Патологическая анатомия : учебник / А. И. Струков, В. В. Серов. - 5-е изд., стер. - М.: Литтерра, 2010. - 848 с. : ил.
|
|
|
|
БОЛЕЗНИ ДЕТСКОГО ВОЗРАСТА ПРЕНАТАЛЬНАЯ ПАТОЛОГИЯ
Понятие о периодизации и закономерностях прогенеза и киматогенеза
В понятие «пренатальная (антенатальная) патология» включаются все патологические процессы и состояния человеческого зародыша, начиная с оплодотворения и кончая рождением ребенка. Пренатальный период человека исчисляется длительностью беременности - 280 днями, или 40 нед, по истечении которых наступают роды.
Учение о внутриутробной патологии (уродствах и пороках развития) возникло очень давно. В арабской медицине XI, XII и XIII веков имелись уже подробные описания различных врожденных пороков. Амбруаз Паре (1510-1590) написал о них книгу. Однако научное изучение этого вопроса началось в начале XX века (Швальбе). При этом большинство исследователей считали, что основную роль в развитии врожденных пороков у человека играет наследственность. Большое значение для понимания влияния факторов внешней среды на формирование пороков развития имело открытие австралийского офтальмолога Грегга, который в 1951 г. опубликовал данные о значении вируса краснухи (рубеолы) в возникновении множественных врожденных пороков развития у человека. Он показал, что при заболевании матери краснухой в первую треть беременности у 12% детей развиваются врожденные пороки и в 7,2% отмечается мертворожденность, во вторую треть беременности - 3,9 и 4,6% соответственно, в последнюю треть беременности пороков развития у плода не возникает, а мертворожденность имеет место в 1,7% случаев. С этого момента сформировалось учение о возможности проявления пороков развития у человека, обусловленных воздействием экзогенных факторов, и подтвердились данные об основном значении времени воздействия этих факторов на развивающийся зародыш.
В настоящее время пренатальная патология человека выросла в проблему, имеющую не меньшее значение в медицине, чем проблема сердечнососудистых заболеваний, опухолей и психических болезней.
Все развитие, начиная от созревания половой клетки (гаметы) до рождения зрелого плода, делят на два периода - период прогенеза и период киматогенеза (от греч. kyema - зародыш) (рис. 291). Периоду прогенеза соответствует созревание гамет (яйцеклетки и сперматозоида) до оплодотворения. В этот период возможно возникновение патологии гамет - гаметопатии. В зависимости от того, в каких структурах наследственного аппарата гаметы произошла мутация, различают генные, хромосомные и геномные мутации. Наследственные болезни, в том числе и пороки раз-
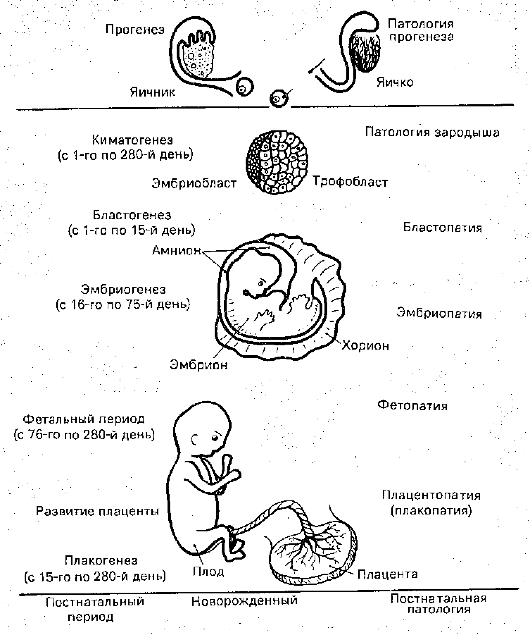 Рис. 291. Киматогенез
и виды патологии зародыша. Слева - нормальный прогенез и киматогенез,
справа - патология прогенеза и киматогенеза (по Герт-леру)
Рис. 291. Киматогенез
и виды патологии зародыша. Слева - нормальный прогенез и киматогенез,
справа - патология прогенеза и киматогенеза (по Герт-леру)
вития, могут быть следствием мутаций гамет родителей ребенка (спонтанные мутации) или его отдаленных предков (унаследованные мутации).
Период киматогенеза исчисляется с момента оплодотворения и образования зиготы до родов и делится на три периода. Первый период - бластогенез - продолжается с момента оплодотворения до 15-го дня беременности, когда идет дробление яйца, заканчивающееся выделением эмбрио- и трофобласта. Второй период - эмбриогенез - охватывает отрезок времени с 16-го по 75-й день беременности, когда осуществляется основной органогенез и образуются амнион и хорион. Третий период - фетогенез - продолжается с 76-го по 280-й день, когда идут дифференцировка и созревание тканей плода, а также образование плаценты, заканчивающиеся рождением плода. Период фетогенеза делят на ранний фетальный (с 76-го по 180-й день), к концу которого незрелый плод приобретает жизнеспособность, и поздний фетальный (со 181-го по 280-й
день), когда завершается созревание плода с одновременным старением плаценты. Патология всего периода киматогенеза называется киматопатией. Соответственно периодам киматогенеза различают: бластопатию, эмбриопатию, раннюю и позднюю фетопатии1.
Этиология. После открытия рубеолярной эмбриопатии расширились представления о влиянии экзогенных факторов, приводящем к киматопатиям. Это нашло свое подтверждение в многочисленных экспериментальных исследованиях.
По современным данным, 20% пороков развития (основная патология периода киматогенеза) связаны с генными мутациями, 10% - с хромосомными аберрациями, около 3-4% - с влиянием экзогенных факторов, более 60% - с невыясненной этиологией. Полагают, что в патологии зародыша преобладает мультифакторная этиология, т.е. комбинация наследственных и экзогенных факторов.
К экзогенным факторам, способным вызывать киматопатии у человека, относятся вирусы и некоторые другие микроорганизмы, а именно вирусы краснухи, иммунодефицита человека (ВИЧ), кори, ветряной оспы, herpes simplex, гепатита, а также микоплазма, листерелла, трепонема, токсоплазма, реже - микобактерия теберкулеза и др. Кроме инфекционных агентов, киматопатии могут быть обусловлены влиянием лучевой энергии (γ-лучи), ионизирующей радиации, некоторыми лекарственными препаратами - талидомидом, гидантоином, фенитоином, цитостатическими средствами, гормонами, витаминами (в частности, витамином D), хинином и др., алкоголем, гипоксией различного генеза, эндокринными заболеваниями матери - сахарным диабетом, тиреотоксическим зобом. Употребление во время беременности алкоголя приводит к развитию алкогольной эмбриофетопатии, характеризующейся общей гипоплазией, умеренной недоношенностью, микроцефалией, сочетающейся с птозом век, эпикантом, микрогенией. Реже встречаются врожденные пороки сердца.
В настоящее время в связи с новыми методами лечения получили особое значение диабетические и тиреотоксические фето- и эмбриопатии. До лечения инсулином у женщин, страдающих сахарным диабетом, беременность наблюдалась редко. В настоящее время способность к деторождению у этих женщин такая же, как и у здоровых. Однако у детей, матери которых больны сахарным диабетом, пороки развития отмечаются чаще. К так называемой диабетической эмбриопатии относятся пороки развития скелета, сердечно-сосудистой, центральной нервной и мочеполовой систем. Диабетическая фетопатия проявляется в виде недоношенности или рождения гигантского плода кушингоидного вида. При повышенной и пониженной функциях щитовидной железы наблюдается склонность к абортам и выкидышам. Среди пороков развития при тиреотоксикозах
1 В узком смысле слова к бласто-, эмбрио- и фетцпатиям относят лишь те болезни зародыша, которые обусловлены экзогенными факторами.
преобладают анэнцефалия, пороки сердца, гипотиреозы с умственной отсталостью - так нызываемые тиреотоксические эмбриофетопатии.
Патогенез. Механизм развития киматопатии в настоящее время широко изучается с помощью многих современных методов. Сложность изучения заключается в том, что приходится иметь дело с двумя биологическими объектами - матерью и зародышем, связь между которыми осуществляется барьерным органом - плацентой.
Первая закономерность, характерная для патологии внутриутробного периода при любом патогенном воздействии, - обязательное искажение нормального хода развития зародыша. Поэтому для понимания патогенеза пренатальной патологии большое значение имеет изучение реактивности зародыша в разные периоды киматогенеза, так как основной жизненной функцией зародыша является беспрерывно идущий процесс морфогенеза. На процессах формообразования в первую очередь и сказывается влияние патогенных факторов.
Нарушение хода морфогенеза называется дизонтогенезом. Однако в разные периоды киматогенеза расстройства дизонтогенеза возникают на разных уровнях - от грубых нарушений развития зародыша, не совместимых с жизнью на ранних его этапах, до изменения тонких ультраструктур клеток и тканей на поздних этапах. По мере созревания зародыша у него постепенно появляется способность реагировать на различные патогенные влияния не только нарушением морфогенеза, но и развитием реактивных патологических процессов - альтерацией, полной и неполной регенерацией тканей, воспалением, иммуно-морфологическими и компенсаторно-приспособительными сдвигами.
Второй закономерностью, характерной для патологии поздних периодов киматогенеза, является сочетание нарушений морфогенеза с другими патологическими процессами, например пороков формирования сердца с гиперплазией мезенхимальных тканей, пороков формирования головного мозга с наличием некрозов, кровоизлияний и др.
Третьей закономерностью, определяющей патогенез любого из периодов киматогенеза и имеющей большое значение в развитии того или иного патологического состояния зародыша, является время воздействия на него патогенного агента.
Так, в период бластогенеза зародыш на любое воздействие отвечает нарушением имплантации оплодотворенного яйца или развития эмбрио- и трофобласта. В период эмбриогенеза, когда осуществляется основной морфогенез внутренних органов и частей тела зародыша, почти любое повреждение приводит к развитию того или иного врожденного порока или к гибели эмбриона.
В период фетогенеза, когда осуществляется тканевая дифференцировка органов, почти любое повреждение ведет к развитию пороков на тканевом уровне.
По данным русских эмбриологов, пренатальная гибель зародыша чаще наблюдается в определенные сроки его внутриутробной жизни. Для эмбриона мле-
копитающих и человека такими периодами особо высокой чувствительности к патогенным агентам являются имплантация оплодотворенного яйца в слизистую оболочку матки, что соответствует 14 дням внутриутробного развития, и плацентация - начало формирования плаценты, что соответствует 3-6-й неделе внутриутробного развития. Эти два периода наибольшей чувствительности зародыша к воздействию повреждающих агентов получили название первого и второго критических периодов.
Факторы, вызывающие пороки развития, получили название тератогенных (от греч. teratos - уродство). Оказалось, что различные тератогенные агенты могут привести к одному и тому же пороку развития в зависимости от времени воздействия на эмбрион; например, влияние лучевой энергии и хинина на 3-й неделе внутриутробного развития приводит к нарушениям формирования нервной трубки зародыша.
Вместе с тем один и тот же тератогенный агент может вызвать разные пороки развития, воздействуя в различные сроки эмбриогенеза.
Известно, что при поражении эмбриона вирусом краснухи возникает рубеолярная эмбриопатия (синдром Грегга), которая заключается в пороках развития глаз, сердца, мозга, зубных зачатков и внутреннего уха. При этом пороки развития глаз (катаракта, микрофтальмия и др.) появляются в тех случаях, если мать переносит краснуху в последнюю декаду I месяца или в первые две декады
II месяца беременности, пороки развития мозга (микроцефалия) - в течение всего II месяца, внутреннего уха - в третью декаду II месяца и в первую декаду
III месяца беременности.
Для каждого органа существует определенный отрезок времени, в течение которого при воздействии тератогенного агента возникает порок развития этого органа. Этот отрезок времени получил название тератогенного терминационного периода (от лат. teratos - уродство и terminus - предел, граница), т.е. предельного срока, в течение которого тератогенный фактор может вызвать врожденный порок (рис. 292). Пользуясь данными эмбриологии, можно судить о сроках возникновения того или иного порока развития и составить так называемые тератологические ка-
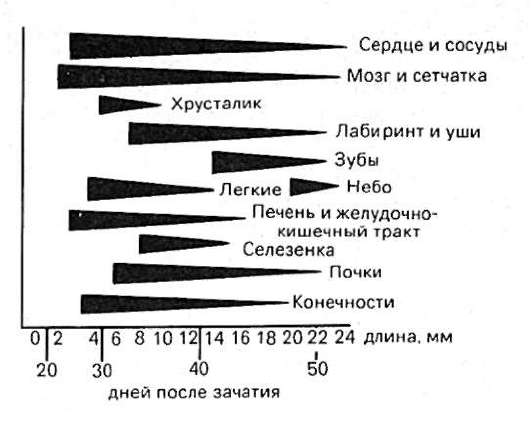 Рис. 292. Схематическое изображение тератогенного терминационного периода отдельных органов и частей тела (по Гертлеру)
Рис. 292. Схематическое изображение тератогенного терминационного периода отдельных органов и частей тела (по Гертлеру)
лендари для пороков развития различных органов. Как показывают данные экспериментальной тератологии, чем выше митотическая активность развивающихся тканей, тем чувствительнее они к воздействию тератогенного агента.
Однако необходимо учитывать, что повреждающий агент может обладать большим или меньшим сродством к тем или иным тканевым зачаткам, что обусловливает иногда некоторые специфические черты, характерные для определенного патогенного агента.
Так, с 1957 по 1964 г. в ФРГ и других странах мира имела место так называемая талидомидная катастрофа. Талидомид применялся в качестве успокаивающего (снотворного) средства. Оказалось, что малые дозы этого преперата опасны для человеческого эмбриона; на животных они не действуют. У многих женщин, принимавших талидомид на II месяце беременности, рождались дети с тяжелыми пороками развития конечностей - амелией, фокомелией. В 40% случаев поражались верхние конечности, в 10% - нижние, в 20% - верхние и нижние конечности, в 20% - конечности (верхние и нижние), органы слуха и зрения (данные 1961 и 1962 гг.). По данным 1964 г., в 45% случаев талидомидные эмбриопатии протекали с пороками развития внутренних органов. Из приведенного наблюдения видно, что талидомид имеет особый тропизм к развивающимся закладкам конечностей.
Кроме нарушений морфогенеза, удалось показать, что у эмбриона могут наблюдаться резорбция его некротизированных тканей, отек тканей, кровоизлияния и в конце эмбриогенеза даже неполная регенерация с образованием рубцов. Следует учитывать, что отмирание тканевых зачатков наблюдается и при нормальном ходе морфогенеза, например при слиянии отдельных зачатков, образовании полостей в них, разрывах мембран (глоточной, клоакальной) и др. Однако по объему и характеру процесс физиологического отмирания клеток отличается от некрозов в условиях патологии, он не сопровождается рубцеванием, а главное не приводит к нарушению процессов формирования. Обширные некрозы тканей эмбриона с рубцеванием появляются, вероятно, при эмбриопатиях, обусловленных действием экзогенных агентов. При генотипических пороках развития значительной альтерации зачатков органов не бывает, а имеется лишь задержка процессов дифференцировки зачатков.
В подавляющем большинстве случаев морфология сложившегося порока развития неспецифична. Поэтому отличить по внешнему виду генотипический порок от фенокопии1 невозможно. Основным проявлением патологии эмбрионального периода является дизонтогенез в виде врожденных пороков развития органов или частей тела зародыша.
К фетальному периоду основной органогенез заканчивается и происходят дальнейший рост и дифференцировка тканей плода.
В раннем фетальном периоде еще продолжается органогенез полушарий большого мозга и центрального органа иммуногенеза - вилочковой
1 Фенокопия - порок развития, возникающий под влиянием экзогенных агентов, морфологически идентичный генотипическому пороку.
железы, поэтому в этом периоде могут возникать пороки развития головного мозга и задержка созревания тканей тимуса.
Кроме дизонтогенеза, у плода иногда встречаются и другие патологические процессы, так как его реактивные возможности по сравнению с эмбрионом возрастают. У плода наблюдаются альтеративные изменения, редуцированное воспаление (см. Воспаление), иммуноморфологические изменения (см. Иммунопатологические процессы), расстройства крово- и лимфообращения, гиперплазия и регенерация. Поэтому в фетальном периоде наблюдаются болезни, сходные с болезнями внеутробного периода. Для болезней плода - фетопатий - характерны следующие особенности.
1. Любая болезненная форма в плодном периоде сочетается с нарушением онтогенеза, но на тканевом уровне. При этом могут быть или неправильные соотношения тканей органов, или задержка их созревания. Например, при megaduodenum, megacolon имеется избыточное развитие мышечной ткани в стенке кишки при отсутствии в ней достаточно развитых нервных приборов; наблюдается задержка созревания почек с обилием зародышевого типа клубочков (рис. 293) и т.д.
2. При инфекционных фетопатиях всегда отмечается генерализованное повреждение тканей и органов плода. Типично наличие множественных очагов преимущественно альтеративного воспаления в паренхиматозных органах или генерализованного гранулематоза (например, при врожденном сифилисе, листериозе).
3. Как правило, развивается выраженный геморрагический синдром с петехиальными сыпями на коже, слизистых оболочках, с кровоизлияниями во внутренних органах.
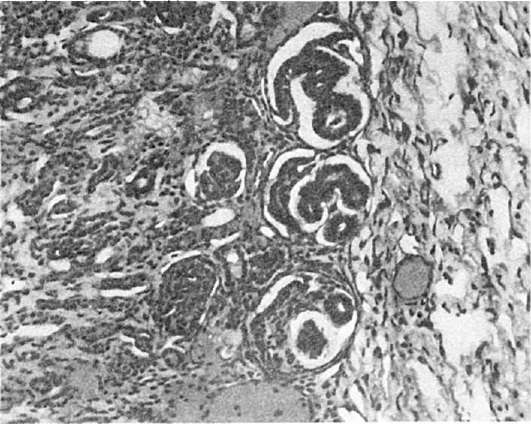 Рис. 293. Клубочки зародышевого типа в коре почки у мальчика в возрасте 7 дней
Рис. 293. Клубочки зародышевого типа в коре почки у мальчика в возрасте 7 дней
4. Наблюдаются задержка инволюции и избыточная пролиферация клеток в очагах экстрамедуллярного кроветворения с появлением их вне факультативно кроветворных органов. У зрелого здорового плода к моменту родов очаги экстрамедуллярного кроветворения редуцируются.
5. Процессы гипертрофии и регенерации идут с преобладанием гиперплазии мезенхимальных тканей, что приводит к избыточному развитию соединительной ткани (например, в мочевыводящих путях при megaureter с одновременной убылью мышечных волокон; при муковисцидозе - в поджелудочной железе; при фиброэластозе сердца - с избыточным развитием эластической и фиброзной тканей в эндокарде и т.д.).
Патогенез перечисленных особенностей болезней плода тесно связан со структурой и функциональной незрелостью его тканей и органов, регулирующих их функции.
Поэтому перечисленные особенности болезней фетального периода заставляют выделить их по сравнению с болезнями внеутробного периода в особую группу фетопатий.
Болезни прогенеза и киматогенеза
Гаметопатии
Гаметопатии - это патология гамет. К ним относятся любые повреждения яйцеклетки и сперматозоида во время ово- и сперматогенеза до оплодотворения. Понятие «гаметопатии» охватывает все виды повреждения мужской и женской гаметы: мутации генов и возникновение наследственных болезней и наследственных пороков развития, хромосомные аберрации с возникновением чаще не наследуемых хромосомных болезней, геномные мутации - изменения числа хромосом гаметы, обычно приводящие к самопроизвольному аборту или хромосомной болезни. Кроме того, необходимо учитывать, что тяжелые повреждения не только ядра, но и цитоплазмы гаметы становятся источником их гибели с развитием стерильности и бесплодия или спонтанных абортов и выкидышей. Из этого следует, что гаметопатии являются одним из факторов внутриутробной летальности, не поддающейся пока точной регистрации.
При повреждении ядра гаметы могут происходить изменения генетического аппарата. Изменения генов, их мутации приводят к закреплению этих изменений в последующих клеточных генерациях. Следует учитывать, что гаметы являются носителями генов, унаследованных ими от всех отдаленных предков. Поэтому в понятие гаметопатии входит поражение не только гамет родителей, но и более отдаленных предков пробанда. Гамета с дефектом гена или генов может стать источником наследственных пороков развития или заболеваний, проявляющихся на разных этапах внутриутробного и внеутробного развития.
Генные пороки и болезни могут наследоваться по аутосомно-рецессивному, аутосомно-доминантному типам или мутантный ген может быть сцеплен с половой Х-хромосомой. При аутосомно-рецессивном типе наследования у пробанда воз-
никает порок только в том случае, если мутантный ген был получен и от отца, и от матери. Родители пробанда сами могут быть здоровы, являясь лишь гетерозиготными носителями мутантного гена. При аутосомно-доминантном типе наследования мутантный ген передается от отца или от матери, которые сами страдают аналогичным пороком.
Пороки, гены которых локализованы в Х-хромосоме, в свою очередь могут наследоваться по рецессивному или доминантному типу. Пороки, сцепленные с Х-хромосомой, передающиеся по рецессивному типу, наблюдаются, как правило, у мальчиков, так как единственная у них Х-хромосома является пораженной. Мутантный ген передает мать, не являющаяся больной. Очень редко носительницей порока может быть девочка. Это бывает в том случае, если отец являлся больным, а мать - носительницей мутантного гена.
Кроме локального поражения генетического аппарата ядра гаметы вследствие мутации генов, в период гаметогенеза может появляться мутация хромосом в виде изменений их числа и структуры. Мутации хромосом получили название хромосомных аберраций. Хромосомные аберрации возникают чаще всего в момент редукционного деления гамет. Их следствием являются хромосомные болезни, которые, однако, в большинстве случаев не наследуются, так как их носители чаще умирают в детстве или являются бесплодными.
Типичными примерами хромосомных болезней являются болезнь Дауна (трисомия по 21-й паре аутосом), синдром Патау (трисомия по 13- 15-й паре аутосом), синдром Шерешевского-Тернера (моносомия половой хромосомы - 45 ХО) и др.
Болезнь Дауна, наблюдающаяся у новорожденных в соотношении 1:600, 1:700, встречается наиболее часто. Клинически у детей с рождения отмечается выраженная задержка умственного и физического развития. Больные имеют типичный внешний вид: косой разрез глаз, западающая спинка носа, высокое небо, низкое расположение маленьких ушных раковин, выраженная гипотония мышц. Дети умирают чаще от интеркуррентных заболеваний. У большинства из них обнаруживаются пороки развития сердца и магистральных сосудов (тетрада Фалло и др.), реже - пороки развития пищеварительной и мочеполовой систем. У этих детей отмечаются недоразвитие полушарий большого мозга, особенно лобных его долей с задержкой дифференцировки нейронов, нарушения процессов миелинизации, архитектоники кровеносных сосудов мозга.
Синдром Патау у новорожденных и мертворожденных встречается с частотой 1 на 5149 рождений. Характерны выраженная общая гипоплазия, аномалии черепа и лица: низкий скошенный лоб, узкие глазные щели, запавшее переносье, широкое основание носа, гипотелоризм, «дефекты скальпа», низко расположенные деформированные ушные раковины, типичные расщелины верхней губы и неба. Отмечаются полидактилия и флексорное положение кистей, микрофтальмия, колобома и помутнение роговицы. Со стороны головного мозга отмечаются микроцефалия, аринэнцефалия (отсутствие обонятельного мозга), аплазия или гипоплазия червя мозжечка и др. Отмечаются также врожденные пороки сердца, органов пищеварения, мочевой системы и др. Дети нежизнеспособны.
Бластопатии
Бластопатия - патология бластоцисты, возникающая в период нидации и дробления в первые 15 дней от момента оплодотворения до выделения эмбрио-и трофобласта.
Этиология и патогенез. Причиной бластопатии чаще всего являются хромосомные аберрации в сочетании с влияниями среды (эндокринные заболевания матери, гипоксия и др.). Патогенез зависит от вида поражения бластоцисты. Так, например, патогенез двойниковых уродств связан с появлением во время дробления двух или более самостоятельно растущих центров. Полагают, что если эти центры разобщены друг с другом, то развиваются два независимо растущих однояйцевых близнеца, нормальное развитие которых не следует относить к бластопатиям. Если центры роста расположены близко и имеют общую для двух близнецов промежуточную зону, то развиваются два сросшихся близнеца. В обоих случаях возможно развитие симметричных и асимметричных близнецов.
Морфология бластопатии разнообразна. К ним относятся нарушения имплантации бластоцисты, а именно эктопическая беременность, поверхностная или очень глубокая имплантация бластоцисты в эндометрий, нарушение ориентации формирующегося эмбриобласта в бластоцисте по отношению к эндометрию, аплазия или гибель развивающегося эмбриобласта с образованием пустого зародышевого мешка, пороки развития всего эмбриона, некоторые одиночные пороки, двойниковые уродства и, наконец, аплазия или гипоплазия формирующегося трофобласта - амниона, амниотической ножки, желточного мешка. Поверхностная или чрезмерно глубокая имплантация бластоцисты приводит к порокам развития формы, локализации, а также приращению плаценты (см. ниже), которые чреваты гибелью плода во время акта родов. Нарушения ориентации эмбриобласта при полной топографической инверсии заканчиваются гибелью эмбриобласта. При неполной инверсии наблюдаются пороки развития пуповины (см. ниже), которые могут приводить к гибели плода во время родов. Пустые зародышевые мешки представляют собой бластоцисты, не содержащие эмбриобласт или содержащие его остатки. Иногда в них можно обнаружить амниотические оболочки, пуповину, желточный мешок.
Патология развития всего эмбриона представляет собой общие тяжелые нарушения, не совместимые с жизнью.
Одиночные и множественные пороки развития, возникающие в период бластулы (в первые 8-12 нед), встречаются в 14,3-22,9% всех спонтанно абортированных зародышей. При этом в 46,2% случаев они сопровождаются аномалиями последа. Такое сочетание часто приводит к гибели зародыша.
Двойниковые уродства встречаются в виде сросшейся двойни. Если сросшаяся двойня состоит из равных симметрично развитых компонентов, она называется диплопагусом (diplopagus от греч. diplos - двойной, agus - соединять); если же она состоит из асимметрично развитых ком-
понентов - гетеропагусом (heteropagus от греч. heteros - другой), при этом недоразвитый близнец, находящийся в зависимости от другого, развитого, получил название паразита. Для обозначения локализации сращения близнецов к анатомическому названию места сращения добавляют также слово пагус; например, сращение в области головы называют краниопагусом, в области груди - торакопагусом, в области таза - ишиопагусом и др.
Двойниковые уродства сочетаются с нежизнеспособностью. В редких случаях описана значительная продолжительность жизни таких близнецов до зрелого возраста. В легких случаях сращений только мягких тканей возможна хирургическая коррекция.
Эмбриопатии
Эмбриопатия - патология эмбрионального периода с 16-го дня беременности до 75-го дня включительно, в течение которого заканчивается основной органогенез и формирование амниона и хориона. К основным видам эмбриопатий относят врожденные пороки развития.
Врожденным пороком развития называют стойкое морфологическое изменение органа, части тела или всего организма, выходящее за пределы вариаций нормального строения определенного биологического вида, возникащее внутриутробно в результате нарушений морфогенеза. Так как органогенез завершается в основном в эмбриональный период, большинство пороков развития появляется именно на этом этапе внутриутробного существования. Однако, кроме врожденных пороков с нарушениями основного морфогенеза органов или частей тела, имеются врожденные пороки, при которых нарушения развития наблюдаются на уровне тканевой дифференцировки. Они часто бывают системными, например пороки развития поперечнополосатой мускулатуры (врожденная миатония Оппенгейма), соединительной ткани (болезнь Марфана), кожи (врожденный ихтиоз), костей хрящевого генеза (врожденная хондродисплазия) и др. Пороки развития могут касаться также тканей одного органа, например гипоплазия гладкой мышечной ткани при megaureter, нервного интрамурального аппарата - при megacolon, легочной ткани - при кистозном легком и др. По срокам возникновения эти пороки относятся к ранним фетопатиям. Ранние фетопатии часто сочетаются с эмбриопатиями; например, врожденный ихтиоз и хондродисплазия - с пороками развития лица, болезнь Марфана - с пороками развития лица и аорты и др. Частота врожденных пороков, по данным ВОЗ, составляет 1,3% от общего числа рождений.
Любой врожденный порок может проявляться в виде: 1) отсутствия какого-либо органа или части тела (агенезия, аплазия); 2) недоразвития органа (гипоплазия); 3) чрезмерного развития (гиперплазия) или наличия избыточного числа органов (удвоение и др.); 4) изменения формы (слияние органов, атрезия, стеноз отверстий, каналов, дизрафия - незаращение эмбриональных щелей, экстрофия - выворот и др.); 5) изменения
в расположении органов (эктопия); 6) персистирования эмбриональных провизорных (предсуществовавших) органов.
Классификация. Врожденные пороки развития разделяют по степени распространенности в организме, по локализации в том или ином органе, по этиологии. По распространенности врожденные пороки могут быть: 1) изолированными - с поражением одного органа; 2) системными - с поражением нескольких органов одной из систем; 3) множественными - с поражением органов разных систем. По локализации различают пороки развития центральной нервной, сердечно-сосудистой, пищеварительной, мочеполовой и других систем. Врожденные пороки развития названной локализации имеют наибольшее значение в патологии. Чаще всего встречаются пороки развития центральной нервной и сердечно-сосудистой систем, так как именно эти системы имеют наибольший тератогенный терминационный период (см. рис. 292). Изолированные пороки развития встречаются чаще множественных, несмотря на то что тератогенный терминационный период для многих органов во времени совпадает.
Наиболее совершенной является классификация пороков развития по этиологии, однако уровень современных знаний пока не позволяет ее придерживаться. Однако известны отдельные виды системных и множественных врожденных пороков, связанных с определенной этиологией, например рубеолярная эмбриопатия, алкогольная, талидомидная эмбриопатии и др., а также наследственно обусловленные генотипические врожденные пороки и врожденные пороки вследствие хромосомных аберраций; последние, как правило, носят характер множественных.
Разграничение генотипических врожденных пороков с их фенокопиями возможно с помощью генеалогического метода изучения родословной, цитогенетического метода, позволяющего изучить кариотип тканей носителя порока при их культивировании, с помощью близнецового метода, основанного на частоте выявления врожденных пороков у однояйцевых близнецов и метода дерматоглифики - изучения комплекса кожных узоров, расположенных на ладонях, подошвах и сгибательной поверхности пальцев, который используется для срочной диагностики хромосомных болезней.
Врожденные пороки центральной нервной системы
Врожденные пороки ЦНС по частоте занимают первое место среди других пороков, встречаются в 30% случаев среди пороков развития, обнаруживаемых у детей.
Этиология и патогенез. Из экзогенных факторов точно установлено значение вируса краснухи, иммунодефицита человека, простого герпеса, предполагается влияние вирусов цитомегалии, Коксаки, лекарственных препаратов (хинин, гидантоин и др.), алкоголя, лучевой энергии, гипоксии. Несомненное значение имеют генные мутации; при хромосомных болезнях в числе множественных пороков они встречаются почти как правило. Развитие порока связано с воздействием повреждающего агента в течение всего эмбрионального периода, включая ранний фетальный.
Наиболее тяжелые пороки возникают при повреждении в начале закладки нервной трубки (3-4-я неделя внутриутробной жизни).
Патологическая анатомия. К основным наиболее тяжелым видам врожденных пороков ЦНС относятся следующие. Анэнцефалия - агенезия головного мозга, при которой отсутствуют передние, средние, иногда и задние его отделы. Продолговатый и спинной мозг сохранены. На месте головного мозга обнаруживается соединительная ткань, богатая сосудами, в которой встречаются отдельные нейроны и клетки нейроглии. Анэнцефалия сочетается с акранией - отсутствием костей свода черепа, покрывающих их мягких тканей и кожи.
Микроцефалия - гипоплазия головного мозга, уменьшение его массы и объема; сочетается с одновременным уменьшением объема черепной коробки и утолщением костей черепа; возможны разные степени тяжести этого порока. Микрогирия - увеличение числа мозговых извилин наряду с уменьшением их величины.
Порэнцефалия - появление кист различных размеров в головном мозге, сообщающихся с боковыми желудочками мозга, выстланных эпендимой. От истинной порэнцефалии следует отличать ложную, при которой кисты не сообщаются с путями оттока ликвора и образуются на месте бывших размягчений ткани головного мозга.
Врожденная гидроцефалия - избыточное накопление ликвора в желудочках мозга (внутренняя гидроцефалия) или в субарахноидальных пространствах (наружная гидроцефалия) (рис. 294) сопровождается увеличением мозгового черепа и резким несоответствием его с лицевым - лицо кажется маленьким, лоб - нависшим. Наблюдаются расхождение и ис-
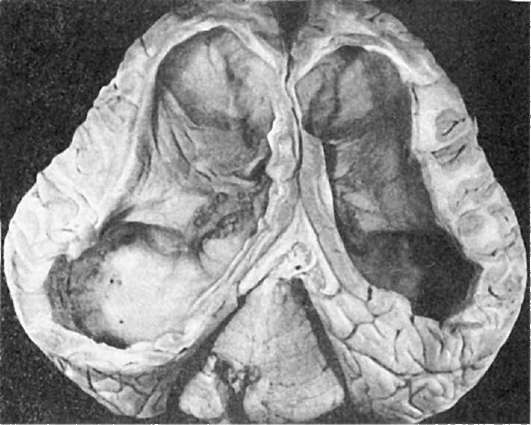 Рис. 294. Гидроцефалия (по А.В. Цинзерлингу)
Рис. 294. Гидроцефалия (по А.В. Цинзерлингу)
тончение костей черепа, выбухание родничков. Нарастает атрофия вещества головного мозга, в большинстве случаев связанная с нарушениями оттока ликвора вследствие стеноза, раздвоения или атрезии водопровода большого мозга (сильвиева водопровода), атрезии срединных и боковых отверстий IV желудочка и межжелудочкового отверстия.
Циклопия - редкий порок, характеризующийся наличием одного или двух глазных яблок, расположенных в одной глазнице, с одновременным пороком развития носа и обонятельной доли головного мозга. Назван изза сходства лица плода с лицом мифического чудовища - циклопа.
Грыжи головного и спинного мозга представляют собой выпячивание вещества мозга и его оболочек через дефекты костей черепа, их швов и позвоночного канала. Грыжи головного мозга: при наличии в грыжевом мешке только оболочек головного мозга и ликвора носят название менингоцеле, оболочек и вещества мозга - менингоэнцефалоцеле, вещества мозга и мозговых желудочков - энцефалоцистоцеле. Чаще встречаются грыжи спинного мозга, связанные с расщеплением дорсальных отделов позвонков, которые называются spina bifida. Грыжи спинного мозга, как и головного, в зависимости от содержимого грыжевого мешка можно разделять на менингоцеле, миелоцеле, менингомиелоцеле. Очень редко встречается рахиосхиз - полный дефект задней стенки позвоночного канала, мягких тканей, кожи и мозговых оболочек; при этом распластанный спинной мозг лежит открытым на передней стенке канала, выпячивания нет.
Прогноз при врожденных пороках ЦНС неблагоприятен, большинство из них несовместимы с жизнью. Хирургическая коррекция эффективна только в некоторых случаях мозговых и спинномозговых грыж. Дети умирают часто от присоединения интеркуррентных инфекционных заболеваний. Мозговые и спинномозговые грыжи осложняются гнойной инфекцией с развитием гнойного менингита и менингоэнцефалита.
Врожденные пороки сердца
Врожденные пороки сердца по частоте занимают второе место после пороков развития ЦНС. По данным разных авторов, они встречаются в 16-40% среди других пороков и в 3-8% случаев по данным вскрытий детей, умерших в перинатальном периоде.
Этиология и патогенез. Причины этих пороков разнообразны и не связаны с влиянием каких-либо определенных экзогенных факторов. Имеют несомненное значение генные мутации и хромосомные аберрации. Среди множественных пороков, наблюдающихся при хромосомных болезнях, пороки сердца встречаются реже, чем пороки ЦНС. Развитие порока связано с воздействием повреждающего агента на эмбрион от 3-й до 11-й недели внутриутробного развития. Различные виды пороков зависят от искажения этапов морфогенеза сердца, из которых основными являются дефекты первоначально парных закладок сердца, неправильные изгибы первичной сердечной трубки, задержка развития или неправильное расположение перегородок сердца, делящих его и артериальный ствол на
правую и левую половины, персистирование предсердно-желудочковых соединений, существующих во время внутриутробной жизни.
Патологическая анатомия. При врожденных пороках сердца в процессе гипертрофии миокарда у детей в возрасте первых 3 мес жизни участвуют не только увеличение объема мышечных волокон с гиперплазией их ультраструктур, но и истинная гиперплазия кардиомиоцитов. Одновременно с этим развивается гиперплазия ретикулиновых аргирофильных волокон стромы сердца. Последующие дистрофические изменения миокарда и стромы, вплоть до развития микронекрозов, приводят к постепенному разрастанию соединительной ткани и возникновению диффузного и очагового кардиосклероза.
Компенсаторная перестройка сосудистого русла гипертрофированного сердца сопровождается увеличением в нем интрамуральных сосудов, артерио-венозных анастомозов, наименьших вен (так называемых сосудов Вьессена-Тебезия) сердца. В связи со склеротическими изменениями в миокарде, а также усилением кровотока в его полостях появляется утолщение эндокарда за счет разрастания в нем эластических и коллагеновых волокон. Перестройка сосудистого русла развивается также и в легких. У детей с врожденными пороками сердца наблюдается отсталость общего физического развития.
Смерть наступает в первые дни жизни от гипоксии при особо тяжелых формах пороков или позже от развития сердечной недостаточности. С прогрессом грудной хирургии стало возможным лечение многих врожденных пороков с использованием хирургической коррекции и протезирования, что заметно изменило течение и исходы врожденных пороков сердца у детей. Благодаря сложности процессов эмбриогенеза сердца врожденные пороки его разнообразны. Однако большинство из них связано с ненормальными сообщениями между малым и большим кругом кровообращения, сужениями в этих системах или с отсутствием нормальных сообщений между ними, вплоть до несовместимого с жизнью полного разобщения малого и большого круга кровообращения. В зависимости от степени гипоксии, обусловленной уменьшением кровотока в малом круге кровообращения и направлением тока крови через ненормальные пути между малым и большим кругом кровообращения, пороки сердца могут быть разделены на два основных типа - синий и белый. При пороках синего типа отмечаются уменьшение кровотока в малом круге кровообращения, гипоксия и направление тока крови по анормальному пути - справа налево. При пороках белого типа гипоксия отсутствует, направление тока крови слева направо. Однако это деление схематично и не всегда применимо ко всем типам врожденных пороков сердца.
Врожденные пороки с нарушением деления полостей сердца. Дефект межжелудочковой перегородки встречается часто, возникновение его зависит от отставания в росте одной из структур, формирующих перегородку, вследствие чего между желудочками развивается ненормальное сообщение. Чаще наблюдается дефект в верхней соединительнотканной (мембранозной) части перегородки (рис. 295). Кровоток через дефект
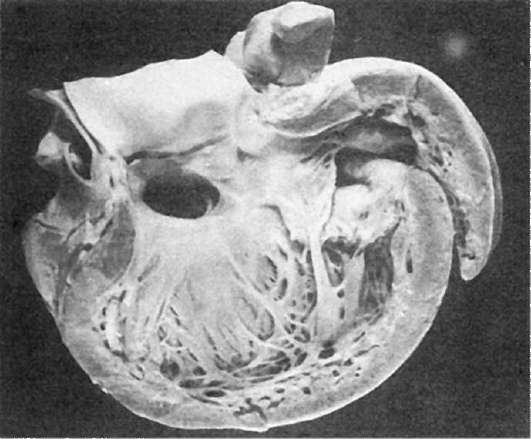 Рис. 295. Дефект в межжелудочковой перегородке сердца (по А.В. Цинзерлингу)
Рис. 295. Дефект в межжелудочковой перегородке сердца (по А.В. Цинзерлингу)
осуществляется слева направо, поэтому цианоза и гипоксии не наблюдается (белый тип порока). Степень дефекта может варьировать, вплоть до полного отсутствия перегородки. При значительном дефекте развивается гипертрофия правого желудочка сердца, при незначительном - существенных изменений гемодинамики не происходит.
Дефект межпредсердной перегородки в виде изолированного порока встречается редко. Он возникает либо при нарушениях развития первичной предсердной перегородки на 5-й неделе эмбриогенеза, либо позднее, при формировании вторичной перегородки и овального отверстия. Дефект первичной перегородки имеет вид отверстия, расположенного непосредственно над клапанами желудочков; при дефекте вторичной перегородки имеется широко открытое овальное отверстие, лишенное заслонки. В том и в другом случае ток крови происходит слева направо, гипоксии и цианоза не бывает (белый тип порока). Переполнение кровью правой половины сердца сопровождается гипертрофией правого желудочка и расширением ствола и ветвей легочной артерии. Полное отсутствие межжелудочковой или межпредсердной перегородок приводит к развитию трехкамерного сердца - тяжелого порока, при котором, однако, в период компенсации не наблюдается полного смешения артериальной и венозной крови, так как основной ток той или другой крови сохраняет свое направление и поэтому степень гипоксии нарастает по мере прогрессирования декомпенсации.
Врожденные пороки сердца с нарушениями деления артериального ствола. Общий артериальный ствол при полном отсутствии деления артериального ствола встречается редко. При этом пороке один общий артериальный
ствол берет свое начало от обоих желудочков, у выхода располагается 4 полулунных клапана или меньше; порок часто сочетается с дефектом межжелудочковой перегородки. Легочные артерии отходят от общего ствола недалеко от клапанов, до ответвления крупных сосудов головы и шеи, они могут совсем отсутствовать и тогда легкие получают кровь из расширенных бронхиальных артерий. При этом пороке наблюдаются резкая гипоксия и цианоз (синий тип порока), дети нежизнеспособны.
Полная транспозиция легочной артерии и аорты возникает при неправильном направлении роста перегородки артериального ствола, когда она растет не по спирали, а в направлении, противоположном остальным, нормально развивающимся отделам сердца. При этом пороке аорта помещается спереди и справа от правого желудочка сердца, легочная артерия лежит позади аорты и отходит от левого желудочка. Артериальная кровь может попасть в большой круг кровообращения только при дефектах в перегородках сердца или при незаращении артериального (боталлова) протока и овального отверстия. Порок сопровождается резкой гипоксией и цианозом (синий тип порока).
Значительно страдает миокард, так как венечные артерии не получают артериальной крови. Дети нежизнеспособны.
Стеноз и атрезия легочной артерии наблюдаются при смещении перегородки артериального ствола вправо, часто сочетаются с дефектом межжелудочковой перегородки и другими пороками. При значительном сужении легочной артерии кровь в легкие попадает через артериальный (боталлов) проток и расширяющиеся бронхиальные артерии. Порок сопровождается гипоксией и выраженным цианозом (синий тип порока).
Стеноз и атрезия аорты являются следствием смещения перегородки артериального ствола влево. Они встречаются реже, чем смещение перегородки вправо, часто сопровождаются гипоплазией левого желудочка сердца. При этом наблюдаются резкая степень гипертрофии правого желудочка сердца, расширение правого предсердия и резкий общий цианоз. Дети нежизнеспособны.
Сужение перешейка аорты (коарктация), вплоть до его атрезии, компенсируется развитием коллатерального кровообращения через межреберные артерии, артерии грудной клетки и резкой гипертрофией левого желудочка сердца.
Незаращение артериального (боталлова) протока можно считать пороком при наличии его с одновременным расширением у детей старше 3 мес жизни. Ток крови осуществляется при этом слева направо (белый тип порока). Изолированный порок хорошо поддается хирургической коррекции.
Комбинированные врожденные пороки сердца. Среди комбинированных пороков чаще встречаются триада, тетрада и пентада Фалло. Триада Фалло имеет 3 признака: дефект межжелудочковой перегородки, стеноз легочной артерии и как следствие этого гипертрофия правого желудочка. Тетрада Фалло имеет 4 признака: дефект межжелудочковой перегородки,
сужение легочной артерии, декстрапозиция аорты (смещение устья аорты вправо) и гипертрофия правого желудочка сердца. Пентада Фалло, кроме этих четырех, включает 5-й признак - дефект межпредсердной перегородки. Чаще всего встречается тетрада Фалло (40-50% всех врожденных пороков сердца). При всех пороках типа Фалло отмечаются ток крови справа налево, уменьшение кровотока в малом круге кровообращения, гипоксия и цианоз (синий тип пороков). К более редким комбинированным врожденным порокам относятся дефект межжелудочковой перегородки со стенозом левого предсердно-желудочкового отверстия (болезнь Лютамбаше), дефект межжелудочковой перегородки и декстрапозиция аорты (болезнь Эйзенменгера) и ответвление левой венечной артерии от легочного ствола (синдром Бланда-Уайта-Гарленда), первичная легочная гипертензия (болезнь Аэрза), зависящая от гипертрофии мышечного слоя сосудов легкого (мелких артерий, вен и венул) и др.
Врожденные пороки органов пищеварения
Врожденные пороки органов пищеварения встречаются в 3-4% вскрытий умерших в перинатальном периоде и составляют 21% всех врожденных пороков этого периода. Они чаще всего представляют собой атрезии и стенозы пищеварительного тракта.
Этиология и патогенез. Атрезия анального отверстия, как и другие пороки каудального конца зародыша, встречаются чаще при диабетической эмбриопатии. В целом происхождение этих пороков различно.
Патогенез связан с нарушением образования отверстий пищеварительной трубки в периоде от 4-й до 8-й недели внутриутробного развития, так как вначале эта трубка заканчивается слепо с обоих концов. Имеет значение и задержка реканализации, так как на 8-й неделе внутриутробной жизни растущий эпителий полностью закрывает просвет кишечной трубки, который в дальнейшем восстанавливается при формировании слизистой оболочки.
Атрезии и стенозы чаще наблюдаются в пищеводе, двенадцатиперстной кишке, проксимальном отрезке тощей и дистальном отрезке подвздошной кишки, в области прямой кишки и анального отверстия. В толстой кишке они встречаются редко. В пищеводе, кроме того, могут наблюдаться трахеопищеводные свищи (рис. 296), образование которых зависит от нарушения деления первичной кишки на пищевод и трахею. Эти свищи приводят к развитию тяжелой аспирационной пневмонии. Атрезии кишки могут быть одиночными и множественными, причем при последних кишечник напоминает «связку сосисок». В области атрезии кишка имеет вид плотного соединительнотканного шнура, который под влиянием перистальтики может растягиваться и разрываться, что приводит к перфоративному перитониту в первые дни жизни новорожденного.
Атрезии и стеноз прямой кишки и анального отверстия могут наблюдаться в разных вариантах: 1) атрезия только анального отверстия - при нормальном развитии прямая кишка отделена от него перепонкой; 2) атрезия только прямой кишки - анальное отверстие ведет в короткий слепой канал,
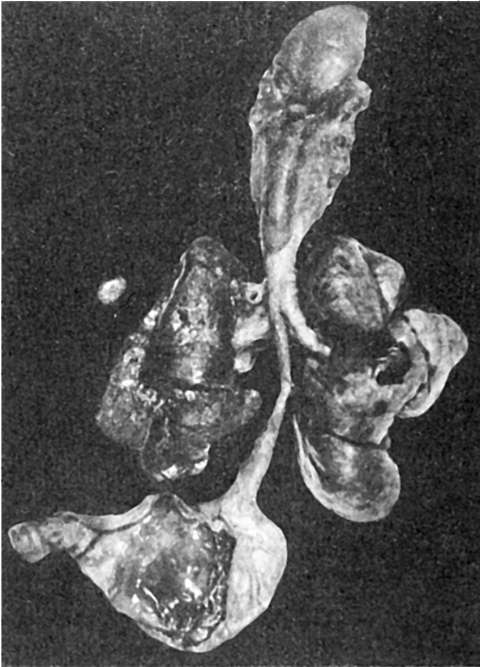 Рис. 296. Трахеопищеводный
свищ. Верхний сегмент заканчивается слепо, резко расширен; нижний
сегмент открывается в трахею в области ее бифуркации (по А.В.
Цинзерлингу)
Рис. 296. Трахеопищеводный
свищ. Верхний сегмент заканчивается слепо, резко расширен; нижний
сегмент открывается в трахею в области ее бифуркации (по А.В.
Цинзерлингу)
выше него лежит тяж недоразвитой прямой кишки; 3) атрезия анального отверстия и прямой кишки - анальное отверстие отсутствует, слепой конец прямой кишки расположен высоко; 4) атрезия со свищами - прямая кишка открывается в мочевой пузырь, мочеиспускательный канал, влагалище, мошонку, промежность и другие органы мочеполовой системы.
Удвоение отдельных участков кишечника касается чаще только слизистой оболочки, мышечная оболочка бывает общей. Удвоенный участок может иметь форму кисты, дивертикула или трубки. Порок осложняется кровотечением, воспалением, некрозом с перфорацией.
Болезнь Гиршпрунга (относится к ранним фетопатиям) - сегментарный аганглиоз, megacolon - отсутствие нейронов межмышечного (так называемого ауэрбахова) сплетения нижнего отрезка сигмовидной и прямой кишки. Вследствие сохранности подслизистого (так
называемого мейсснерова) сплетения аганглионарный участок кишки спастически сокращен, выше него происходит растяжение кишки меконием или калом с развитием последующей компенсаторной гипертрофии мышечной оболочки; в растянутом отрезке иногда наблюдаются изъязвления. Больные страдают запорами, развивается непроходимость.
Гипертрофический пилоростеноз (относится к ранним фетопатиям) - врожденная гипертрофия мускулатуры пилорического отдела желудка с сужением его просвета; самый частый врожденный порок желудка, этиология и патогенез которого не выяснены. Встречается у 0,3% новорожденных, у мальчиков - в 5-7 раз чаще. Отмечается семейный характер заболевания. Вместо нормального пилорического кольца имеется трубка с узким просветом и толстыми плотными стенками, вдающаяся в двенадцатиперстную кишку. Наблюдается упорная рвота, начинающаяся с 3-4-й недели жизни, вплоть до развития комы от потери хлоридов. Хирургическое лечение приводит к полному излечению.
Пороки пищеварительного тракта, связанные с сохранением некоторых эмбриональных структур. К ним относятся грыжа пупка, кисты и свищи пупочного кольца и меккелев дивертикул.
Грыжа пупка - дефект передней брюшной стенки в области пупка с выпячиванием полупрозрачного грыжевого мешка, образованного пуповиной и амнионом, содержащего петли тонкой кишки. Порок возникает вследствие того, что петли кишечника не переходят в брюшную полость на 8-10-й неделе внутриутробного развития. Поддается хирургическому лечению. От него следует отличать эвентрацию органов брюшной полости с ее гипоплазией, при которой брюшная стенка широко открыта, грыжевой мешок отсутствует, хирургическое лечение неэффективно. Кисты и свищи области пупочного кольца образуются вследствие персистирования желточного протока, соединяющего кишечную трубку с желточным мешком на ранних этапах внутриутробного развития. Если проток сохранен полностью, появляется пупочно-кишечный свищ, из которого наружу выделяется кал. Если он сохранен частично, в области пупка формируется кишечная киста - энтерокистома, частичное сохранение протока в области кишки приводит к образованию меккелева дивертикула - пальцевидного выпячивания стенки подвздошной кишки, расположенного обычно на 25 см выше подвздошно-слепокишечной (так называемой баугиниевой) заслонки. Между ними и пупочным кольцом иногда сохраняется остаток желточного протока в виде соединительнотканного тяжа. Все эти пороки могут приводить к кровотечениям, воспалению, в том числе перитониту, непроходимости, инвагинации; подлежат хирургическому лечению.
Врожденные пороки печени и желчных путей. Поликистоз печени - множественные кисты разных размеров, выстланные эпителием и заполненные прозрачной жидкостью, - встречается редко, часто сочетается с поликистозом почек и поджелудочной железы.
Атрезия и стеноз внепеченочных желчных протоков может наблюдаться в одном или во всех трех протоках. Агенезия и гипоплазия внутрипеченочных желчных протоков (относится к ранним фетопатиям) - уменьшение числа или полное отсутствие желчных протоков в портальном тракте в области триад. Этиология связана с вирусом гепатита. Отмечаются семейные случаи. Рассматривается как следствие нарушений образования протоков из печеночного дивертикула (5-8-я неделя) или задержки реканализации (8-й неделя внутриутробного развития). Желтуха очень интенсивная, развивается на 3-5-й день жизни, порок приводит к билиарному циррозу печени. При агенезии и гипоплазии внутрипеченочных протоков возможно развитие врожденного, билиарного цирроза. Дети доживают до 6-7 мес. При врожденном циррозе они погибают в первые дни жизни от печеночной недостаточности.
Врожденная гиперплазия внутрипеченочных желчных протоков (относится к ранним фетопатиям) - причудливое разрастание желчных протоков в области портального тракта с избыточным развитием соединительной ткани - комбинируется с мелкокистозными почками. Желтуха нехарактерна, она появляется в случае присоединения вторичного гнойного холангита. Этот порок встречается и у взрослых. При комбинации с мелким кистозом почек смерть наступает от почечной недостаточности в первые дни жизни.
Врожденные пороки почек, мочевыводящих путей и половых органов
Этиология. Развитие пороков не связано с действием определенных экзогенных агентов. Многие из них являются наследственными или семейными. Встречаются при хромосомных синдромах. Пороки эти многообразны и возникают в период 4-8-й недели киматогенеза.
Врожденные пороки почек. Агенезия почек - врожденное отсутствие одной или обеих почек (арения) - встречается редко, при этом у новорожденного выражена складчатость кожи, лицо одутловатое, старческое, ушные раковины расположены низко, нос широкий и плоский, выступают лобные бугры; наблюдаются врожденные пороки и других органов. Дети нежизнеспособны.
Гипоплазия почек - врожденное уменьшение их массы и объема, может быть одно- и двусторонним; при односторонней гипоплазии наблюдается викарная гипертрофия второй нормальной почки.
Дисплазия почек - гипоплазия с одновременным наличием в почках эмбриональных тканей. Микроскопически в ткани почки обнаруживаются очажки нефробластомы, примитивные канальцы и клубочки, кисты, островки хряща и гладкой мышечной ткани. Сочетаются с аплазией, гипоплазией или атрезией и стенозом мочеточников. При двусторонней резко выраженной гипоплазии и дисплазии почек дети нежизнеспособны.
Крупнокистозные почки (поликистоз почек взрослого типа) - двустороннее значительное увеличение почек с образованием в их корковом слое большого числа крупных кист с прозрачным содержимым, между кистами - участки нормальной почечной ткани (рис. 297). Порок комбинируется с кистами печени и поджелудочной железы, наследуется по доминантному типу. Патогенез его связывают с нарушением соединения зачатков метанефрогенной ткани и уретральной трубки, образуются ретенционные кисты.
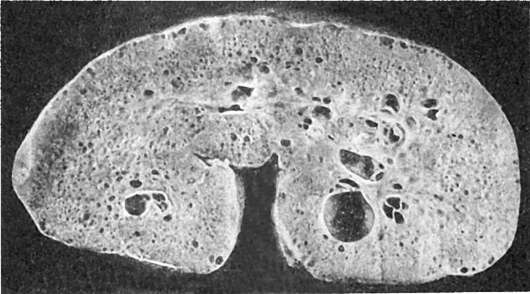 Рис. 297. Поликистозная почка (макрокистоз)
Рис. 297. Поликистозная почка (макрокистоз)
Мелкокистозные почки (поликистоз «инфантильного» типа) - двустороннее значительное увеличение почек с образованием многочисленных мелких кист в корковом и мозговом слоях, тесно прилежащих друг к другу. Почки имеют вид крупнопористой губки. При микроскопическом исследовании вся почечная ткань выглядит сплошь замещенной кистами, выстланными кубическим эпителием. Порок сочетается с кистами печени и гиперплазией желчных ходов. Дети нежизнеспособны.
Сращение почек (подковообразная почка) и дистопия клинически не проявляются.
Врожденные пороки мочевыводящих путей. Основные из них: 1) удвоение лоханок и мочеточников; 2) агенезия, атрезия, стеноз мочеточников, эктопия их устьев; 3) мегалоуретер относится к ранним фетопатиям - резкое расширение мочеточника (одного или двух) вследствие гипоплазии мышечной ткани или нервного аппарата; 4) экстрофия мочевого пузыря в результате аплазии его передней стенки, брюшины и кожи в области лобка; 5) агенезия мочевого пузыря; 6) атрезия, стеноз уретры (чаще у девочек) и гипоспадия - дефект нижней стенки, эписпадия - дефект верхней стенки мочеиспускательного канала у мальчиков.
Все пороки мочевыводящих путей ведут к нарушению оттока мочи и без своевременного хирургического лечения, которое в настоящее время с успехом выполняется, приводят к почечной недостаточности. Наиболее тяжелые из них (например, агенезия, атрезия мочевых путей) приводят к смерти от уремии вскоре после рождения, другие долго могут не проявляться клинически, однако постепенно приводят к гидронефрозу, иногда к образованию камней, возникновению восходящего хронического пиелонефрита, что угрожает развитием почечной недостаточности, заканчивающейся смертью больного (чаще в 20-30 лет) от уремии.
Врожденные пороки половых органов. Они часто связаны с болезнью эндокринных желез (надпочечников, гипофиза) матери и плода, с приемом гормональных препаратов во время беременности; установлена наследственная передача по рецессивному типу. К ним относятся: крипторхизм - задержка яичек в брюшной полости или в паховом канале (у новорожденных не следует считать пороком, так как у них встречается крипторхизм в 30% случаев, к 12-16 годам наблюдается только в 2-3% случаев и тогда расценивается как порок); атрезия шейки матки и влагалища, удвоение матки; гермафродитизм - наличие признаков обоего пола у одного лица. Различают истинный гермафродитизм - одновременное наличие женских и мужских половых желез и ложный - наружные половые органы пола, противоположного половым железам. Пороки развития половых органов жизни не угрожают, в некоторых случаях возможно хирургическое лечение.
Врожденные пороки органов дыхания
Врожденные пороки органов дыхания часто сочетаются с другими пороками и встречаются у 4,2% умерших в перинатальном периоде, у 3% детей, умерших до 1 года.
Аплазия и гипоплазия бронхов и легких, одного легкого или его доли встречаются чаще. Гипоплазия легких бывает преимущественно вторичной, когда имеет место дисплазия грудной клетки.
Кисты легких (относятся к ранним и поздним фетопатиям) могут быть множественными (поликистоз легких), располагаться в одном легком, в одной доле или быть единичными. Кисты имеют разное происхождение - образуются при агенезии одного из порядков ветвления бронхов. В первом случае газообмен не осуществляется, так как слепо заканчивающиеся разветвления бронхов окружены соединительной тканью. Во втором случае крупные и средние бронхи непосредственно переходят в легочную ткань или бронхиолы. В постнатальном периоде установившийся акт дыхания приводит к эктазии кист бронхов с развитием так называемых врожденных бронхоэктазов (рис. 298).
Врожденная эмфизема (относится к ранним и поздним фетопатиям) - резкое вздутие чаще верхней доли левого легкого в связи с гипоплазией хрящей, эластической и мышечной тканей бронхов. Она вызывает смещение органов средостения в противоположную сторону. Порок выявляется только в постнатальном периоде.
Врожденные пороки легких, если они совместимы с жизнью, приводят к осложнениям в виде вторичной инфекции с развитием хронического бронхита и пневмонии, следствием которых являются пневмосклероз, облитерация плевральных полостей, легочное сердце с последующей его недостаточностью. Смерть от этих осложнений чаще наблюдается у взрослых.
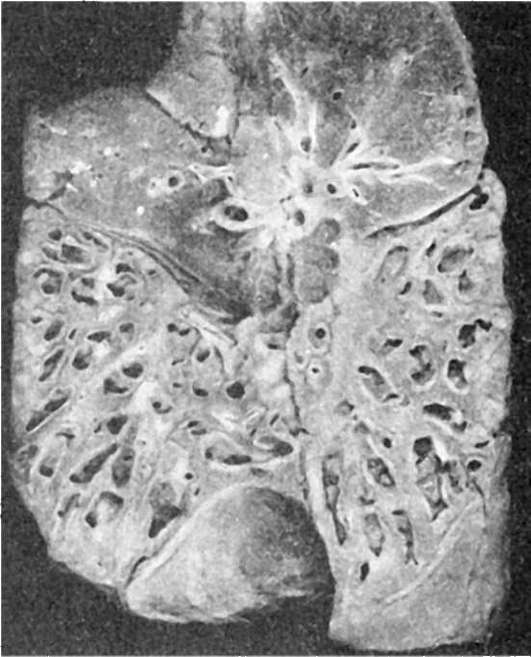 Рис. 298. Врожденные бронхоэктазы (по А.В. Цинзерлингу)
Рис. 298. Врожденные бронхоэктазы (по А.В. Цинзерлингу)
Врожденные пороки костно-суставной и мышечной систем
Врожденные пороки скелета и мышечной системы встречаются, по данным вскрытий, в 0,5-0,8 и 13,5% случаев соответственно среди всех пороков у умерших в перинатальном периоде. В их этиологии из экзогенных факторов особое значение имеет талидомид. Различают системные и изолированные пороки скелета.
Системные пороки костно-суставной системы. Хондродисплазия (относится к ранним фетопатиям) - группа врожденных пороков, характеризующихся значительным укорочением и утолщением конечностей. Хондродисплазия плода, или летальная микромелия (рис. 299), - укорочение и утолщение конечностей, кожа их образует крупные складки, головка новорожденного увеличена, нос седловидный, рот приоткрыт, язык толстый, шея короткая, тела позвонков тоже утолщены, грудная клетка гипоплазирована; порок сочетается с гипоплазией легких. Другим видом хондродисплазии является ахондроплазия, характеризующаяся только укорочением и утолщением конечностей и нарушением развития костей лицевого скелета. Порок проявляется позже, когда становится заметным отставание ребенка в росте; наследуется по доминантному типу, возможны спонтанные мутации генов. Сущность порока состоит в нарушении развития костей хрящевого генеза, кости соединительного происхождения развиваются нормально.
При микроскопическом исследовании обнаруживается изменение энхондрального костеобразования при сохранности периостального, что ведет к нарушению роста трубчатых костей в длину. Прогноз для жизни благоприятный, умственной осталости не наблюдается.
Несовершенный остеогенез (ранняя фетопатия) - врожденная ломкость костей, наследуется по доминантному типу. Порок характеризуется множественными, часто врожденными переломами с искривлениями конечностей и ребер. Свод черепа построен только из соединительной ткани, наблюдается отосклероз, голубые склеры, гидроцефалия.
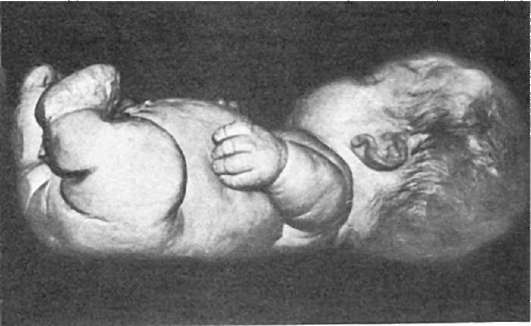 Рис. 299. Хондродисплазия (по А.В. Цинзерлингу)
Рис. 299. Хондродисплазия (по А.В. Цинзерлингу)
Врожденная мраморная болезнь (ранняя фетопатия) - выраженный остеосклероз с одновременным нарушением развития кроветворной ткани наследственного характера. Дети умирают в первые месяцы, реже - в первые годы жизни.
Изолированные пороки костно-суставной системы. К ним относятся врожденный вывих и дисплазия тазобедренного сустава одноили двусторонний (ранняя фетопатия), врожденная ампутация и аплазия (амелия) конечностей, фокомелия - недоразвитие проксимальных отделов конечностей, когда стопы и кисти начинаются непосредственно от туловища, полидактилия - увеличение числа пальцев, синдактилия - сращение пальцев и др.
Системная гипоплазия мышечной ткани. Примером ее может быть врожденная миатония Оппенгейма (относится к ранним фетопатиям), при которой наблюдается гипоплазия поперечнополосатых мышц. В первые месяцы жизни дети умирают от пневмонии, развитие которой связано с гипоплазией дыхательных мышц, за исключением диафрагмы.
Изолированные пороки мышечной системы. К важнейшим из них относятся: врожденные истинные и ложные диафрагмальные грыжи. При ложных грыжевой мешок отсутствует, имеется дефект диафрагмы, объем которого варьирует, через него органы брюшной полости, чаще петли кишок, могут проникать в грудную полость. Врожденная кривошея характеризуется укорочением грудиноключично-сосцевидной мышцы вследствие ее очагового фиброза, отчего головка ребенка наклоняется в пораженную сторону.
Врожденные пороки лица. Основными врожденными пороками являются: хейлосхиз - расщепление верхней губы, палатосхиз - расщепление твердого неба, микрогнатия - гипоплазия нижней челюсти, гипертелоризм - широкое расстояние между глазами. Эти пороки часто сочетаются с другими множественными пороками развития.
Фетопатии
Фетопатии - патология фетального периода с 76-го по 280-й день беременности, в течение которого заканчивается основная тканевая дифференцировка органов и формирование плаценты. Характерной особенностью фетопатии является сочетание поражений двух типов - нарушений тканевого морфогенеза с реактивными изменениями в виде расстройств кровообращения, дистрофии, некрозов, воспаления, иммунных реакций, компенсаторно-приспособительных процессов, регенерации. При ранних фетопатиях преобладают нарушения тканевого морфогенеза, при поздних - реактивные процессы. Следует различать инфекционные и неинфекционные фетопатии.
Инфекционные фетопатии
Этиология и патогенез. Инфекционные фетопатии могут быть связаны с воздействием вирусов, многих бактерий и других возбудителей. В плаценте при этом часто возникает воспаление.
Инфицирование плода осуществляется чаще всего гематогенным путем. Возбудитель через плаценту по пупочной вене попадает в организм плода. При переходе воспалительного процесса с плаценты на плодные оболочки возможно инфицирование околоплодных вод с последующим заглатыванием или аспирацией плодом возбудителя. Реже инфицирование осуществляется восходящим путем через влагалище в канал шейки матки или нисходящим путем через трубы, если у матери в брюшной полости имеется очаг воспаления. Источником заражения чаще являются вялотекущие хронические или латентные инфекции матери, так как при таких формах течения инфекционных болезней содержание иммуноглобулинов и титр соответствующих иммунных антител бывают недостаточными как для завершения процесса у самой матери, так и для предотвращения заболевания плода. Такие соотношения наблюдаются, например, при токсоплазмозе, сывороточном гепатите.
Патологическая анатомия. При всех инфекционных фетопатиях наблюдается генерализованный, а при бактериальных и грибковых септический тип изменений с образованием множественных очагов ареактивного некроза в паренхиматозных органах и головном мозге (при врожденной ветряной оспе, простом герпесе, цитомегалии, инфицировании вирусом Коксаки) или продуктивных диффузных воспалительных инфильтратов в сочетании с ареактивными некротическими очагами (врожденный сывороточный гепатит, цитомегалия, краснуха, токсоплазмоз), или с образованием гранулем во многих органах (врожденный сифилис, листериоз, туберкулез, поражение грибами). При этом на фоне генерализованных поражений могут преобладать изменения в определенных органах, например при токсоплазмозе - в головном мозге, при сывороточном гепатите - в печени, при инфекции вирусом Коксаки - в миокарде и головном мозге и др. Как правило, наблюдается выраженный геморрагический синдром в виде петехий на коже, слизистых и серозных оболочках, кровоизлияний во внутренних органах, склонность к которым при инфекционном процессе возрастает вследствие развития генерализованных васкулитов. Иммунные реакции плода выражаются в задержке созревания вилочковой железы, в его атрофии с уменьшением его объема и массы, в наличии у доношенных плодов очагов экстрамедуллярного кроветворения, а у недоношенных - в увеличении их объема, что сопровождается гепато- и спленомегалией. Часто наблюдаются конъюгационная желтуха, тканевая незрелость органов у доношенных или недоношенность и общая гипотрофия плода.
Прогноз в большинстве случаев неблагоприятный, смерть наступает в первые дни или в первые 3 мес жизни. При выздоровлении остаются стойкие изменения в органах, приводящие к инвалидности или к смерти от недостаточности жизненно важных органов в другие периоды жизни.
Неинфекционные фетопатии
К основным формам неинфекционных фетопатии относятся гемолитическая болезнь новорожденных, фетальный муковисцидоз, фиброэластоз
эндокарда, диабетическая фетопатия и многие, преимущественно ранние, фетопатии. Ранние фетопатии проявляются в виде изолированных врожденных пороков (гипертрофический пилоростеноз, мегаколон, мегалоуретер, агенезия, гипоплазия и гиперплазия желчных протоков, кистоз легких и др.), а также системных врожденных пороков костно-суставной и мышечной тканей, кожи и др.
Фетальный муковисцидоз - перинатально возникающая форма муковисцидоза (кистозного фиброза поджелудочной железы). Заболевание сопровождается изменением характера слизи и других секретов, выделяемых эпителием экскреторных желез, что встречается, по данным вскрытий детей, в 0,1-0,2%. Наиболее часто встречается легочно-кишечная форма, которой болеют дети первых месяцев жизни, реже отмечается изолированная легочная или кишечная форма, наблюдающаяся у детей в любом возрасте. Совсем редко обнаруживают формы с развитием билиарного цирроза печени (встречается у детей старшего возраста и у взрослых). Фетальный муковисцидоз развивается внутриутробно или в первые дни жизни.
Этиология и патогенез. Болезнь наследуется по аутосомно-рецессивному типу. В основе патогенеза лежит, вероятно, ферментопатия, характер которой не раскрыт, приводящая к нарушению структуры гликопротеидов (мукоидов). Секрет многих желез становится густым, вязким, что приводит к задержке его эвакуации, развитию ретенционных кист и к нарушению проходимости по естественным каналам. Поражаются прежде всего экскреторный аппарат поджелудочной железы, слизистые железы дыхательного и пищеварительного трактов, желчные пути, слюнные, потовые и слезные железы.
Патологическая анатомия. При макроскопическом исследовании поджелудочная железа может быть без изменений, в редких случаях в ней отмечаются уплотнение, подчеркнутый рисунок долек, появление мелких кист. Микроскопически в кистозно расширенных протоках и в ацинусах наблюдается сгущение секрета. Железистая паренхима атрофична, островковый аппарат сохранен, в интерстиции отмечаются диффузный фиброз и лимфогистиоцитарные инфильтраты (рис. 300). Изменения могут колебаться от кистозного расширения единичных протоков и ацинусов до кистозного превращения всей экскреторной железистой паренхимы. В результате сгущения слизи в бронхах возникают обтурационные ателектазы с неизбежным вторичным инфицированием и развитием хронического бронхита, пневмонии с бронхоэктазами и абсцедированием. В кишечнике отмечается сгущение каловых масс с развитием копростаза, перфорации и калового перитонита. Изменению свойств кала способствует не только сгущение слизи, но и недостаточность поджелудочной железы (отсутствие липазы, липокаина и трипсина). В печени имеется жировая инфильтрация. Сгущение желчи ведет к холестазу и билиарному циррозу. Фетальный муковисцидоз проявляется в виде мекониальной кишечной непроходимости (мекониальный илеус). В поджелудочной железе
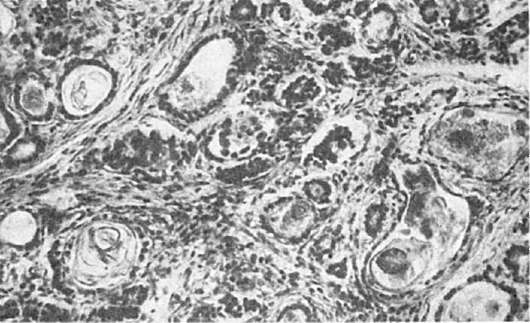 Рис. 300. Кистозный фиброз поджелудочной железы при муковисцидозе
Рис. 300. Кистозный фиброз поджелудочной железы при муковисцидозе
выраженные кистозные изменения могут отсутствовать. Вся тонкая кишка до подвздошно-слепокишечной (баугиниевой) заслонки заполнена зеленовато-оливковым густым, вязким меконием, толстая кишка спавшаяся, имеет вид так называемой микроколон. После перфорации между петлями кишки видны массы мекония и фибринозно-гнойные наложения на брюшине. При внутриутробном мекониальном перитоните между петлями кишки образуются спайки с замурованными в них зеленоватыми комочками мекония. Такие бляшкообразные плотные наложения встречаются на париетальной брюшине, на капсуле селезенки и печени.
Осложнения. Кроме осложнений, связанных непосредственно с основным заболеванием (хроническая пневмония, каловый и мекониальный перитонит, цирроз печени), у больных наблюдается прогрессирующее общее истощение, которое зависит от нарушений липидного, белкового, витаминного обмена (витаминов A, D, Е и К, растворимых в липидах) в результате нарастающей недостаточности поджелудочной железы.
Смерть наступает от легочно-сердечной недостаточности, перитонита, печеночной комы. При мекониальном илеусе дети погибают в первые дни жизни.
Фиброэластоз эндокарда - врожденное заболевание, при котором в эндокарде и в субэндокардиальном слое миокарда наблюдается склероз с обилием эластических волокон. Встречается редко.
Этиология и патогенез. Отмечается семейный характер заболевания, предполагают влияние вируса цитомегалии, белкового голодания матери, авитаминозов, гипоксии плода. Патогенез не ясен. Возможно, что ведущая роль принадлежит повреждению миокарда, в ответ на которое компенсаторно разрастаются эластическая и коллагеновая ткани эндокарда.
Патологическая анатомия. Сердце увеличено в 2,5-4 раза по сравнению с нормой за счет значительной гипертрофии преимущественно левого желудочка, эндокард его резко утолщен, беловато-желтый. Воз-
можно одновременное поражение эндокарда остальных отделов сердца. В половине случаев отмечаются утолщение и деформация митрального и аортальных клапанов, в 1/3 наблюдений - комбинация с врожденными пороками, чаще с сужением аорты.
Значительный склероз эндокарда и кардиосклероз ведут к снижению сократительной способности миокарда.
Смерть наступает от острой сердечной недостаточности (молниеносная форма) в первые дни жизни или от нарастающей недостаточности сердца при интеркуррентных заболеваниях (пневмония) в первые месяцы жизни.
Диабетическая фетопатия - заболевание плода, обусловленное предиабетом и диабетом матери.
Этиология и патогенез. Основное значение имеют нарушения углеводного обмена плода под влиянием постоянного изменения уровня глюкозы в крови матери, особенно значительных при плохо леченном диабете беременных. В связи с попыткой компенсации уровня глюкозы в крови у плода развиваются гипертрофия инсулярного аппарата с последующим истощением его и дистрофией β-клеток, а также синдром Иценко-Кушинга. После рождения, когда снижается влияние материнского диабета, могут наступить восстановление функции поджелудочной железы плода и нормализация обмена. Если этого не происходит, развивается тяжелое страдание - диабет новорожденного. Однако диабет новорожденного не всегда связан с диабетом матери, так как может зависеть от повреждений инсулярного аппарата плода другого происхождения. В противоположность этому диабетическая фетопатия связана только с диабетом и предиабетом матери.
Патологическая анатомия. При этой фетопатии имеется склонность к рождению крупных плодов - с массой тела 4-6 кг, хотя это и необязательно. Тело плода покрыто обильной сыровидной смазкой, кожа багровосинюшная с петехиями, шея короткая, лицо одутловатое, отечное, мягкие ткани туловища и конечностей пастозные (рис. 301), имеются признаки незрелости - отсутствие ядра окостенения бедра или уменьшение его размеров и др. Отмечается гепато- и кардиомегалия. При микроскопическом исследовании в поджелудочной железе наблюдается гипертрофия островков поджелудочной железы с увеличением числа β-клеток. Наряду с этим отмечаются дегрануляция, вакуолизация и пикноз ядер этих клеток, свидетельствующие об истощении их секреции. В печени имеются диффузная жировая инфильтрация, обширные очаги экстрамедуллярного кроветворения, иногда некрозы. В миокарде отмечаются вакуольная дистрофия, микронекрозы, в почках - отложение гликогена в извитых канальцах, в селезенке - экстрамедуллярное кроветворение. В сосудах микроциркуляторного русла почек, кожи, сетчатки глаза наблюдаются утолщение стенок за счет отложений ШИК-положительного материала, пролиферация эндотелия наряду со значительной извитостью и эктазией сосудистого русла.
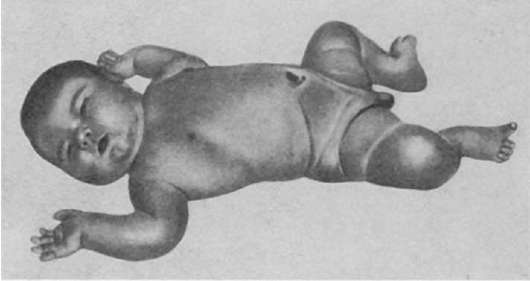 Рис. 301. Диабетическая фетопатия
Рис. 301. Диабетическая фетопатия
Из осложнений у плодов и новорожденных с диабетической фетопатией часто развивается гипоксия во время родов, образование гиалиновых мембран в легких, что зависит от дефицита антиателектатического фактора - сурфактанта, вещества фосфолипидной природы - в результате нарушений при диабетической фетопатии не только углеводного, но и липидного обмена.
Смерть наступает от асфиксии плода или новорожденного, а также от гипогликемии, наступающей после родового стресса.