеобразен (хотя и не всегда). У новорожденных и детей грудного возраста короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп (рис. 5.16), голеней, кистей рук и предплечий. В школьном и особенно в подростковом возрасте выявляется отставание в росте,
в развитии вторичных половых признаков (рис. 5.17). У взрослых отмечают нарушения скелета, черепно-лицевые дизморфии, вальгусную девиацию коленных и локтевых суставов, укорочение метакарпальных и метатарзальных костей, остеопороз, бочкообразную грудную клетку, низкий рост волос на шее, антимонголоидный разрез глазных щелей, птоз, эпикант, ретрогению, низкое расположение ушных раковин. Рост взрослых больных на 20-30 см ниже среднего. Тяжесть клинических (фенотипических) проявлений зависит от многих пока неизвестных факторов, в том числе от типа хромосомной патологии (трисомия, делеция, изохромосома). Мозаичные формы болезни, как правило, имеют более слабые проявления в зависимости от соотношения клонов 46ХХ:45Х.
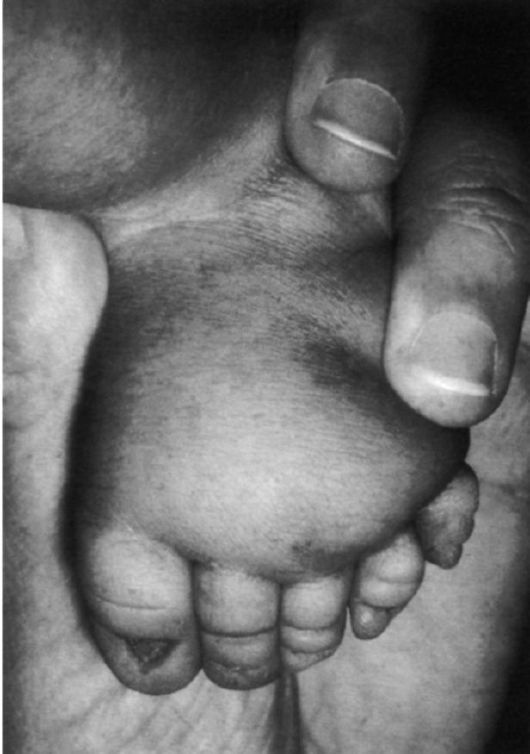 Рис. 5.16. Лимфатический отек стопы у новорожденного с синдромом Шерешевского-Тернера. Маленькие выпуклые ногти.
Рис. 5.16. Лимфатический отек стопы у новорожденного с синдромом Шерешевского-Тернера. Маленькие выпуклые ногти.
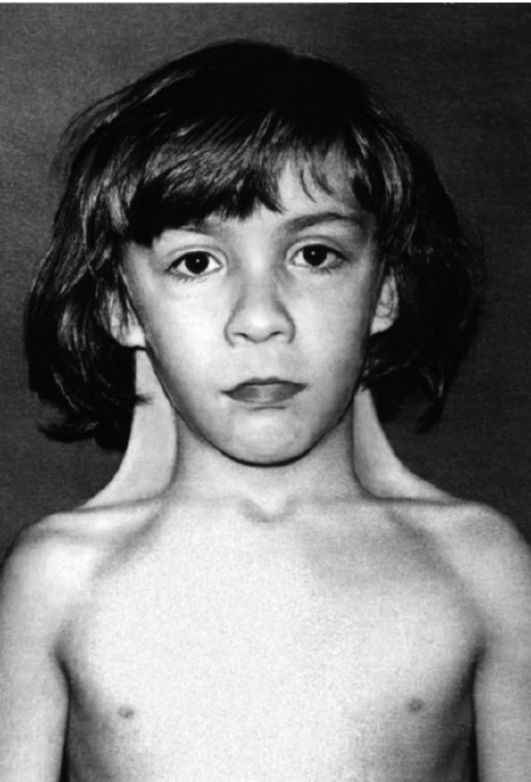 Рис. 5.17. Девочка с синдромом Шерешевского-Тернера (шейные крыловидные складки, широко расположенные и недоразвитые соски молочных желез).
Рис. 5.17. Девочка с синдромом Шерешевского-Тернера (шейные крыловидные складки, широко расположенные и недоразвитые соски молочных желез).
В табл. 5.8 представлены данные о частоте основных симптомов при синдроме Шерешевского-Тернера.
Лечение больных с синдромом Шерешевского-Тернера комплексное: 1) реконструктивная хирургия (врожденные пороки внут-
ренних органов); 2) пластическая хирургия (удаление крыловидных складок и т.п.); 3) гормональное лечение (эстрогены, гормон роста); 4) психотерапия. Своевременное применение всех методов лечения, включая применение генно-инженерного гормона роста, дает больным возможность достичь приемлемого роста и вести полноценную жизнь.
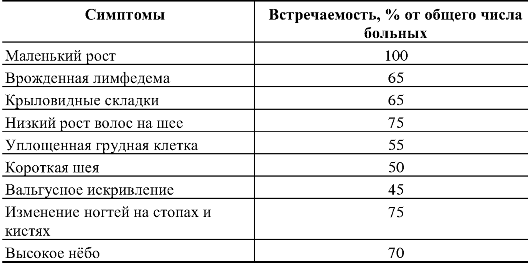
Таблица 5.8. Клинические симптомы синдрома Шерешевского-Тернера и их встречаемость
 Синдромы частичных анеуплоидий
Синдромы частичных анеуплоидий
Эта многочисленная группа синдромов обусловлена хромосомными мутациями. Какой бы вид хромосомной мутации ни был исходно (инверсия, транслокация, дупликация, делеция), возникновение клинического хромосомного синдрома определяется либо избытком (частичная трисомия), либо недостатком (частичная моносомия) генетического материала или одновременно тем и другим эффектом разных измененных участков хромосомного набора. К настоящему времени обнаружено около 1000 разных вариантов хромосомных мутаций, унаследованных от родителей или возникших в раннем эмбриогенезе. Однако клиническими формами хромосомных синдромов считают только те перестройки (их около 100), по которым описано несколько пробандов с совпадением характера цитогенетических изменений и клинической картины (корреляция кариотипа и фенотипа).
Частичные анеуплоидии возникают главным образом в результате неточного кроссинговера в хромосомах с инверсиями или транслокациями. Лишь в небольшом числе случаев возможно первичное возникновение делеций в гамете или в клетке на ранних стадиях дробления.
Частичные анеуплоидии, как и полные, вызывают резкие отклонения в развитии, поэтому относятся к группе хромосомных болезней. Большинство форм частичных трисомий и моносомий не повторяют клиническую картину полных анеуплоидий. Они являются самостоятельными нозологическими формами. Лишь у небольшого числа пациентов клинический фенотип при частичных анеуплоидиях совпадает с таковым при полных формах (синдром Шерешевского-Тернера, синдром Эдвардса, синдром Дауна). В этих случаях речь идет о частичной анеуплоидии по так называемым критическим для развития синдрома районам хромосом.
Какой-либо зависимости тяжести клинической картины хромосомного синдрома от формы частичной анеуплоидии или от индивидуальной хромосомы нет. Величина вовлеченного в перестройку участка хромосомы может иметь значение, но случаи подобного рода (меньшая или большая длина) должны рассматриваться как разные синдромы. Общие закономерности корреляций клинической картины и характера хромосомных мутаций выявить трудно, потому что многие формы частичных анеуплоидий элиминируются в эмбриональном периоде.
Фенотипические проявления любых аутосомных делеционных синдромов состоят из двух групп аномалий: неспецифических находок, общих для многих различных форм частичных аутосомных анеуплоидий (задержка пренатального развития, микроцефалия, гипертелоризм, эпикант, явно низко распололженные уши, микрогнатия, клинодактилия и т. д.); комбинации находок, типичных для данного синдрома. Наиболее подходящее объяснение причин неспецифических находок (большинство из которых не имеют клинического значения) - это неспецифические эффекты аутосомного дисбаланса как такового, а не результаты делеций или дупликаций специфических локусов.
Хромосомным синдромам, обусловленным частичными анеуплоидиями, присущи общие свойства всех хромосомных болезней: врожденные нарушения морфогенеза (врожденные пороки развития, дизморфии), нарушение постнатального онтогенеза, тяжесть клинической картины, сокращенная продолжительность жизни.
Синдром «кошачьего крика» - частичная моносомия по короткому плечу хромосомы 5 (5р-). Синдром моносомии 5р- был первым описанным синдромом, обусловленным хромосомной мутацией (делецией). Это открытие сделал Дж. Лежен в 1963 г.
У детей с такой хромосомной аномалией отмечается необычный плач, напоминающий требовательное кошачье мяуканье или крик. По этой причине синдром сначала был назван синдромом «кошачьего крика». Частота синдрома достаточно высокая для делеционных синдромов - 1 : 45 000. Описано несколько сотен больных, поэтому цитогенетика и клиническая картина этого синдрома изучены хорошо.
Цитогенетически в большинстве случаев выявляется делеция с утратой от 1/3 до 1/2 длины короткого плеча хромосомы 5. Потеря всего короткого плеча или, наоборот, незначительного участка встречается редко. Для развития клинической картины синдрома 5р- имеет значение не величина утраченного участка, а конкретный фрагмент хромосомы. За развитие полного синдрома ответствен лишь незначительный участок в коротком плече хромосомы 5 [5р- (15,1-15,2)]. Помимо простой делеции, при этом синдроме обнаружены и другие цитогенетические варианты: кольцевая хромосома 5 (естественно, с делецией соответствующего участка короткого плеча); мозаицизм по делеции; реципрокная транслокация короткого плеча хромосомы 5 (с потерей критического участка) с другой хромосомой.
Клиническая картина синдрома 5р- довольно сильно различается у отдельных больных по сочетанию врожденных пороков развития органов. Наиболее характерный признак - «кошачий крик» - обусловлен изменением гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки). Практически у всех больных имеются те или иные изменения мозговой части черепа и лица: лунообразное лицо, микроцефалия, гипертелоризм, микрогения, эпикант, антимонголоидный разрез глаз, высокое нёбо, плоская спинка носа (рис. 5.18, 5.19). Ушные раковины деформированы и расположены низко. Кроме того, встречаются врожденные пороки сердца и некоторых других внутренних органов, изменения костно-мышечной системы (синдактилия стоп, клинодактилия V пальца кисти, косолапость). Выявляют мышечную гипотонию, а иногда и диастаз прямых мышц живота.
Выраженность отдельных признаков и клинической картины в целом меняется с возрастом. Так, «кошачий крик», мышечная гипо-
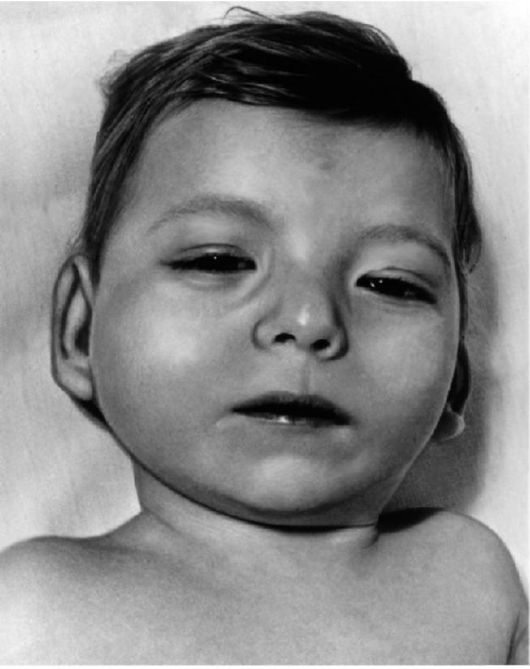 Рис. 5.18. Ребенок
с выраженными признаками синдрома «кошачьего крика» (микроцефалия,
лунообразное лицо, эпикант, гипертелоризм, широкая плоская спинка носа,
низко расположенные ушные раковины).
Рис. 5.18. Ребенок
с выраженными признаками синдрома «кошачьего крика» (микроцефалия,
лунообразное лицо, эпикант, гипертелоризм, широкая плоская спинка носа,
низко расположенные ушные раковины).
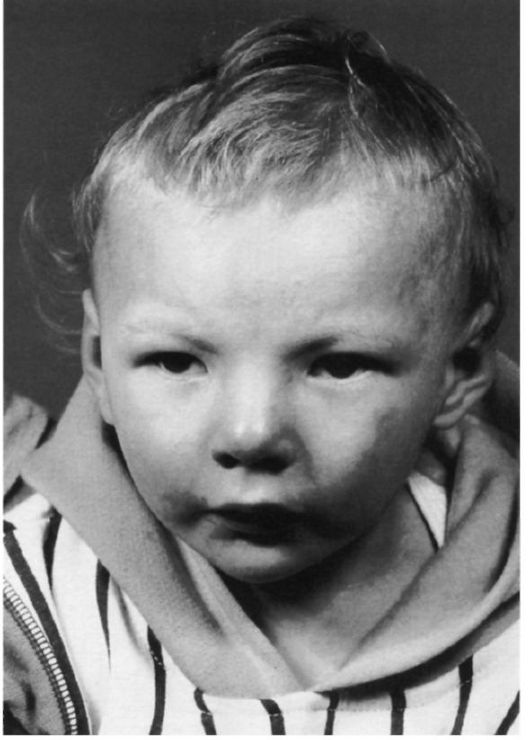 Рис. 5.19. Ребенок с маловыраженными признаками синдрома «кошачьего крика».
Рис. 5.19. Ребенок с маловыраженными признаками синдрома «кошачьего крика».
тония, лунообразное лицо с возрастом исчезают почти полностью, а микроцефалия выявляется более отчетливо, становятся заметнее психомоторное недоразвитие, косоглазие. Продолжительность жизни больных с синдромом 5р- зависит от тяжести врожденных пороков внутренних органов (особенно сердца), выраженности клинической картины в целом, уровня медицинской помощи и повседневной жизни. Большинство больных умирают в первые годы, около 10% больных достигают 10-летнего возраста. Имеются единичные описания больных в возрасте 50 лет и старше.
Во всех случаях больным и их родителям показано цитогенетическое обследование, потому что у одного из родителей может быть реципрокная сбалансированная транслокация, которая при прохождении через стадию мейоза может обусловливать делецию участка 5р- (15,1-15,2).
Синдром Вольфа-Хиршхорна (частичная моносомия 4р-) обусловлен делецией сегмента короткого плеча хромосомы 4. Клинически синдром Вольфа-Хиршхорна проявляется многочисленны-
ми врожденными пороками с последующей резкой задержкой физического и психомоторного развития. Уже внутриутробно отмечается гипоплазия плода. Средняя масса тела детей при рождении от доношенной беременности составляет около 2000 г, т.е. пренатальная гипоплазия выражена больше, чем при других частичных моносомиях. У детей с синдромом Вольфа-Хиршхорна отмечаются следующие признаки (симптомы): микроцефалия, клювовидный нос, гипертелоризм, эпикант, аномальные ушные раковины (часто с преаурикулярными складками), расщелины верхней губы и нёба, аномалии глазных яблок, антимонголоидный разрез глаз, маленький рот, гипоспадия, крипторхизм, сакральная ямка, деформация стоп и др. (рис. 5.20). Наряду с пороками развития наружных органов более чем у 50% детей имеются пороки внутренних органов (сердца, почек, ЖКТ).
Жизнеспособность детей резко снижена, большинство умирают в возрасте до 1 года. Описан лишь 1 больной в возрасте 25 лет.
Цитогенетика синдрома довольно характерная, как и многих делеционных синдромов. Примерно в 80% случаев у пробанда выявляется делеция части короткого плеча хромосомы 4, а у родителей кариотипы нормальные. Остальные случаи обусловлены транслокационными комбинациями или кольцевыми хромосомами, но всегда при этом отмечается потеря фрагмента 4р16.
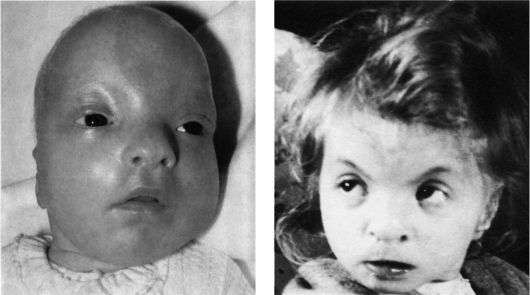 Рис. 5.20. Дети
с синдромом Вольфа-Хиршхорна (микроцефалия, гипертелоризм, эпикант,
аномальные ушные раковины, косоглазие, микрогения, птоз).
Рис. 5.20. Дети
с синдромом Вольфа-Хиршхорна (микроцефалия, гипертелоризм, эпикант,
аномальные ушные раковины, косоглазие, микрогения, птоз).
Цитогенетическое обследование больного и его родителей показано для уточнения диагноза и прогноза здоровья будущих детей, поскольку родители могут иметь сбалансированные транслокации. Частота рождения детей с синдромом Вольфа-Хиршхорна невысокая (1:100 000).
Синдром частичной трисомии по короткому плечу хромосомы 9 (9р+) - наиболее частая форма частичных трисомий (опубликовано около 200 сообщений о таких больных), синдром клинически выражен.
Клиническая картина многообразна и включает внутриутробные и постнатальные нарушения развития: задержку роста, умственную отсталость, микробрахицефалию, антимонголоидный разрез глаз, энофтальм (глубоко посаженные глаза), гипертелоризм, округлый кончик носа, опущенные углы рта, низко расположенные оттопыренные ушные раковины с уплощенным рисунком, гипоплазию (иногда дисплазию) ногтей (рис. 5.21). Врожденные пороки сердца обнаружены у 25% больных.
Реже встречаются другие врожденные аномалии, свойственные всем хромосомным болезням: эпикант, косоглазие, микрогнатия, высокое арковидное нёбо, сакральный синус, синдактилии.
Больные с синдромом 9р+ рождаются в срок. Пренатальная гипоплазия выражена умеренно (средняя масса тела новорожденных 2900-3000 г). Жизненный прогноз сравнительно благоприятный. Больные доживают до пожилого и преклонного возраста.
Цитогенетика синдрома 9р+ многообразна. Большая часть случаев - результат несбалансированных транс-
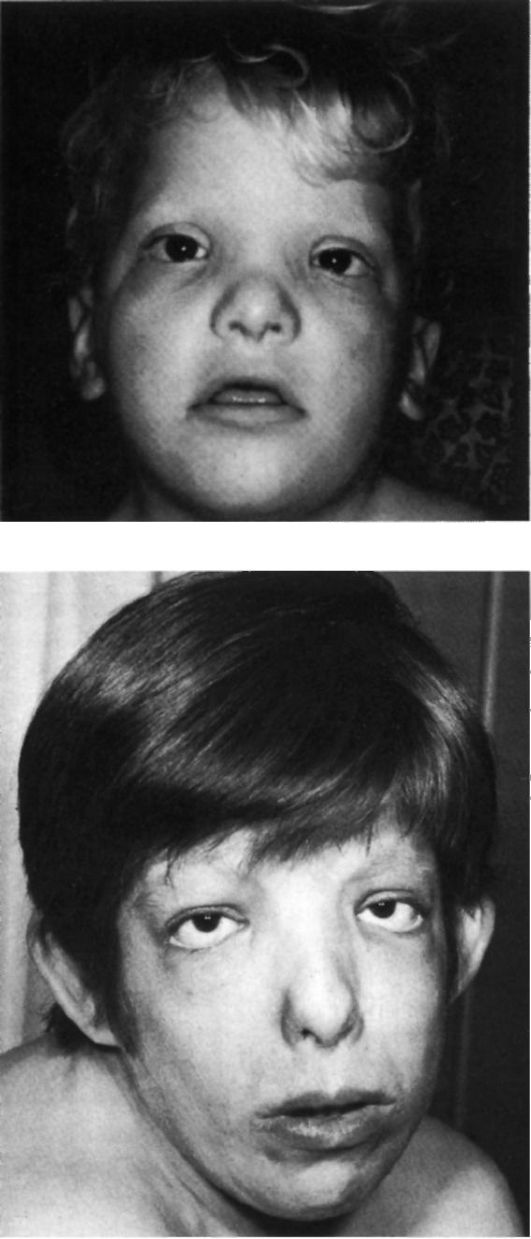 Рис. 5.21. Синдром
трисомии 9р+ (гипертелоризм, птоз, эпикант, луковицеобразный нос,
короткий фильтр, большие, низко расположенные ушные раковины, толстые
губы, короткая шея).
Рис. 5.21. Синдром
трисомии 9р+ (гипертелоризм, птоз, эпикант, луковицеобразный нос,
короткий фильтр, большие, низко расположенные ушные раковины, толстые
губы, короткая шея).
а - ребенок 3-х лет; б - женщина 21 года.
локаций (семейных или спорадических). Описаны и простые дупликации, изохромосомы 9р. Клинические проявления синдрома однотипны при разных цитогенетических вариантах, что вполне объяснимо, поскольку во всех случаях имеется тройной набор генов части короткого плеча хромосомы 9.
Микроцитогенетические синдромы
В эту недавно выделенную группу входят синдромы, обусловленные незначительными делециями или дупликациями строго определенных участков хромосом. Соответственно их называют микроделеционными и микродупликационными синдромами. Многие из этих синдромов первоначально были описаны как доминантные заболевания (точечные мутации), но с помощью современных высокоразрешающих цитогенетических методов (особенно молекулярно-цитогенетических) установлена истинная этиология синдромов. Стало возможным обнаруживать делеции и дупликации протяженностью до одного гена с примыкающими областями.
На примере расшифровки микроцитогенетических синдромов можно видеть взаимное проникновение цитогенетических методов в генетический анализ, молекулярно-генетических методов в цитогенетику. Это позволяет расшифровывать природу ранее непонятных наследственных синдромов (болезней), а также выяснять функциональные зависимости между генами. Термин «микроцитогенетика» уже вошел в литературу. Еще не установлено, что лежит в основе развития микроцитогенетических синдромов - отсутствие структурного гена или более протяженного участка, включающего конкретный ген, как влияет на проявление микроделеционного синдрома состояние локуса в гомологичной хромосоме. По-видимому, природа клинических проявлений разных микроделеционных синдромов различна. Патологический процесс при некоторых из них развертывается через активацию онкогенов, клиника других синдромов обусловлена не только делециями как таковыми, но и явлениями хромосомного импринтинга и однородительских дисомий. Клинические и цитогенетические характеристики микроделеционных синдромов постоянно уточняются. В табл. 5.9 суммированы сведения о микроцитогенетических синдромах (микроделеционных и микродупликационных).
Большинство микроцитогенетических синдромов встречается редко (1:50 000-1:100 000 новорожденных). Их клиническая картина,
Таблица 5.9. Общие сведения о микроцитогенетических синдромах

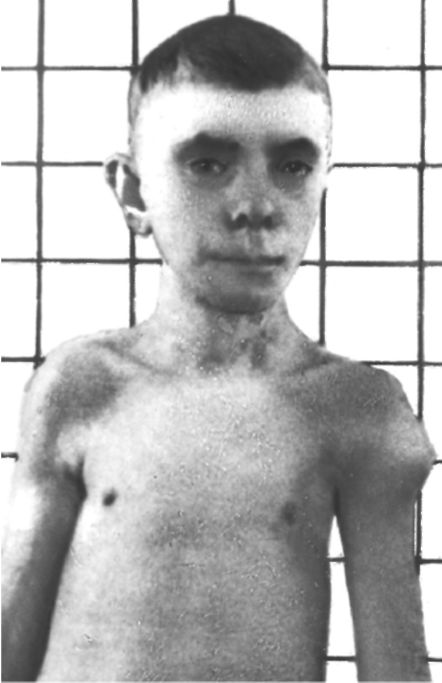 Рис. 5.22. Синдром Лан- гера-Гидеона. Множественные экзостозы.
Рис. 5.22. Синдром Лан- гера-Гидеона. Множественные экзостозы.
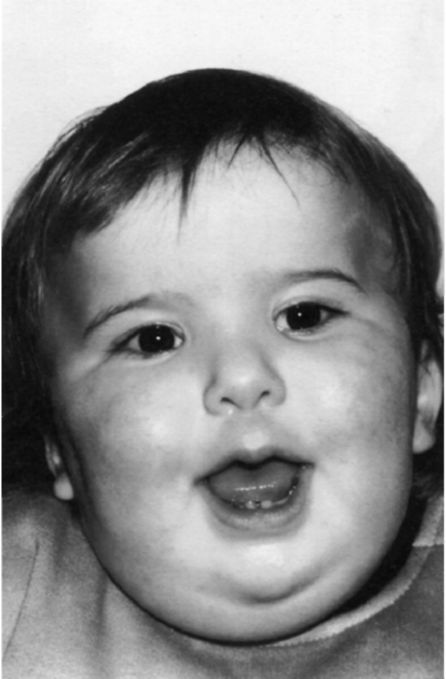 Рис. 5.23. Мальчик с синдромом Прадера-Вилли.
Рис. 5.23. Мальчик с синдромом Прадера-Вилли.
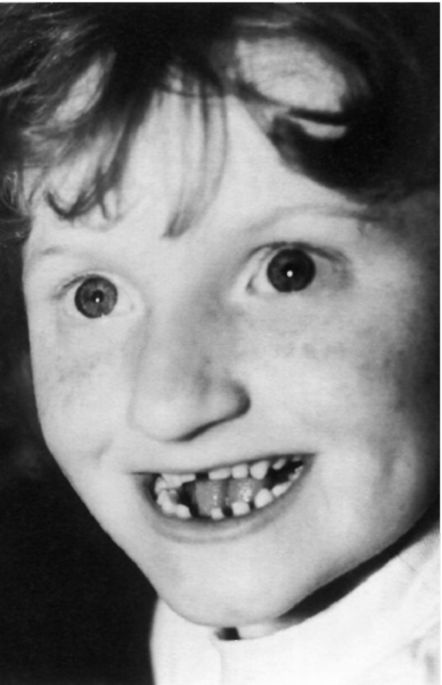 Рис. 5.24. Девочка с синдромом Ангельмана.
Рис. 5.24. Девочка с синдромом Ангельмана.
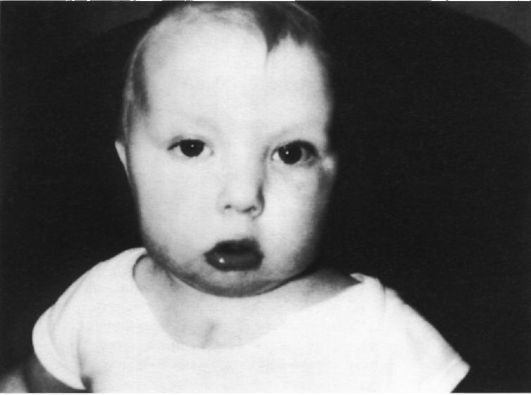 Рис. 5.25. Ребенок с синдромом ДиДжорджи.
Рис. 5.25. Ребенок с синдромом ДиДжорджи.
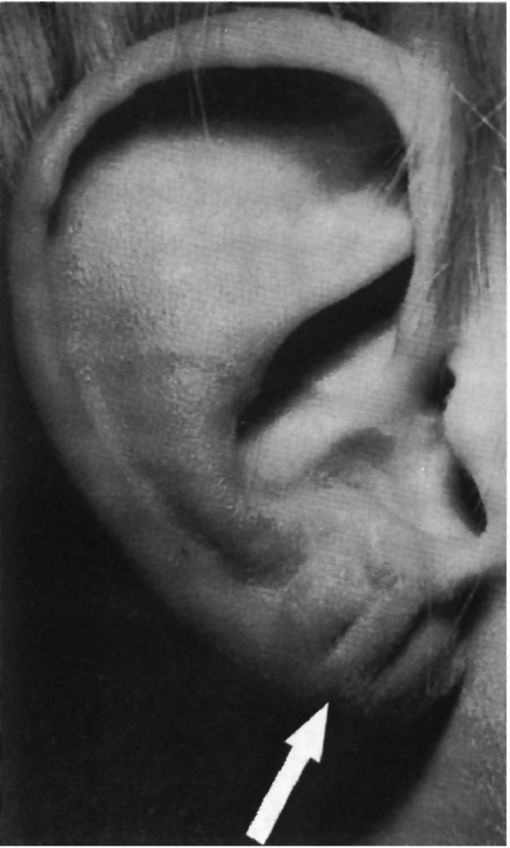 Рис. 5.26. Поперечные насечки на мочке уха (при синдроме Беквита-Видемана - типичный симптом (указаны стрелкой).
Рис. 5.26. Поперечные насечки на мочке уха (при синдроме Беквита-Видемана - типичный симптом (указаны стрелкой).
как правило, отчетливая. Диагноз можно поставить по совокупности симптомов. Однако в связи с прогнозом здоровья будущих детей в семье, в том числе у родственников родителей пробанда, необходимо провести высокоразрешающее цитогенетическое исследование у пробанда и его родителей.
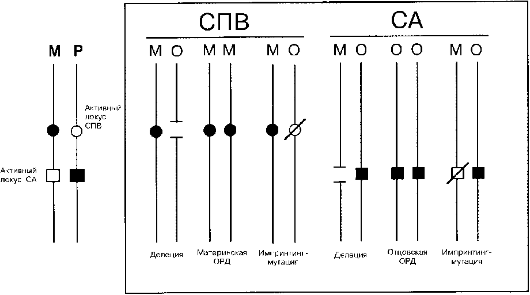 Рис. 5.27. Три класса мутаций при синдроме Прадера-Вилли (СПВ) и Ангельмана (СА). М - мать; О - отец; ОРД - однородительская дисомия.
Рис. 5.27. Три класса мутаций при синдроме Прадера-Вилли (СПВ) и Ангельмана (СА). М - мать; О - отец; ОРД - однородительская дисомия.
Клинические проявления микроцитогенетических синдромов сильно варьируют в связи с разной протяженностью делеции или дупликации, а также в связи с родительской принадлежностью микроперестройки - унаследована ли она от отца или от матери. В последнем случае речь идет об импринтинге на хромосомном уровне. Это явление было открыто при цитогенетическом изучении двух клинически различающихся синдромов (Прадера-Вилли и Ангельмана). В обоих случаях микроделеция наблюдается в хромосоме 15 (участок q11-q12). Лишь молекулярно-цитогенетическими методами установлена истинная природа синдромов (см. табл. 5.9). Участок q11-q12 в хромосоме 15 дает настолько выраженный эффект импринтинга, что синдромы могут быть вызваны однородительскими дисомиями (рис. 5.27) или мутациями с эффектом импринтинга.
Как видно из рис. 5.27, дисомия по материнским хромосомам 15 вызывает синдром Прадера-Вилли (потому что отсутствует участок q11-q12 отцовской хромосомы). Такой же эффект дает делеция этого же участка или мутация в отцовской хромосоме при разнородительской дисомии. Прямо противоположная ситуация наблюдается при синдроме Ангельмана.
Факторы повышенного риска рождения детей с хромосомными болезнями
В последние десятилетия многие исследователи обращались к причинам возникновения хромосомных болезней. Не вызывало со-
мнений, что образование хромосомных аномалий (и хромосомных, и геномных мутаций) происходит спонтанно. Экстраполировались результаты экспериментальной генетики и предполагался индуцированный мутагенез у человека (ионизирующая радиация, химические мутагены, вирусы). Однако реально причины возникновения хромосомных и геномных мутаций в зародышевых клетках или на ранних стадиях развития зародыша до сих пор не расшифрованы.
Проверялись многие гипотезы нерасхождения хромосом (сезонность, расово-этническая принадлежность, возраст матери и отца, задержанное оплодотворение, порядок рождения, семейное накопление, лекарственное лечение матерей, вредные привычки, негормональная и гормональная контрацепция, флюридины, вирусные болезни у женщин). В большинстве случаев эти гипотезы не подтвердились, но генетическая предрасположенность к болезни не исключается. Хотя в большинстве случаев нерасхождение хромосом у человека спорадическое, можно предполагать, что оно в определенной степени генетически детерминировано. Об этом свидетельствуют следующие факты:
- потомство с трисомией появляется у одних и тех же женщин повторно с частотой не менее 1%;
- родственники пробанда с трисомией 21 или другими анеуплоидиями имеют несколько повышенный риск рождения ребенка с анеуплоидией;
- кровное родство родителей может повышать риск трисомии у потомства;
- частота зачатий с двойной анеуплоидией может быть выше, чем предсказывается, в соответствии с частотой отдельных анеуплоидий.
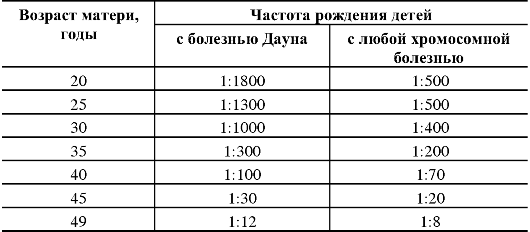
К биологическим факторам повышения риска нерасхождения хромосом относится возраст матери, хотя механизмы этого явления неясны (табл. 5.10, рис. 5.28). Как видно из табл. 5.10, риск рождения ребенка с хромосомной болезнью, обусловленной анеуплоидией, с возрастом матери постепенно повышается, но особенно резко после 35 лет. У женщин старше 45 лет каждая 5-я беременность завершается рождением ребенка с хромосомной болезнью. Наиболее четко возрастная зависимость проявляется для трисомии 21 (болезнь Дауна). Для анеуплоидий по половым хромосомам возраст родителей либо совсем не имеет значения, либо его роль очень незначительна.
Из рис. 5.28 видно, что с возрастом повышается также частота спонтанных абортов, которая к 45 годам увеличивается в 3 раза и более. Такое положение можно объяснить тем, что спонтанные
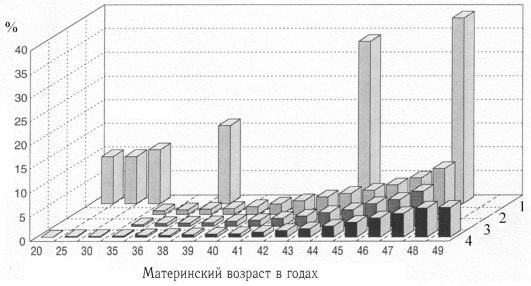 Рис. 5.28. Зависимость частоты хромосомных аномалий от возраста метери.
Рис. 5.28. Зависимость частоты хромосомных аномалий от возраста метери.
1 - спонтанные аборты при зарегистрированных беременностях; 2 - общая частота хромосомных аномалий во II триместре; 3 - синдром Дауна во II триместре; 4 - синдром Дауна среди живорожденных.
аборты во многом обусловлены (до 40-45%) хромосомными аномалиями, частота которых имеет возрастную зависимость.
Таблица 5.10. Зависимость частоты рождения детей с хромосомными болезнями от возраста матери
 Выше
были рассмотрены факторы повышенного риска анеуплоидий у детей от
кариотипически нормальных родителей. По существу, из многочисленных
предполагаемых факторов
Выше
были рассмотрены факторы повышенного риска анеуплоидий у детей от
кариотипически нормальных родителей. По существу, из многочисленных
предполагаемых факторов
только два имеют значение для планирования беременности, а точнее, являются строгими показаниями для пренатальной диагностики. Это рождение ребенка с анеуплоидией по аутосомам и возраст матери старше 35 лет.
Цитогенетическое исследование у супружеских пар позволяет выявить кариотипические факторы риска: анеуплоидию (в основном в мозаичной форме), робертсоновские транслокации, сбалансированные реципрокные транслокации, кольцевые хромосомы, инверсии. Повышение риска зависит от типа аномалии (от 1 до 100%): например, если у одного из родителей в робертсоновскую транслокацию вовлечены гомологичные хромосомы (13/13, 14/14, 15/15, 21/21, 22/22), то здорового потомства у носителя таких перестроек быть не может. Беременности будут заканчиваться либо спонтанными абортами (во всех случаях транслокаций 14/14, 15/15, 22/22 и частично при транслокациях 13/13, 21/21), либо рождением детей с синдромом Патау (13/13) или синдромом Дауна (21/21).
Для расчета риска рождения ребенка с хромосомной болезнью в случае аномального кариотипа у родителей были составлены таблицы эмпирического риска. Теперь в них почти нет необходимости. Методы пренатальной цитогенетической диагностики позволили перейти от оценки риска к установлению диагноза у эмбриона или плода.
Ключевые слова и понятия Изохромосомы
Импринтинг на хромосомном уровне История открытия хромосомных болезней Классификация хромосомных болезней Корреляция фено- и кариотипа Микроделеционные синдромы Общие клинические черты хромосомных болезней Однородительские дисомии Патогенез хромосомных болезней Показания для цитогенетической диагностики Робертсоновские транслокации Сбалансированные реципрокные транслокации Типы хромосомных и геномных мутаций Факторы риска при хромосомных болезнях Хромосомные аномалии и спонтанные аборты Частичные анеуплоидии
Частичные моносомии Частичные трисомии Частота хромосомных болезней Эффекты хромосомных аномалий
Контрольные вопросы
1. Какие виды хромосомных аномалий не встречаются у живорожденных:
а) трисомии по аутосомам;
б) трисомии по половым хромосомам;
в) моносомии по аутосомам;
г) моносомия по Х-хромосоме;
д) нуллисомия по Х-хромосоме.
2. Какие мутации относятся к геномным:
а) инверсии, транслокации, дупликации, делеции;
б) полиплоидии, анеуплоидии;
в) триплоидии, тетраплоидии;
г) внутрихромосомные и межхромосомные перестройки.
3. Выберите основные показания для исследования кариотипа:
а) в анамнезе умершие дети с множественными пороками развития;
б) хроническое прогредиентное течение болезни с началом в детском возрасте;
в) неврологические проявления (судороги, снижение или повышение мышечного тонуса, спастические парезы);
г) олигофрения в сочетании с пороками развития.
4. Укажите формулы кариотипа при синдроме Шерешевского-Тернера:
а) 46,ХХ/45,Х0;
б) 47,ХХХ;
в) 45,Х0;
г) 47,ХХY;
д) 46,ХХ.
5. Метод точной диагностики хромосомных болезней:
а) клинический;
б) дерматоглифический;
в) цитогенетический;
г) клинико-генеалогический;
д) специфическая биохимическая диагностика.
6. Риск рождения ребенка с хромосомными аномалиями существенно повыша-
ется в возрастных интервалах:
а) 20-25 лет;
б) 25-30 лет;
г) 30-35 лет;
д) 35-40 лет.
7. К хромосомным относятся мутации:
а) делеция;
б) триплоидия;
в) инверсия;
г) изохромосома.
8. Формула кариотипа при синдроме «кошачьего крика»:
а) 45,Х0;
б) 46,ХХ, 9р+;
в) 46,ХХ, 5р-;
г) 46,ХХ/45,Х0.
9. Показания для проведения цитогенетического анализа:
а) гепатоспленомегалия, катаракта, умственная отсталость;
б) привычное невынашивание беременности и мертворождения в анамнезе;
в) непереносимость некоторых пищевых продуктов, гемолитические кризы;
г) умственная отсталость, микроаномалии развития или врожденные пороки развития.
10. Формулы хромосомного набора у больного с синдромом Клайнфелтера:
а) 45,Х;
б) 47,ХХХ;
в) 47,ХYY;
г) 46,ХУ,5р-;
д) 48,ХХYY;
е) 47,ХХY.
11. Полиплоидия - это:
а) уменьшение числа хромосом в наборе на несколько пар;
б) диплоидный набор хромосом в гамете;
в) увеличение числа хромосом, кратное гаплоидному набору.
12. В основе хромосомных болезней лежат хромосомные и геномные мутации, возникающие:
а) только в половых клетках;
б) в соматических и половых клетках;
в) только в соматических клетках.
13. Укажите формулу кариотипа при синдроме Патау:
а) 47,ХХ, 18+;
б) 47,ХY, 13+;
в) 46,ХХ, 5р-;
г) 47,ХХY;
д) 45,Х.
14. Летальные нарушения кариотипа:
а) моносомии по Х-хромосоме;
б) трисомии по половым хромосомам;
в) моносомии по аутосомам;
г) трисомии по аутосомам.
15. Набор симптомов, включающий умственную отсталость, долихоцефалию, деформированные ушные раковины, флексорное положение пальцев рук, врожденный порок сердца, указывает на:
а) синдром Эдвардса;
б) синдром Патау;
в) синдром Дауна;
г) синдром «кошачьего крика».
16. Показания для проведения кариотипирования:
а) задержка физического и полового развития, гипогонадизм, гипогенитализм;
б) задержка психомоторного развития в сочетании с диспластичным фенотипом;
в) приобретенные деформации позвоночника и грудины, помутнение роговицы, гепатоспленомегалия;
г) прогредиентная утрата приобретенных навыков, судорожный синдром, спастические параличи.
17. Анеуплоидия - это:
а) увеличение хромосомного набора на целый гаплоидный набор;
б) изменение числа хромосом в результате добавления одной или нескольких хромосом;
в) изменение числа хромосом в результате утери одной или нескольких хромосом;
г) изменение числа хромосом в результате утери или добавления одной или нескольких хромосом.
18. Правильная формула кариотипа при синдроме Эдвардса:
а) 46,ХY, 21 + ;
б) 47,ХХY;
в) 47,ХХ, 13+;
г) 47,ХХ, 18+;
д) 46,ХХ, 9р+;
е) 45,t (13/21).
19. Наиболее тяжелые последствия вызывают:
а) моносомии по половым хромосомам;
б) трисомии по половым хромосомам;
в) моносомии по аутосомам;
г) трисомии по аутосомам.
20. Симптомокомплекс, включающий микроцефалию, расщелину губы и нёба, полидактилию и поликистоз почек, наиболее характерен для:
а) синдрома Эдвардса;
б) синдрома Дауна;
в) синдрома Вольфа-Хиршхорна;
г) синдрома Патау.
21. Клинически хромосомные болезни проявляются:
а) множественными признаками дизморфогенеза;
б) врожденными пороками развития;
в) отставанием в умственном развитии;
г) необычным цветом и запахом мочи.
22. Возможные формулы кариотипа при синдроме Дауна:
а) 47,XX, 13+;
б) 47,ХХ, 22+;
в) 46,ХY, 14-,t (21/14);
г) 47,ХХХ;
д) 47,ХХ, 21+;
23. Более тяжелые клинические проявления имеют хромосомные болезни, обусловленные:
а) недостатком генетического материала;
б) избытком генетического материала.
24. Признаки синдрома Беквита-Видемана:
а) макроглоссия;
б) гипогликемия;
в) эпилепсия;
г) экзостозы;
д) большие рост и масса тела новорожденных.
25. Причины возникновения трисомий:
а) отставание хромосом в анафазе;
б) нерасхождение хромосом;
в) точечные мутации.
26. Возможные формулы кариотипа при симптомокомплексе, включающем низкий рост, короткую шею, бочкообразную грудную клетку, задержку полового развития:
а) 47,XXY;
б) 45,Х;
в) 46,ХХ/45,Х;
г) 47,XYY.
27. Исследование кариотипа показано:
а) у женщины с 1 спонтанным абортом в анамнезе;
б) у родителей ребенка с простой формой трисомии 21;
в) у супружеской пары с мертворождением и 3 спонтанными абортами в анамнезе.
28. Носители робертсоновских транслокаций:
а) клинически здоровы;
б) имеют кариотип, состоящий из 45 хромосом;
в) имеют риск развития опухолей;
г) имеют кариотип, состоящий из 46 хромосом, одна из которых является результатом слияния длинных плеч акроцентрических хромосом, а другая - коротких;
д) имеют риск рождения ребенка с хромосомной болезнью.
29. Выберите термин, соответствующий описанной ситуации:
а) премутация;
б) геномный импринтинг;
в) однородительская дисомия.
1) У 7-летнего мальчика с умственной отсталостью, низким ростом, маленькими кистями и стопами, полифагией (синдром Прадера-Вилли) при молекулярно-генетическом исследовании обнаружили 2 материнские хромосомы 15 и ни одной отцовской.
2) При цитогенетическом обследовании 6-летней девочки с тяжелой умственной отсталостью, судорогами, атаксией, прогенией (синдром Ангельмана) обнаружили интерстициальную микроделецию материнской хромосомы 15.
3) При ДНК-исследовании гена FMR-I у 32-летней женщины, имеющей сына с синдромом ломкой Х-хромосомы, обнаружили 1 аллель с 21 CGG- повтором и 1 аллель с 92 CGG-повторами.