Эндокринология : учебник. И.И. Дедов - 2009. - 432 с.: ил.
|
|
|
|
ГЛАВА 7 САХАРНЫЙ ДИАБЕТ
7.1. КЛАССИФИКАЦИЯ САХАРНОГО ДИАБЕТА
Сахарный диабет (СД) - группа обменных заболеваний, характеризующихся гипергликемией вследствие нарушения секреции и/или эффективности действия инсулина. Хроническая гипергликемия, развивающаяся при СД, сопровождается развитием осложнений со стороны многих органов и систем, в первую очередь, со стороны сердца, кровеносных сосудов, глаз, почек и нервов. СД в общей сложности страдают 5-6 % населения. В экономически развитых странах мира каждые 10-15 лет число больных СД возрастает в 2 раза. Ожидаемая продолжительность жизни при СД снижается на 10-15 %.
Причины развития СД широко варьируют. В подавляющем большинстве случаев СД развивается либо вследствие абсолютного дефицита инсулина (сахарный диабет 1 типа - СД-1), либо вследствие снижения чувствительности периферических тканей к инсулину в сочетании с секреторной дисфункцией β-клеток поджелудочной железы (сахарный диабет 2 типа - СД-2). В ряде случаев отнесение пациента к СД-1 или СД-2 затруднено, тем не менее на практике более значима компенсация СД, а не точное установление его типа. Этиологическая классификация выделяет четыре основных клинических класса СД (табл. 7.1).
Наиболее часто встречающиеся СД-1 (п. 7.5), СД-2 (п. 7.6) и гестационный СД (п. 7.9) обсуждаются в отдельных главах. На другие специфические типы приходится всего около 1 % случаев СД. Этиология и патогенез этих типов СД представляется более изученной по сравнению с СД-1 и особенно СД-2. Ряд вариантов СД обусловлено моногенно наследуемыми генетическими дефектами функции β-клеток. Сюда относятся различные варианты аутосомно-доминантно наследуемого синдрома MODY (англ. maturity onset diabetes of the young - диабет взрослого типа у молодых), которые характеризуются нарушением, но не отсутствием секреции инсулина при нормальной чувствительности к нему периферических тканей.
Табл. 7.1. Классификация сахарного диабета
 Казуистически редко встречаются генетические дефекты действия инсулина, связанные с мутацией рецептора инсулина (лепречаунизм, синдром Рабсона-Мандехолла). СД закономерно развивается при заболеваниях экзокринной части поджелудочной железы, приводящих
к деструкции β-клеток (панкреатит, панкреатэктомия, кистозный фиброз,
гемохроматоз), а также при ряде эндокринных заболеваний, при которых
происходит избыточная продукция контроинсулярных гормонов (акромегалия,
синдром Кушинга). Лекарственные препараты и химикаты (вакор,
пентамидин, никотиновая кислота, диазоксид и др.) редко являются
причиной СД, но могут способствовать манифестации и декомпенсации
заболевания у лиц с инсулинорезистентностью. Ряд инфекционных заболеваний (краснуха,
цитомегалия, коксаки- и аденовирусная инфекция) могут сопровождаться
деструкцией β-клеток, при этом у большинства пациентов определяются
иммуногенетические маркеры СД-1. К редким формам иммуноопосредованного диабета относят
СД, развивающийся у пациентов со «stiff-rnan»-синдромом (аутоиммунное
неврологическое заболевание), а также СД вследствие воздействия
аутоантител к рецепторам инсулина. Различные варианты СД с повышенной
частотой встречаются при
Казуистически редко встречаются генетические дефекты действия инсулина, связанные с мутацией рецептора инсулина (лепречаунизм, синдром Рабсона-Мандехолла). СД закономерно развивается при заболеваниях экзокринной части поджелудочной железы, приводящих
к деструкции β-клеток (панкреатит, панкреатэктомия, кистозный фиброз,
гемохроматоз), а также при ряде эндокринных заболеваний, при которых
происходит избыточная продукция контроинсулярных гормонов (акромегалия,
синдром Кушинга). Лекарственные препараты и химикаты (вакор,
пентамидин, никотиновая кислота, диазоксид и др.) редко являются
причиной СД, но могут способствовать манифестации и декомпенсации
заболевания у лиц с инсулинорезистентностью. Ряд инфекционных заболеваний (краснуха,
цитомегалия, коксаки- и аденовирусная инфекция) могут сопровождаться
деструкцией β-клеток, при этом у большинства пациентов определяются
иммуногенетические маркеры СД-1. К редким формам иммуноопосредованного диабета относят
СД, развивающийся у пациентов со «stiff-rnan»-синдромом (аутоиммунное
неврологическое заболевание), а также СД вследствие воздействия
аутоантител к рецепторам инсулина. Различные варианты СД с повышенной
частотой встречаются при
многих генетических синдромах, в частности, при синдромах Дауна, Клайнфелтера, Тернера, Вольфрама, Прадера-Вилли и ряде других.
7.2. КЛИНИЧЕСКИЕ АСПЕКТЫ ФИЗИОЛОГИИ УГЛЕВОДНОГО ОБМЕНА
Инсулин синтезируется и секретируется β-клетками островков Лангерганса поджелудочной железы (ПЖЖ). Кроме того, островки Лангерганса секретируют глюкагон (α-клетки), соматостатин (δ-клетки) и панкреатический полипептид (РР-клетки). Гормоны островковых клеток взаимодействуют между собой: глюкагон в норме стимулирует секрецию инсулина, а соматостатин подавляет секрецию инсулина и глюкагона. Молекула инсулина состоит из двух полипептидных цепей (А-цепь - 21 аминокислота; В-цепь - 30 аминокислот) (рис. 7.1). Синтез инсулина начинается с образования препроинсулина, который расщепляется протеазой с образованием проинсулина. В секреторных гранулах аппарата Гольджи проинсулин расщепляется на инсулин и С-пептид, которые высвобождаются в кровь в процессе экзоцитоза (рис. 7.2).
Основным стимулятором секреции инсулина является глюкоза. Высвобождение инсулина в ответ на повышение уровня глюкозы в крови происходит двухфазно (рис. 7.3). Первая, или острая, фаза длится несколько минут, и она связана с высвобождением накопив-
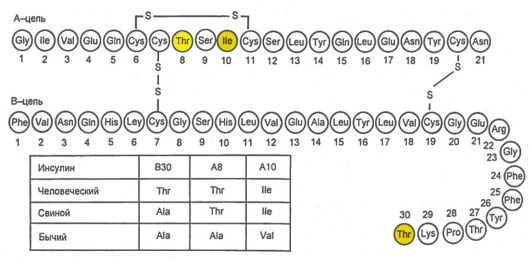 Рис. 7.1. Схема первичной структуры молекулы инсулина
Рис. 7.1. Схема первичной структуры молекулы инсулина
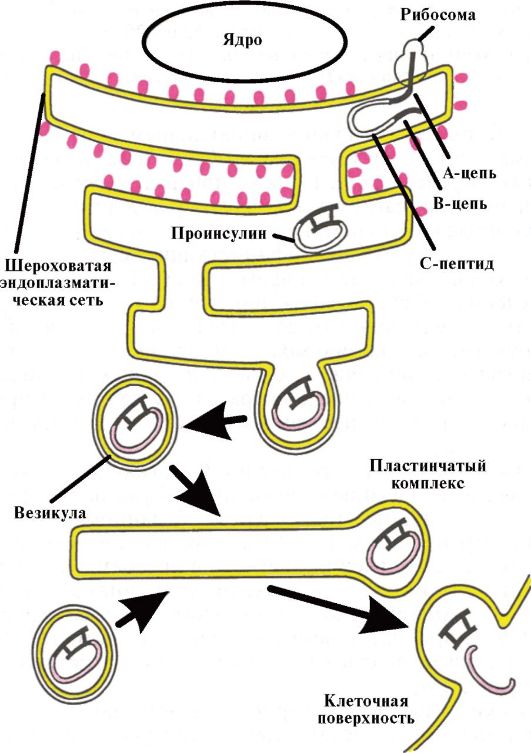 Рис. 7.2. Схема биосинтеза инсулина
Рис. 7.2. Схема биосинтеза инсулина
шегося в β-клетке инсулина в период между приемами пищи. Вторая фаза продолжается до тех пор, пока уровень гликемии не достигнет нормального тощакового (3,3-5,5 ммоль/л). Сходным образом на β- клетку воздействуют препараты сульфонилмочевины.
По портальной системе инсулин достигает печени - своего главного органа-мишени. Печеночные рецепторы связывают половину секретированного гормона. Другая половина, попадая в системный кровоток, достигает мышц и жировой ткани. Большая часть инсулина (80 %) подвергается протеолитическому распаду в печени, остальная - в почках, и лишь незначительное количество метаболизируется непосредственно мышечными и жировыми клетками. В норме ПЖЖ
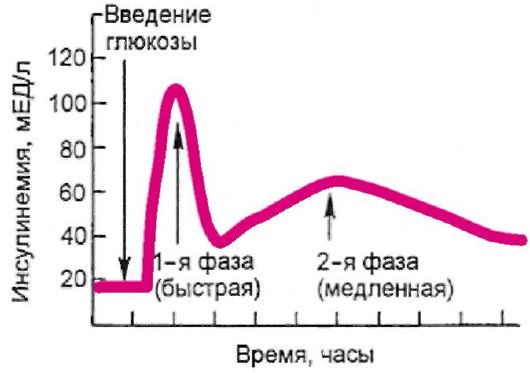 Рис. 7.3. Двухфазное высвобождение инсулина под воздействием глюкозы
Рис. 7.3. Двухфазное высвобождение инсулина под воздействием глюкозы
взрослого человека секретирует 35-50 Ед инсулина в сутки, что составляет 0,6-1,2 Ед на 1 кг массы тела. Эта секреция подразделяется на пищевую и базальную. Пищевая секреция инсулина со ответствует постпрандиальному подъему уровня глюкозы, т.е. за счет нее обеспечивается нейтрализация гипергликемизирующего действия пищи. Количество пищевого инсулина примерно соответствует количеству принятых углеводов - около 1-2,5 Ед
на 10-12 г углеводов (1 хлебная единица - ХЕ). Базальная секреция инсулина обеспечивает оптимальный уровень гликемии и анаболизма в интервалах между едой и во время сна. Базальный инсулин секретируется со скоростью примерно 1 Ед/ч, при длительной физической нагрузке или длительном голодании она существенно уменьшается. На пищевой инсулин приходится не менее 50-70 % суточной продукции инсулина (рис. 7.4).
Секреция инсулина подвержена не только пищевым, но и суточ-
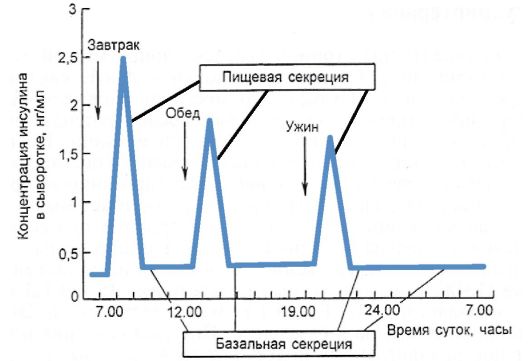 Рис. 7.4. Схема суточной продукции инсулина в норме
Рис. 7.4. Схема суточной продукции инсулина в норме
ным колебаниям: потребность в инсулине повышается в ранние утренние часы, а в дальнейшем постепенно падает в течение дня. Так, на завтрак на 1 ХЕ секретируется 2,0-2,5 Ед инсулина, на обед - 1,0-1,5 Ед, а на ужин - 1,0 ЕД. Одной из причин такого изменения чувствительности к инсулину является высокий уровень ряда контринсулярных гормонов (в первую очередь кортизола) в утренние часы, который постепенно падает до минимального в начале ночи.
Основными физиологическими эффектами инсулина являются стимуляция переноса глюкозы через мембраны клеток инсулинзависимых тканей. Основными органами-мишенями инсулина являются печень, жировая ткань и мышцы. К инсулиннезависимым тканям, поступление глюкозы в которые не зависит от эффектов инсулина, в первую очередь относятся центральная и периферическая нервная система, эндотелий сосудов, клетки крови и др. Инсулин стимулирует синтез гликогена в печени и мышцах, синтез жиров в печени и жировой ткани, синтез белков в печени, мышцах и других органах. Все эти изменения направлены на утилизацию глюкозы, что приводит к снижению ее уровня в крови. Физиологическим антагонистом инсулина является глюкагон, который стимулирует мобилизацию гликогена и жиров из депо; в норме уровень глюкагона меняется реципрокно продукции инсулина.
Биологические эффекты инсулина опосредованы его рецепторами, которые расположены на клетках-мишенях. Рецептор инсулина представляет собой гликопротеин, состоящий из четырех субъединиц. При высоком уровне инсулина в крови число его рецепторов по принципу понижающей регуляции снижается, что сопровождается снижением чувствительности клетки к инсулину. После связывания инсулина с клеточным рецептором образовавшийся комплекс поступает внутрь клетки. Далее внутри мышечной и жировой клетки инсулин вызывает мобилизацию внутриклеточных везикул, которые содержат транспортер глюкозы GLUT-4. В результате этого везикулы перемещаются к клеточной поверхности, где GLUT-4 выполняет функцию входного отверстия для глюкозы. Аналогичное действие на GLUT-4 оказывает физическая нагрузка.
7.3. ЛАБОРАТОРНАЯ ДИАГНОСТИКА И КРИТЕРИИ КОМПЕНСАЦИИ САХАРНОГО ДИАБЕТА
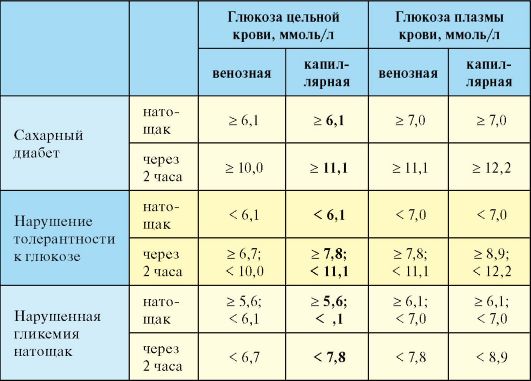
Лабораторная диагностика СД базируется на определении уровня глюкозы крови, при этом критерии диагностики едины для всех
типов и вариантов СД (табл. 7.2). Данные других лабораторных исследований (уровень глюкозурии, определение уровня гликированного гемоглобина) для верификации диагноза СД использоваться не должны. Диагноз СД может быть установлен на основании двукратного обнаружения одного из трех критериев:
1. При явных симптомах СД (полиурия, полидипсия) и уровне глюкозы в цельной капиллярной крови более 11,1 ммоль/л вне зависимости от времени суток и предшествовавшего приема пищи.
2. При уровне глюкозы в цельной капиллярной крови натощак более 6,1 ммоль/л.
3. При уровне глюкозы в цельной капиллярной крови через 2 часа после приема 75 грамм глюкозы (оральный глюкозотолерантный тест) более 11,1 ммоль/л.
Табл. 7.2. Критерии диагностики сахарного диабета
 Наиболее
важным и значимым тестом в диагностике СД является определение уровня
гликемии натощак (минимум 8 часов голодания). В РФ уровень гликемии, как
правило, оценивается в цельной крови. Во многих странах широко
используется определение уровня глюкозы
Наиболее
важным и значимым тестом в диагностике СД является определение уровня
гликемии натощак (минимум 8 часов голодания). В РФ уровень гликемии, как
правило, оценивается в цельной крови. Во многих странах широко
используется определение уровня глюкозы
в плазме крови. Оральному глюкозотолерантному тесту (ОГТТ; определение уровня глюкозы через 2 часа после приема внутрь растворенных в воде 75 граммов глюкозы) в этом плане придается меньшее значение. Тем не менее на основании ОГТТ диагностируется нарушение толерантности к глюкозе (НТГ). НТГ диагностируется если уровень гликемии цельной капиллярной крови натощак не превышает 6,1 ммоль/л, а через 2 часа после нагрузки глюкозой оказывается выше 7,8 ммоль/ л, но ниже 11,1 ммоль/л. Другим вариантом нарушения углеводного обмена является нарушенная гликемия натощак (НГНТ). Последняя устанавливается если уровень гликемии цельной капиллярной крови натощак находится в пределах 5,6-6,0 ммоль/л, а через 2 часа после нагрузки глюкозой меньше 7,8 ммоль/л). НТГ и НГНТ в настоящее время объединяют термином предиабет, поскольку у обеих категорий пациентов высок риск манифестации СД и развития диабетической макроангиопатии.
Для диагностики СД уровень гликемии должен определяться стандартными лабораторными методами. При интерпретации показателей гликемии следует иметь в виду, что натощак уровень глюкозы в цельной венозной крови соответствует ее уровню в цельной капиллярной. После приема пищи или ОГТТ ее уровень в венозной крови примерно на 1,1 ммоль/л ниже, чем в капиллярной. Содержание глюкозы в плазме примерно на 0,84 ммоль/л выше, чем в цельной крови. С целью оценки компенсации и адекватности терапии СД уровень гликемии оценивается в капиллярной крови при помощи портативных глюкометров самими пациентами, их родственниками или медицинским персоналом.
При любом типе СД, а также при значительной нагрузке глюкозой может развиваться глюкозурия, которая является следствием превышения порога реабсорбции глюкозы из первичной мочи. Порог реабсорбции глюкозы значительно индивидуально варьирует (≈ 9-10 ммоль/л). Как отдельно взятый показатель глюкозурия для постановки диагноза СД использоваться не должна. В норме, за исключением случаев значительной пищевой нагрузки рафинированными углеводами, глюкозурия не встречается.
Продукция кетоновых тел (ацетон, ацетоацетат, β-гидроксибутират) значительно интенсифицируется при абсолютном дефиците инсулина. При декомпенсации СД-1 может определяться выраженная кетонурия (исследуется при помощи тест-полосок, которые опускаются в мочу). Легкая (следовая) кетонурия может определяться у здоровых людей при голодании и безуглеводной диете.
Важным лабораторным показателем, который используется для дифференциальной диагностики типов СД, а также для выявления формирования дефицита инсулина у пациентов с СД-2, является уровень С-пептида. По уровню С-пептида в крови можно косвенно судить об инсулинсекретирующей способности β-клеток ПЖЖ. Последние продуцируют проинсулин, от которого перед секрецией отщепляется С-пептид, попадающий в кровь в одинаковых количествах с инсулином. Инсулин на 50 % связывается в печени и имеет время полужизни в периферической крови около 4 мин. С-пептид из кровотока печенью не удаляется и имеет время полужизни в крови около 30 мин. Кроме того, он не связывается клеточными рецепторами на периферии. Поэтому определение уровня С-пептида является более надежным тестом для оценки функции инсулярного аппарата. Уровень С-пептида наиболее информативно исследовать на фоне стимуляционных проб (после приема пищи или введения глюкагона). Тест неинформативен, если он проводится на фоне выраженной декомпенсации СД, поскольку выраженная гипергликемия оказывает токсическое действие на β-клетки (глюкозотоксичность). Инсулинотерапия в течение нескольких предшествовавших дней на результаты теста никак не повлияет.
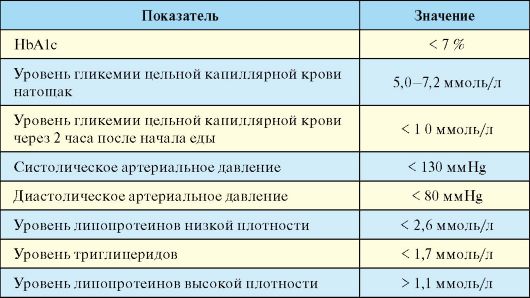
Основной целью лечения любого типа СД является предотвращение его поздних осложнений, которое может быть достигнуто на фоне его стабильной компенсацией по ряду параметров (табл. 7.3). Основным критерием качества компенсации углеводного обмена при СД является уровень гликированного (гликозилированного) гемоглобина (HbA1c). Последний представляет собой гемоглобин, нековалентно связанный с глюкозой. В эритроциты глюкоза поступает независимо от инсулина, и гликозилирование гемоглобина является необратимым процессом, а его степень прямо пропорциональна концентрации глюкозы, с которой он контактировал на протяжении 120 дней своего существования. Небольшая часть гемоглобина гликозилируется и в норме; при СД она может быть значительно повышена. Уровень HbA1c, в отличие от уровня глюкозы, который постоянно меняется, интегрально отражает гликемию на протяжении последних 3-4 месяцев. Именно с таким интервалом и рекомендуется определение уровня HbA1c с целью оценки компенсации СД.
Хроническая гипергликемия является далеко не единственным фактором риска развития и прогрессирования поздних осложнений СД. В связи с этим оценка компенсации СД базируется на комплексе
лабораторных и инструментальных методов исследования (табл. 7.3). Помимо показателей, характеризующих состояние углеводного обмена, наиболее важное значение имеют уровень артериального давления и липидный спектр крови.
Табл. 7.3. Критерии компенсации сахарного диабета
 Помимо
приведенных критериев компенсации, при планировании целей лечения СД
необходим индивидуальный подход. Вероятность развития и прогрессирования
поздних осложнений СД (особенно микроангиопатии) возрастает с
увеличением длительности заболевания. Таким образом, если у детей и
молодых пациентов, стаж диабета которых в дальнейшем может достигнуть
нескольких десятков лет, необходимо добиваться оптимальных показателей
гликемии, то у пациентов, у которых СД манифестировал в пожилом и
старческом возрасте, жесткая эугликемическая компенсация, значительно
повышающая риск гипогликемий, не всегда целесообразна.
Помимо
приведенных критериев компенсации, при планировании целей лечения СД
необходим индивидуальный подход. Вероятность развития и прогрессирования
поздних осложнений СД (особенно микроангиопатии) возрастает с
увеличением длительности заболевания. Таким образом, если у детей и
молодых пациентов, стаж диабета которых в дальнейшем может достигнуть
нескольких десятков лет, необходимо добиваться оптимальных показателей
гликемии, то у пациентов, у которых СД манифестировал в пожилом и
старческом возрасте, жесткая эугликемическая компенсация, значительно
повышающая риск гипогликемий, не всегда целесообразна.
7.4. ПРЕПАРАТЫ ИНСУЛИНА И ИНСУЛИНОТЕРАПИЯ
Препараты инсулина жизненно необходимы пациентам с СД-1; кроме того, их получает до 40 % пациентов с СД-2. К общим показаниям для назначения инсулинотерапии при СД, многие из которых фактически перекрываются одно другим, относятся:
1. Сахарный диабет 1 типа
2. Панкреатэктомия
3. Кетоацидотическая и гиперосмолярная кома
4. При сахарном диабете 2 типа:
- явные признаки дефицита инсулина, такие как прогрессирующееснижение массы тела и кетоз, выраженная гипергликемия;
- большие хирургические вмешательства;
- острые макроваскулярные осложнения (инсульт, инфаркт миокарда, гангрена и пр.) и тяжелые инфекционные заболевания, сопровождающиеся декомпенсацией углеводного обмена;
- уровень гликемии натощак более 15-18 ммоль/л;
- отсутствие стойкой компенсации, несмотря на назначение максимальных суточных доз различных таблетированных сахароснижающих препаратов;
- поздние стадии поздних осложнений СД (тяжелая полинейропатия и ретинопатия, хроническая почечная недостаточность).
5. Невозможность добиться компенсации гестационного СД с помощью диетотерапии.
По происхождению препараты инсулина могут быть классифицированы на три группы:
• животные инсулины (свиные);
• человеческие инсулины (полусинтетические, генно-инженерные);
• аналоги инсулинов (лизпро, аспарт, гларгин, детемир).
Прогресс технологий производства человеческих инсулинов привел к тому, что использование свиных инсулинов (отличается от человеческого одной аминокислотой) в последнее время существенно сократилось. Свиной инсулин может быть использован для производства человеческого инсулина полусинтетическим методом, который подразумевает замену одной отличающейся аминокислоты в его молекуле. Наиболее высоким качеством отличаются генно-инженерные человеческие инсулины. Для их получения участок генома человека, ответственный за синтез инсулина, ассоциируют с геномом E.coli или дрожжевой культуры, в результате чего последние начинают продуцировать человеческий инсулин. Создание аналогов инсулина при помощи перестановок различных аминокислот преследовало цель получения препаратов с заданной и наиболее благоприятной фармакокинетикой. Так, инсулин лизпро (Хумалог) является аналогом
инсулина ультракороткого действия, при этом его сахароснижающий эффект развивается уже спустя 15 минут после инъекции. Аналог инсулина гларгин (Лантус), напротив, характеризуется длительным действием, которое продолжается на протяжении суток, при этом особенностью кинетики препарата является отсутствие выраженных пиков концентрации в плазме. Большинство использующихся в настоящее время препаратов инсулина и его аналогов выпускаются в концентрации 100 Ед/мл. По длительности действия инсулины подразделяются на 4 основные группы (табл. 7.4):
Табл. 7.4. Фармакокинетика препаратов и аналогов инсулина
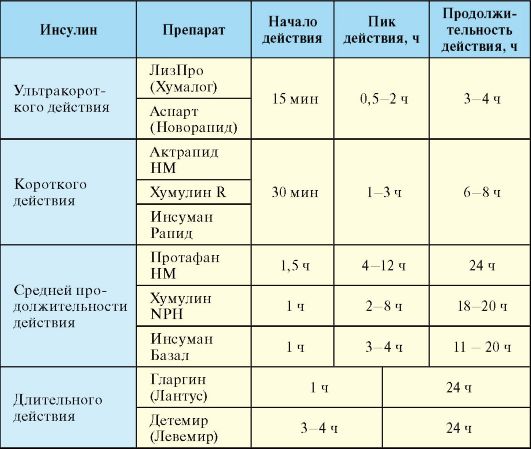 1. Ультракороткого действия (лизпро, аспарт).
1. Ультракороткого действия (лизпро, аспарт).
2. Короткого действия (простой человеческий инсулин).
3. Средней продолжительности действия (инсулины на нейтральном протамине Хагедорна).
4. Длительного действия (гларгин, детемир).
5. Смеси инсулинов различной продолжительности действия (Новомикс-30, Хумулин-МЗ, Хумалог-Микс-25).
Препараты ультракороткого действия [лизпро (Хумалог), аспарт (Новорапид)] являются аналогами инсулина. Их преимуществами являются быстрое развитие сахароснижающего эффекта после инъекции (через 15 минут), что позволяет делать инъекцию непосредственно перед едой или даже сразу после еды, а также короткая продолжительность действия (менее 3 часов), что снижает риск развития гипогликемии. Препараты короткого действия (простой инсулин, инсулин-регуляр) представляют собой раствор, содержащий инсулин в концентрации 100 Ед/мл. Инъекция простого инсулина делается за 30 минут до еды; длительность действия составляет порядка 4-6 часов. Препараты ультракороткого и короткого действия могут вводиться подкожно, внутримышечно и внутривенно.
Среди препаратов средней продолжительности действия чаще всего используются препараты на нейтральном протамине Хагедорна (НПХ). НПХ представляет собой белок, который нековалентно адсорбирует инсулин, замедляя его всасывание из подкожного депо. Эффективная продолжительность действия инсулинов НПХ обычно составляет около 12 часов; они вводятся только подкожно. Инсулин НПХ представляет собой суспензию, в связи с чем в отличие от простого инсулина во флаконе он мутный, а при длительной стоянии образуется взвесь, которую необходимо тщательно перемешать перед инъекцией. Инсулины НПХ в отличие от других препаратов пролонгированного действия можно в любых соотношениях смешивать с инсулином короткого действия (простым инсулином), при этом фармакокинетика компонентов смеси не изменится, поскольку НПХ не будет связывать дополнительные количества простого инсулина (рис. 7.5). Кроме того, протамин используется для приготовления стандартных смесей аналогов инсулина (Новомикс-30, Хумалог-Микс-25).
Среди препаратов длительного действия в настоящее время активно используют аналоги инсулина гларгин (Лантус) и детемир (Левемир). Благоприятная особенность фармакокинетики этих препаратов заключается в том, что в отличие от инсулинов НПХ они обеспечивают более равномерное и длительное поступление препарата из подкожного депо. В связи с этим гларгин может назначаться всего один раз в день, при этом практически независимо от времени суток.
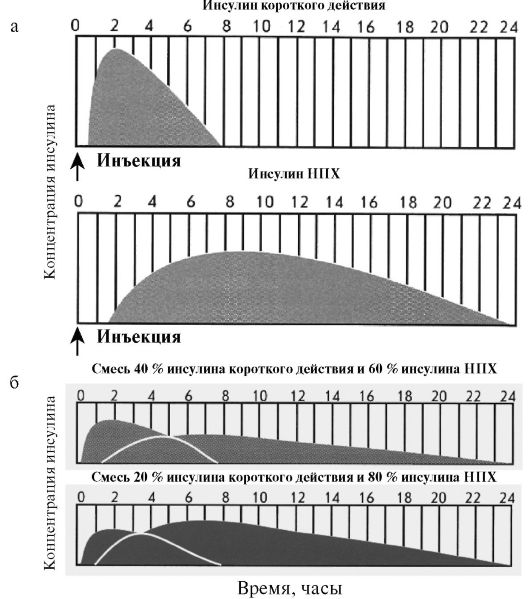 Рис. 7.5. Фармакококинетика различных препаратов инсулина:
Рис. 7.5. Фармакококинетика различных препаратов инсулина:
а) монокомпонентных; б) стандартных смесей инсулинов
Помимо монокомпонентных препаратов инсулина, в клинической практике широко используются стандартные смеси. Как правило, речь идет о смесях короткого или ультракороткого инсулина с инсулином средней продолжительности действия. Например, препарат «Хумулин-МЗ» содержит в одном флаконе 30 % простого инсулина и 70 % инсулина НПХ; препарат «Новомикс-30» содержит 30 % инсулина аспарт и 70 % кристаллической протаминовой суспензии инсулина аспарт; препарат «Хумалог-Микс-25» содержит 25 % инсулина лизпро и 75 % протаминовой суспензии инсулина лизпро. Преимуществом
стандартных смесей инсулинов является замена двух инъекций одной и несколько большая точность дозировки компонентов смеси; недостатком - невозможность индивидуального дозирования отдельных компонентов смеси. Это определяет предпочтительность использования стандартных смесей инсулинов для терапии СД-2 или при так называемой традиционной инсулинотерапии (назначение фиксированных доз инсулинов), тогда как для интенсивной инсулинотерапии (гибкий подбор дозы в зависимости от показателей гликемии и количества углеводов в пище) предпочтительнее использование монокомпонентных препаратов.
Залогом успешной инсулинотерапии является четкое соблюдение техники инъекций. Существует несколько способов введения инсулина. Наиболее простой и при этом надежный метод - инъекции при помощи инсулинового шприца. Более удобным способом введения инсулина являются инъекции при помощи шприц-ручки, которая представляет собой комбинированное устройство, содержащее резервуар с инсулином (картридж), систему дозирования и иглу с инжектором.
Для поддерживающей терапии (когда речь не идет о выраженной декомпенсации СД или о критических состояниях) инсулин вводится подкожно. Инъекции инсулина короткого действия рекомендуется делать в подкожную жировую клетчатку живота, инсулина пролонгированного действия - в клетчатку бедра или плеча (рис. 7.6 а). Инъекции делаются глубоко в подкожную клетчатку через широко сжатую кожу под углом в 45° (рис. 7.6 б). Пациенту необходимо рекомендовать ежедневную смену мест введения инсулина в пределах одной области в целях предупреждения развития липодистрофий.
К факторам, влияющим на скорость абсорбции инсулина из подкожного депо, следует отнести дозу инсулина (увеличение дозы увеличивает продолжительность абсорбции), место инъекции (абсорбция быстрее из клетчатки живота), температура окружающей среды (согревание и массаж места инъекции ускоряет абсорбцию).
Более сложным методом введения, который, тем не менее, у многих пациентов позволяет достигнуть хороших результатов лечения, является использование дозатора инсулина, или системы для непрерывного подкожного введения инсулина. Дозатор представляет собой портативный прибор, состоящий из компьютера, который задает режим подачи инсулина, а также системы подачи инсулина, осуществляющейся по катетеру и миниатюрной игле в подкожную
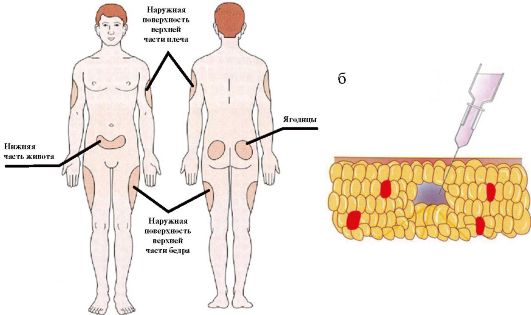 Рис. 7.6. Инъекции инсулина: а) типичные места инъекций; б) положение иглы инсулинового шприца при инъекции
Рис. 7.6. Инъекции инсулина: а) типичные места инъекций; б) положение иглы инсулинового шприца при инъекции
жировую клетчатку. При помощи дозатора осуществляется непрерывное базальное введение инсулина короткого или ультракороткого действия (скорость порядка 0,5-1 Ед/час), а перед приемом пищи в зависимости от содержания в ней углеводов и уровня гликемии пациент вводит необходимую болюсную дозу того же инсулина короткого действия. Преимуществом инсулинотерапии с помощью дозатора является введение одного только инсулина короткого (или даже ультракороткого) действия, что само по себе несколько более физиологично, поскольку абсорбция пролонгированных препаратов инсулина подвергается большим колебаниям; в связи с этим непрерывное введение инсулина короткого действия оказывается более управляемым процессом. Недостатком инсулинотерапии при помощи дозатора является необходимость постоянного ношения прибора, а также длительное нахождение инъекционной иглы в подкожной клетчатке, требующее периодического контроля за процессом подачи инсулина. Инсулинотерапия при помощи дозатора в первую очередь показана пациентам с СД-1, которые готовы овладеть техникой ее ведения. Особенно в этом плане следует обращать внимание на пациентов с выраженным феноменом «утренней зари», а также на беременных и планирующих беременность пациенток с СД-1 и паци-
ентов с неупорядоченным образом жизни (возможность более гибкого режима питания).
7.5. САХАРНЫЙ ДИАБЕТ 1 ТИПА
СД-1 - органоспецифическое аутоиммунное заболевание, приводящее к деструкции инсулинпродуцирующих β-клеток островков ПЖЖ, проявляющееся абсолютным дефицитом инсулина. В ряде случаев у пациентов с явным СД-1 отсутствуют маркеры аутоиммунного поражения β-клеток (идиопатический СД-1).
Этиология
СД-1 является заболеванием с наследственной предрасположенностью, но ее вклад в развитие заболевания невелик (определяет его развитие примерно на 1/з). Конкордантность у однояйцевых близнецов по СД-1 составляет всего 36 %. Вероятность развития СД-1 у ребенка при больной матери составляет 1-2 %, отце - 3-6 %, брате или сестре - 6 %. Одни или несколько гуморальных маркеров аутоиммунного поражения β-клеток, к которым относятся антитела к островкам ПЖЖ, антитела к глутамат-декарбоксилазе (GAD65) и антитела к тирозин-фосфатазе (IA-2 и ΙΑ-2β), обнаруживаются у 85-90 % пациентов. Тем не менее основное значение в деструкции β-клеток придается факторам клеточного иммунитета. СД-1 ассоциирован с такими гаплотипами HLA, как DQA и DQB, при этом одни аллели HLA-DR/DQ могут быть предрасполагающими к развитию заболевания, тогда как другие - протективными. С повышенной частотой СД-1 сочетается с другими аутоиммунными эндокринными (аутоиммунный тиреоидит, болезнь Аддисона) и неэндокринными заболеваниями, такими как алопеция, витилиго, болезнь Крона, ревматические заболевания (табл. 7.5).
Патогенез
СД-1 манифестирует при разрушении аутоиммунным процессом 80-90 % β-клеток. Скорость и интенсивность этого процесса может существенно варьировать. Наиболее часто при типичном течении заболевания у детей и молодых людей этот процесс протекает достаточно быстро с последующей бурной манифестацией заболевания, при которой от появления первых клинических симптомов до развития кетоацидоза (вплоть до кетоацидотической комы) может пройти всего несколько недель.
Табл. 7.5. Сахарный диабет 1 типа
 Продолжение табл. 7.5
Продолжение табл. 7.5
 В других, значительно более редких случаях, как правило, у взрослых старше 40 лет, заболевание может протекать латентно (латентный аутоиммунный диабет взрослых - LADA), при
этом в дебюте заболевания таким пациентам нередко устанавливается
диагноз СД-2, и на протяжении нескольких лет компенсация СД может
достигаться назначением препаратов сульфонилмочевины. Но в дальнейшем,
обычно спустя 3 года, появляются признаки абсолютного дефицита инсулина
(похудение, кетонурия, выраженная гипергликемия, несмотря на прием
таблетированных сахароснижающих препаратов).
В других, значительно более редких случаях, как правило, у взрослых старше 40 лет, заболевание может протекать латентно (латентный аутоиммунный диабет взрослых - LADA), при
этом в дебюте заболевания таким пациентам нередко устанавливается
диагноз СД-2, и на протяжении нескольких лет компенсация СД может
достигаться назначением препаратов сульфонилмочевины. Но в дальнейшем,
обычно спустя 3 года, появляются признаки абсолютного дефицита инсулина
(похудение, кетонурия, выраженная гипергликемия, несмотря на прием
таблетированных сахароснижающих препаратов).
В основе патогенеза СД-1, как указывалось, лежит абсолютный дефицит инсулина. Невозможность поступления глюкозы в инсулинзависимые ткани (жировая и мышечная) приводит к энергетической недостаточности в результате чего интенсифицируется липолиз и протеолиз, с которыми связана потеря массы тела. Повышение уровня гликемии вызывает гиперосмолярность, что сопровождается осмотическим диурезом и выраженным обезвоживанием. В условиях дефицита инсулина и энергетической недостаточности растормаживается продукция контринсулярных гормонов (глюкагон, кортизол, гормон роста), которая, несмотря на нарастающую гликемию, обусловливает стимуляцию глюконеогенеза. Усиление липолиза в жировой ткани приводит к значительному увеличению концентрации свободных жирных кислот. При дефиците инсулина липосинтетическая способность печени оказывается подавленной, и свобод-
ные жирные кислоты начинают включаться в кетогенез. Накопление кетоновых тел приводит к развитию диабетического кетоза, а в дальнейшем - кетоацидоза. При прогрессирующем нарастании обезвоживания и ацидоза развивается коматозное состояние (см. п. 7.7.1), которое при отсутствии инсулинотерапии и регидратации неизбежно заканчивается смертью.
Эпидемиология
На СД-1 приходится порядка 1,5-2 % всех случаев диабета, и этот относительный показатель в дальнейшем будет уменьшаться в силу быстрого роста заболеваемости СД-2. Риск развития СД-1 на протяжении жизни у представителя белой расы составляет около 0,4 %. Заболеваемость СД-1 увеличивается на 3 % в год: на 1,5 % - за счет новых случаев и еще на 1,5 % - за счет увеличения продолжительности жизни пациентов. Распространенность СД-1 варьирует в зависимости от этнического состава популяции. На 2000 год она составила 0,02 % в Африке, 0,1 % в Южной Азии, а также в Южной и Центральной Америке и 0,2 % в Европе и Северной Америке. Наиболее высока заболеваемость СД-1 в Финляндии и Швеции (30-35 случаев на 100 тысяч населения в год), а наиболее низка в Японии, Китае и Корее (соответственно 0,5-2,0 случая). Возрастной пик манифестации СД-1 соответствует примерно 10-13 годам. В подавляющем большинстве случаев СД-1 манифестирует до 40 лет.
Клинические проявления
В типичных случаях, особенно у детей и молодых людей, СД-1 дебютирует яркой клинической картиной, которая развивается на протяжении нескольких месяцев или даже недель. Манифестацию СД-1 могут спровоцировать инфекционные и другие сопутствующие заболевания. Характерны общие для всех типов СД симптомы, связанные с гипергликемией: полидипсия, полиурия, кожный зуд, но при СД-1 они очень ярко выражены. Так, на протяжении дня пациенты могут выпивать и выделять до 5-10 литров жидкости. Специфичным для СД-1 симптомом, который обусловлен абсолютным дефицитом инсулина, является похудение, достигающее 10-15 кг на протяжении 1-2 месяцев. Характерна выраженная общая и мышечная слабость, снижение работоспособности, сонливость. В начале заболевания у некоторых пациентов может отмечаться повышение аппетита, которое сменяется анорексией по мере развития кетоацидоза. Последний характеризуется появлением запаха ацетона (или фруктового запаха) изо рта, тош-
нотой, рвотой, нередко болями в животе (псевдоперитонит), тяжелым обезвоживанием и заканчивается развитием коматозного состояния (см. п. 7.7.1). В ряде случаев первым проявлением СД-1 у детей является прогрессирующее нарушение сознания вплоть до комы на фоне сопутствующих заболеваний, как правило, инфекционных или острой хирургической патологии.
В относительно редких случаях развития СД-1 у лиц старше 35-40 лет (латентный аутоиммунный диабет взрослых) заболевание может манифестировать не столь ярко (умеренная полидипсия и полиурия, отсутствие потери массы тела) и даже выявляться случайно при рутинном определении уровня гликемии. В этих случаях пациенту нередко в начале устанавливается диагноз СД-2 и назначаются таблетированные сахароснижающие препараты (ТСП), которые какое-то время обеспечивают приемлемую компенсацию СД. Тем не менее на протяжении нескольких лет (часто в течение года) у пациента появляются симптомы, обусловленные нарастающим абсолютным дефицитом инсулина: похудение, невозможность поддержания нормальной гликемии на фоне ТСП, кетоз, кетоацидоз.
Диагностика
Учитывая, что СД-1 имеет яркую клиническую картину, а также является относительно редким заболеванием, скрининговое определение уровня гликемии с целью диагностики СД-1 не показано. Вероятность развития заболевания у ближайших родственников пациентов невысока, что вместе с отсутствием эффективных методов первичной профилактики СД-1 определяет нецелесообразность изучения у них иммуногенетических маркеров заболевания. Диагностика СД-1 в подавляющем большинстве случаев базируется на выявлении значительной гипергликемии у пациентов с выраженными клиническими проявлениями абсолютного дефицита инсулина. ОГТТ с целью диагностики СД-1 приходится проводить очень редко.
Дифференциальная диагностика
В сомнительных случаях (выявление умеренной гипергликемии при отсутствии явных клинических проявлений, манифестация в относительно немолодом возрасте), а также с целью дифференциальной диагностики с другими типами СД используется определение уровня С-пептида (базального и через 2 часа после приема пищи). Косвенное диагностическое значение в сомнительных случаях может иметь определение иммунологических маркеров СД-1 - антитела к островкам
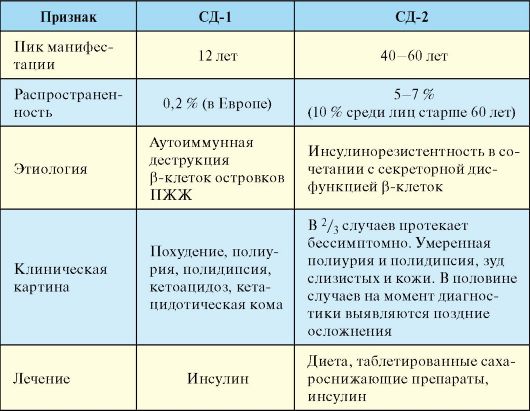
ПЖЖ, к глутаматдекарбоксилазе (GAD65) и тирозинфосфатазе (IA-2 и IA-2β). Дифференциальная диагностика СД-1 и СД-2 представлена в табл. 7.6.
Табл. 7.6. Дифференциальная диагностика и отличия СД-1 и СД-2
 Лечение
Лечение
Лечение любого типа СД базируется на трех основных принципах: сахароснижающая терапия (при СД-1 - инсулинотерапия), диета и обучение пациентов. Инсулинотерапия при СД-1 носит заместительный характер и ее целью является максимальная имитация физиологической продукции гормона с целью достижения принятых критериев компенсации (табл. 7.3). К физиологической секреции инсулина наиболее приближена интенсивная инсулинотерапия. Потребность в инсулине, соответствующая его базальной секреции, обеспечивается двумя инъекциями инсулина средней продолжительности действия (утром и вечером) или одной инъекцией инсулина длительного действия (гларгин). Суммарная доза базального инсу-
лина не должна превышать половины всей суточной потребности в препарате. Пищевая или болюсная секреция инсулина замещается инъекциями инсулина короткого или ультракороткого действия перед каждым приемом пищи, при этом его доза рассчитывается, исходя из количества углеводов, которое предполагается принять во время предстоящего приема пищи, и имеющегося уровня гликемии, определяемого пациентом с помощью глюкометра перед каждой инъекцией инсулина (рис. 7.7).
Ориентировочная схема интенсивной инсулинотерапии, которая будет меняться практически каждый день, может быть представлена следующим образом. Исходят из того, что суточная потребность в инсулине составляет около 0,5-0,7 Ед на 1 кг массы тела (для пациента с массой тела 70 кг около 35-50 Ед). Около 1/з - 1/2 этой дозы составит инсулин пролонгированного действия (20-25 Ед), 1/2 - 2/з инсулин короткого или ультракороткого действия. Доза инсулина НПХ делится на 2 инъекции: утром 2/з его дозы (12 Ед), вечером - 1/з (8-10 Ед).
Целью первого этапа подбора инсулинотерапии является нормализация уровня глюкозы натощак. Вечерняя доза инсулина НПХ обычно вводится в 22-23 часа, утренняя вместе с инъекцией инсулина короткого действия перед завтраком. При подборе вечерней дозы инсулина НПХ необходимо иметь в виду возможность развития ряда
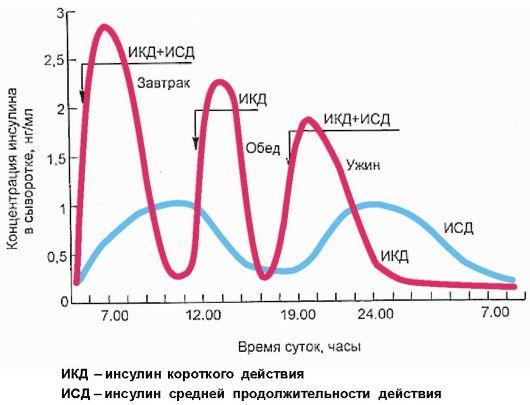 Рис. 7.7. Схема интенсивной инсулинотерапии
Рис. 7.7. Схема интенсивной инсулинотерапии
достаточно типичных феноменов. Причиной утренней гипергликемии может быть недостаточнсть дозы инсулина пролонгированного действия, поскольку к утру потребность в инсулине существенно возрастает (феномен «утренней зари»). Помимо недостаточности дозы к утренней гипергликемии может привести ее избыток - феномен Сомоджи (Somogyi), постгипогликемическая гипергликемия. Этот феномен объясняется тем, что максимальная чувствительность тканей к инсулину отмечается между 2 и 4 часами ночи. Именно в это время уровень основных контринсулярных гормонов (кортизол, гормон роста и др.) в норме наиболее низок. Если вечерняя доза инсулина пролонгированного действия избыточна, то в это время развивается гипогликемия. Клинически она может проявляться плохим сном с кошмарными сновидениями, бессознательными действиями во сне, утренней головной болью и разбитостью. Развитие в это время гипогликемии вызывает значительный компенсаторный выброс глюкагона и других контринсулярных гормонов с последующей гипергликемией в утренние часы. Если в этой ситуации не снизить, а увеличить дозу пролонгированного инсулина, вводимого вечером, ночная гипогликемия и утренняя гипергликемия будут усугубляться, что в итоге может привести к синдрому хронической передозировки инсулина (синдром Сомоджи), который представляет собой сочетание ожирения с хронической декомпенсацией СД, частыми гипогликемиями и прогрессирующими поздними осложнениями. Для диагностики феномена Сомоджи необходимо исследование уровня гликемии около 3 ч ночи, которое является неотъемлемым компонентом подбора инсулинотерапии. Если снижение вечерней дозы НПХ до безопасной в плане развития ночной гипогликемии сопровождается гипергликемией утром (феномен утренней зари), пациенту необходимо рекомендовать более ранний подъем (6-7 утра), в то время, когда введенный на ночь инсулин еще продолжает поддерживать нормальный уровень гликемии.
Вторая инъекция инсулина НПХ обычно делается перед завтраком вместе с утренней инъекцией инсулина короткого (ультракороткого) действия. В данном случае доза подбирается преимущественно исходя из показателей уровня гликемии перед основными дневными приемами пищи (обед, ужин); кроме того, ее может лимитировать развитие гипогликемий в промежутках между приемами пищи, например в полдень, между завтраком и обедом.
Вся доза инсулина пролонгированного действия (гларгин) вводится одни раз в день, при этом не принципиально, в какое время. Кинетика
инсулинов гларгин и детемир более благоприятна в плане риска развития гипогликемий, в том числе ночных.
Доза инсулина короткого или ультракороткого действия даже в первый для пациента день назначения инсулина будет зависеть от количества употребляемых углеводов (хлебных единиц) и уровня гликемии перед инъекцией. Условно, исходя из суточного ритма секреции инсулина в норме, около 1/4 дозы инсулина короткого действия (6-8 ЕД) отводится на ужин, оставшаяся доза примерно поровну разделится на завтрак и обед (10-12 ЕД). Чем выше исходный уровень гликемии, тем меньше он будет снижаться на единицу вводимого инсулина. Инъекция инсулина короткого действия делается за 30 минут до еды, ультракороткого действия непосредственно перед едой или даже сразу после еды. Адекватность дозы инсулина короткого действия оценивается по показателям гликемии через 2 часа после еды и перед очередным приемом пищи.
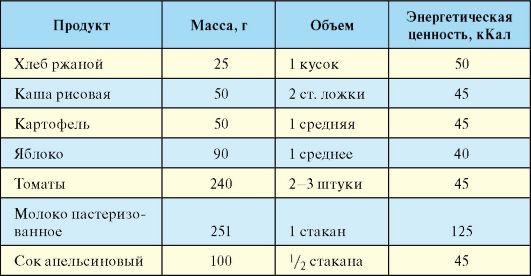
Для расчета дозы инсулина при интенсивной инсулинотерапии достаточно подсчета числа ХЕ, исходя только из углеводного компонента. При этом в расчет берутся не все углеводсодержащие продукты, а только так называемые подсчитываемые. К последним относятся картофель, зерновые продукты, фрукты, жидкие молочные и сладкие продукты. Продукты, содержащие неусваиваемые углеводы (большинство овощей), в расчет не берутся. Разработаны специальные обменные таблицы, с помощью которых, выражая количество углеводов в ХЕ, можно рассчитать необходимую дозу инсулина. Одной ХЕ соответствует 10-12 г углеводов (табл. 10.7).
После приема пищи, содержащей 1 ХЕ, уровень гликемии увеличивается на 1,6-2,2 ммоль/л, т.е. примерно на столько, на сколько снижается уровень глюкозы при введении 1 Ед инсулина. Другими словами, на каждую ХЕ, содержащуюся в пище, которую планируется съесть, необходимо заранее ввести (в зависимости от времени суток) около 1 Ед инсулина. Кроме того, необходим учет результатов самоконтроля уровня гликемии, который производится перед каждой инъекцией, и времени суток (около 2 Ед инсулина на 1 ХЕ утром и в обед, 1 Ед на 1 ХЕ - на ужин). Так, если выявлена гипергликемия, дозу инсулина, рассчитанную в соответствии с предстоящим приемом пищи (по числу ХЕ), нужно увеличить, и наоборот, если выявлена гипогликемия, инсулина вводится меньше.
Табл. 7.7. Эквивалентная замена продуктов, составляющих 1 ХЕ
 Например,
если у пациента за 30 мин до планируемого ужина, содержащего 5 ХЕ,
уровень гликемии составляет 7 ммоль/л, ему необходимо ввести 1 Ед
инсулина для того, чтобы гликемия снизилась до нормального уровня: с 7
ммоль/л примерно до 5 ммоль/л. Кроме того, 5 Ед инсулина необходимо
ввести на покрытие 5 ХЕ. Таким образом, пациент в данном случае введет 6
Ед инсулина короткого или ультракороткого действия.
Например,
если у пациента за 30 мин до планируемого ужина, содержащего 5 ХЕ,
уровень гликемии составляет 7 ммоль/л, ему необходимо ввести 1 Ед
инсулина для того, чтобы гликемия снизилась до нормального уровня: с 7
ммоль/л примерно до 5 ммоль/л. Кроме того, 5 Ед инсулина необходимо
ввести на покрытие 5 ХЕ. Таким образом, пациент в данном случае введет 6
Ед инсулина короткого или ультракороткого действия.
После манифестации СД-1 и начала инсулинотерапии на протяжении достаточно длительного времени потребность в инсулине может быть небольшой и составлять менее 0,3-0,4 Ед/кг. Этот период обозначается как фаза ремиссии, или «медовый месяц». После периода гипергликемии и кетоацидоза, которые подавляют секрецию инсулина 10-15 % сохранившимися β-клетками, компенсация гормональнометаболических нарушений введением инсулина восстанавливает функцию этих клеток, которые затем берут на себя обеспечение организма инсулином на минимальном уровне. Этот период может продолжаться от нескольких недель до нескольких лет, но в конечном счете, вследствие аутоиммунной деструкции оставшихся β-клеток, «медовый месяц» заканчивается.
Диета при СД-1 у обученных пациентов, которые владеют навыками самоконтроля и подбора дозы инсулина, может быть либерализованной, т.е. приближающейся к свободной. Если у пациента отсутствует избыток или дефицит массы тела, диета должна быть
изокалорийной. Основным компонентом пищи при СД-1 являются углеводы, на которые должно приходиться около 65 % суточного калоража. Предпочтение следует отдавать продуктам, содержащим сложные, медленно всасывающиеся углеводы, а также продуктам, богатым пищевой клетчаткой. Продукты, содержащие легкоусваемые углеводы (мучное, сладкое), следует избегать. Доля белков должна быть снижена до 10-35 %, что способствует снижению риска развития микроангиопатии, а доля жиров - до 25-35 %, при этом на предельные жиры должно приходиться до 7 % калоража, что снижает риск развития атеросклероза. Кроме того, необходимо избегать приема алкогольных напитков, особенно крепких.
Неотъемлемым компонентом работы с пациентом с СД-1 и залогом его эффективной компенсации является обучение пациентов. На протяжении всей жизни пациент ежедневно должен самостоятельно в зависимости от многочисленных факторов изменять дозу инсулина. Очевидно, что это требует владения определенными навыками, которым пациента необходимо обучить. «Школа пациента с СД-1» организуется в эндокринологических стационарах или амбулаторно и представляет собой 5-7 структурированных занятий, на которых врач или специально обученная медсестра в интерактивном режиме с использованием различных наглядных пособий проводит обучение пациентов принципам самоконтроля.
Прогноз
При отсутствии инсулинотерапии больной СД-1 неизбежно погибает от кетоацидотической комы. При неадекватной инсулинотерапии, на фоне которой не достигаются критерии компенсации СД и пациент находится в состоянии хронической гипергликемии (табл. 7.3), начинают развиваться и прогрессировать поздние осложнения (п. 7.8). При СД-1 наибольшее клиническое значение в этом плане имеют проявления диабетической микроангиопатии (нефропатия и ретинопатия) и нейропатии (синдром диабетической стопы). Макроангиопатия при СД-1 на первый план выходит относительно редко.
7.6. САХАРНЫЙ ДИАБЕТ 2 ТИПА
Сахарный диабет 2 типа - хроническое заболевание, проявляющееся нарушением углеводного обмена с развитием гипергликемии вследствие инсулинорезистентности и секреторной дисфункции β-клеток,
а также липидного обмена с развитием атеросклероза. Поскольку основной причиной смерти и инвалидизации пациентов являются осложнения системного атеросклероза, СД-2 иногда называют сердечно-сосудистым заболеванием.
Табл. 7.8. Сахарный диабет 2 типа
 Этиология
Этиология
СД-2 является многофакторным заболеванием с наследственной предрасположенностью. Конкордатность по СД-2 у однояйцевых близнецов достигает 80 % и более. Большинство пациентов с СД-2 указывают на наличие СД-2 у ближайших родственников; при наличии СД-2 у одного из родителей вероятность его развития у потомка на протяжении жизни составляет 40 %. Какого-то одного гена, полиморфизм которого определяет предрасположенность к СД-2, не обнаружено. Большое значение в реализации наследственной предрасположенности к СД-2 играют факторы окружающей среды, в первую очередь, особенности образа жизни. Факторами риска развития СД-2 являются:
- ожирение, особенно висцеральное (см. п. 11.2);
- этническая принадлежность (особенно при смене традиционного образа жизни на западный);
- СД-2 у ближайших родственников;
- малоподвижный образ жизни;
- особенности диеты (высокое потребление рафинированных углеводов и низкое содержание клетчатки);
- артериальная гипертензия.
Патогенез
Патогенетически СД-2 представляет собой гетерогенную группу нарушений обмена веществ, именно это и определяет его значительную клиническую неоднородность. В основе его патогенеза лежит инсулинорезистентность (снижение опосредованной инсулином утилизации глюкозы тканями), которая реализуется на фоне секреторной дисфункции β-клеток. Таким образом, происходит нарушение баланса чувствительности к инсулину и инсулиновой секреции. Секреторная дисфункция β-клеток заключается в замедлении «раннего» секреторного выброса инсулина в ответ на увеличение уровня глюкозы в крови. При этом 1-я (быстрая) фаза секреции, которая заключается в опорожнении везикул с накопленным инсулином, фактически отсутствует; 2-я (медленная) фаза секреции осуществляется в ответ на стабилизирующуюся гипергликемию постоянно, в тоническом режиме, и, несмотря на избыточную секрецию инсулина, уровень гликемии на фоне инсулинорезистентности не нормализуется (рис. 7.8).
Следствием гиперинсулинемии является снижение чувствительности и числа инсулиновых рецепторов, а также подавление
пострецепторных механизмов, опосредующих эффекты инсулина (инсулинорезистентность). Содержание основного транспортера глюкозы в мышечных и жировых клетках (GLUT-4) снижено на 40 % у лиц с висцеральных ожирением и на 80 % - у лиц с СД-2. Вследствие инсулинорезистентности гепатоцитов и портальной гиперинсулинемией происходит гиперпродукция глюкозы печенью, и развивается гипергликемия натощак, которая выявляется у большинства пациентов с СД-2, в том числе и на ранних этапах заболевания.
Сама по себе гипергликемия неблагоприятно влияет на характер и уровень секреторной активности β-клеток (глюкозотоксичность). Длительно, на протяжении многих лет и десятилетий существующая гипергликемия в конечном счете приводит к истощению продукции инсулина β-клетками и у пациента могут появиться некоторые симптомы дефицита инсулина - похудение, кетоз при сопутствующих инфекционных заболеваниях. Тем не менее, остаточная продукция инсулина, которой оказывается достаточно для предотвращения кетоацидоза, при СД-2 практически всегда сохраняется.
Эпидемиология
СД-2 определяет эпидемиологию СД в целом, поскольку на него приходится около 98 % случаев этого заболевания. Распространенность СД-2 варьирует в разных странах и этнических группах. В европейских
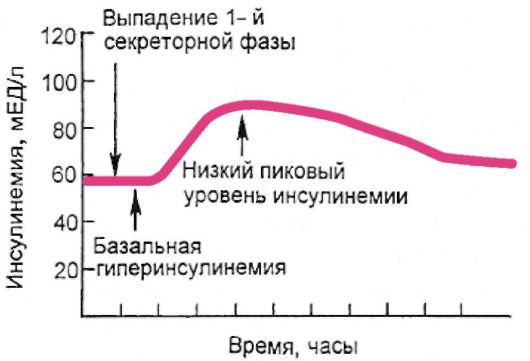 Рис. 7.8. Секреторная дисфункция β-клеток при сахарном диабете 2 типа (выпадение 1-й быстрой фазы секреции инсулина)
Рис. 7.8. Секреторная дисфункция β-клеток при сахарном диабете 2 типа (выпадение 1-й быстрой фазы секреции инсулина)
странах, США и Российской Федерации она составляет около 5-6 % населения. С возрастом заболеваемость СД-2 увеличивается: среди взрослых распространенность СД-2 составляет 10 %, среди лиц старше 65 лет достигает 20 %. Заболеваемость СД-2 в 2,5 раза выше среди коренных жителей Америки и Гавайских островов; среди индейцев племени Пима (штат Аризона) она достигает 50 %. Среди сельского населения Индии, Китая, Чили и Африканских стран, которые ведут традиционный образ жизни, распространенность СД-2 очень низка (менее 1 %). С другой стороны, среди переселенцев в западные индустриальные страны она достигает значительного уровня. Так, среди выходцев из Индии и Китая, проживающих в США и Великобритании, распространенность СД-2 достигает 12-15 %.
ВОЗ предсказывает увеличение числа больных диабетом в мире на 122 % в течение ближайших 20 лет (с 135 до 300 миллионов). Это связано как с прогрессирующим старением населения, так и с распространением и усугублением урбанизированного образа жизни. В последние годы отмечается значительное «омоложение» СД-2 и рост его заболеваемости среди детей.
Клинические проявления
В большинстве случаев, выраженные клинические проявления отсутствуют, и диагноз устанавливается при рутинном определении уровня гликемии. Заболевание обычно манифестирует в возрасте старше 40 лет, при этом у подавляющего большинства пациентов имеет место ожирение и другие компоненты метаболического синдрома (см. п. 11.2). Пациенты не предъявляют жалоб на снижение работоспособности, если для этого отсутствуют другие причины. Жалобы на жажду и полиурию редко достигают значительной выраженности. Достаточно часто пациентов беспокоит кожный и влагалищный зуд, в связи с чем они обращаются к дерматологам и гинекологам. Поскольку от реальной манифестации СД-2 до постановки диагноза зачастую проходят многие годы (в среднем около 7 лет), у многих пациентов на момент выявления заболевания в клинической картине доминируют симптомы и проявления поздних осложнений СД. Более того, первое обращение пациента с СД-2 за медицинской помощью очень часто происходит в связи с поздними осложнениями. Так, пациенты могут госпитализироваться в хирургические стационары с язвенным поражением ног (синдром диабетической стопы), обращаться в связи с прогрессирующим снижением зрения к офтальмологам (диабетическая ретинопатия), госпитализироваться с инфарктами, инсуль-
тами, облитерирующим поражением сосудов ног в учреждения, где у них впервые обнаруживается гипергликемия.
Диагностика
Критерии диагностики, единые для всех типов СД, представлены в п. 7.3. Диагноз СД-2 в подавляющем большинстве случаев базируется на выявлении гипергликемии у лиц с типичными клиническими признаками СД-2 (ожирение, возраст старше 40-45 лет, положительный семейный анамнез СД-2, другие компоненты метаболического синдрома), при отсутствии клинических и лабораторных признаков абсолютного дефицита инсулина (выраженное похудение, кетоз). Сочетание высокой распространенности СД-2, свойственного ему длительного бессимптомного течения и возможности предотвращения его тяжелых осложнений при условии ранней диагностики предопределяют необходимость скрининга, т.е. проведения обследования с целью исключения СД-2 среди лиц без каких-либо симптомов заболевания. Основным тестом, как указывалось, является определение уровня гликемии натощак. Оно показано в следующих ситуациях:
1. У всех людей в возрасте старше 45 лет, особенно при избытке массы тела (ИМТ более 25 кг/м2) с интервалом раз в 3 года.
2. В более молодом возрасте при наличии избытка массы тела (ИМТ более 25 кг/м2) и дополнительных факторов риска, к которым относятся:
- малоподвижный образ жизни;
- СД-2 у ближайших родственников;
- принадлежность к национальностям высокого риска развития СД-2 (афроамериканцы, латиноамериканцы, коренные американцы и др.);
- женщины, родившие ребенка весом более 4 кг и/или при наличии гестационного СД в анамнезе;
- артериальная гипертензия (≥ 140/90 мм Hg);
- уровень ЛПВП > 0,9 ммоль/л и/или триглицеридов > 2,8 ммоль/л;
- синдром поликистозных яичников;
- НТГ и НГНТ;
- сердечно-сосудистые заболевания.
Значительный рост заболеваемости СД-2 среди детей диктует необходимость скринингового определения уровня гликемии среди детей и подростков (начиная с 10 лет с интервалом в 2 года или с началом
пубертата, если он произошел в более раннем возрасте), относящихся к группам повышенного риска, к которым относятся дети с избытком массы тела (ИМТ и/или масса тела > 85 перцентиля, соответствующего возрасту, или вес более 120 % по отношению к идеальному) в сочетании с любыми двумя перечисленными дополнительными факторами риска:
• СД-2 среди родственников первой или второй линии родства;
• принадлежность к национальностям высокого риска;
• клинические проявления, ассоциированные с инсулинорезистентностью (acanthosis nigricans, артериальная гипертензия, дислипидемия);
• СД, в том числе гестационный, у матери.
Дифференциальная диагностика
Наибольшее клиническое значение имеет дифференциальная диагностика СД-2 и СД-1, принципы которой описаны в п. 7.5 (табл. 7.6). Как указывалось, в большинстве случаев она базируется на данных клинической картины. В тех случаях, когда установление типа СД встречает затруднения, или есть подозрение на какой-то редкий вариант СД, в том числе в рамках наследственных синдромов, наиболее важный практический вопрос, на который необходимо ответить, состоит в том, нуждается ли пациент в инсулинотерапии.
Лечение
Основными компонентами лечения СД-2 являются: диетотерапия, расширение физической активности, сахароснижающая терапия, профилактика и лечение поздних осложнений СД. Поскольку большинство пациентов с СД-2 страдают ожирением, диета должна быть направлена на снижение веса (гипокалорийная) и профилактику поздних осложнений, в первую очередь макроангиопатии (атеросклероза). Гипокалорийная диета необходима всем пациентам с избытком массы тела (ИМТ 25-29 кг/м2) или ожирением (ИМТ > 30 кг/м2). В большинстве случаев следует рекомендовать снижение суточного калоража пищи до 1000-1200 ккал для женщин и до 1200-1600 ккал для мужчин. Рекомендуемое соотношение основных пищевых компонентов при СД-2 аналогично таковому при СД-1 (углеводы - 65 %, белки 10-35 %, жиры до 25-35 %). Употребление алкоголя необходимо ограничить в связи с тем, что он является существенным источником дополнительных калорий, кроме того, прием алкоголя на фоне тера-
пии препаратами сульфонилмочевины и инсулином может спровоцировать развитие гипогликемии (см. п. 7.7.3).
Рекомендации по расширению физической активности должны быть индивидуализированы. В начале рекомендуются аэробные нагрузки (ходьба, плаванье) умеренной интенсивности продолжительностью 30-45 минут 3-5 раз в день (около 150 минут в неделю). В дальнейшем необходимо постепенное увеличение физических нагрузок, что в существенной мере способствует снижению и нормализации массы тела. Кроме того, физические нагрузки способствуют снижению инсулинорезистентности и оказывают гипогликемизирующее действие. Сочетание диетотерапии и расширения физических нагрузок без назначения сахароснижающих препаратов позволяет поддерживать компенсацию СД в соответствии с установленными целями (табл. 7.3) примерно у 5 % пациентов с СД-2.
Препараты для сахароснижающей терапии при СД-2 могут быть подразделены на четыре основные группы.
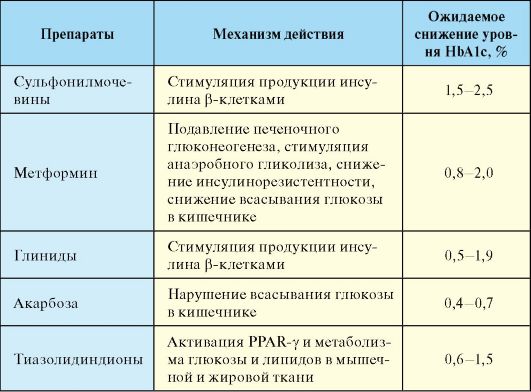
I. Препараты, способствующие снижению инсулинорезистентности (сенситайзеры). К этой группе относится метформин и тиазолидиндионы. Метформин является единственным использующимся в настоящее время препаратом из группы бигуанидов. Основными компонентами механизма его действия являются:
1. Подавление глюконеогенеза в печени (снижение продукции глюкозы печенью), которое приводит к снижению уровня гликемии натощак.
2. Снижение инсулинорезистентности (увеличение утилизации глюкозы периферическими тканями, прежде всего мышцами).
3. Активация аннаэробного гликолиза и уменьшение всасывания глюкозы в тонкой кишке.
Метформин является препаратом первого выбора сахароснижающей терапии у пациентов с СД-2, ожирением и гипергликемией натощак. Начальная доза составляет 500 мг на ночь или во время ужина. В дальнейшем доза постепенно повышается до 2-3 грамм на 2-3 приема. Среди побочных эффектов относительно часто встречаются диспепсические явления (диарея), которые, как правило, транзиторны и проходят самостоятельно через 1-2 недели приема препарата. Поскольку метформин не оказывает стимулирующего эффекта на продукцию инсулина, на фоне монотерапии этим препаратом гипогликемии не
развиваются (его действие обозначатся как антигипергликемическое, а не как гипогликемическое). Противопоказаниями к назначению метформина являются беременность, тяжелая сердечная, печеночная, почечная и другая органная недостаточность, а также гипоксические состояния другого генеза. Крайне редким осложнением, которое встречается при назначении метформина без учета приведенных противопоказаний, является лактатацидоз, являющийся следствием гиперактивации анаэробного гликолиза.
Тиазолидиндионы (пиоглитазон, розиглитазон) являются агонистами γ-рецепторов, активируемых пролифератором пероксисом (PPAR-γ). Тиазолидиндионы активируют метаболизм глюкозы и липидов в мышечной и жировой тканях, что приводит к повышению активности эндогенного инсулина, т.е. К устранению инсулинорезистентности (сенситайзеры инсулина). Суточная доза пиоглитазона составляет 15-30 мг/сут, розиглитазона - 4-8 мг (на 1-2 приема). Весьма эффективна комбинация тиазолидиндионов с метформином. Противопоказанием к назначению тиазолидиндионов является повышение (в 2,5 раза и более) уровня печеночных трансаминаз. Помимо гепатотоксичности, к побочным эффектам тиазолидиндионов относятся задержка жидкости и отеки, которые чаще развиваются при комбинации препаратов с инсулином.
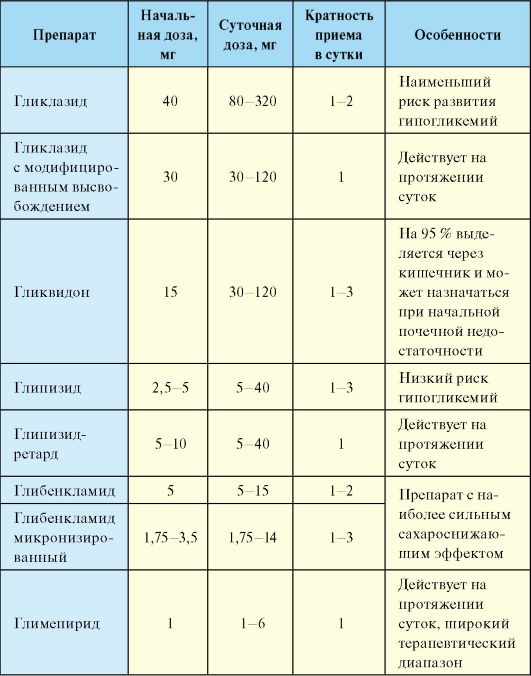
II. Препараты, воздействующие на β-клетку и способствующие усилению секреции инсулина. К этой группе относятся препараты сульфонилмочевины и глиниды (прандиальные регуляторы гликемии), которые используют преимущественно для нормализации уровня гликемии после еды. Основной мишенью препаратов сульфонилмочевины (ПСМ) являются β-клетки панкреатических островков. ПСМ связываются на мембране β-клеток со специфическими рецепторами. Это приводит к закрытию АТФ-зависимых калиевых каналов и деполяризации клеточной мембраны, что в свою очередь способствует открытию кальциевых каналов. Поступление кальция внутрь β -клеток приводит к их дегрануляции и выбросу инсулина в кровь. В клинической практике используется достаточно много ПСМ, которые отличаются по продолжительности и выраженности сахароснижающего эффекта (табл. 7.9).
Табл. 7.9. Препараты сульфонилмочевины
 Основным
и достаточно частым побочным эффектом ПСМ является гипогликемия (см. п.
7.7.3). Она может возникать при передозировке препаратом, его кумуляции
(почечная недостаточность),
Основным
и достаточно частым побочным эффектом ПСМ является гипогликемия (см. п.
7.7.3). Она может возникать при передозировке препаратом, его кумуляции
(почечная недостаточность),
несоблюдении диеты (пропуск приема пищи, прием алкоголя) или режима (значительная физическая нагрузка, перед которой не снижена доза ПСМ или не приняты углеводы).
К группе глинидов (прандиальные регуляторы гликемии) относятся репаглинид (производное бензоевой кислоты; суточная доза 0,5-16 мг/сут) и натеглинид (производное D-фенилаланина; суточная доза 180-540 мг/сут). После приема препараты быстро и обратимо взаимодействуют с рецептором сульфонилмочевины на β-клетке, в результате чего происходит короткое повышение уровня инсулина, которое имитирует первую фазу его секреции в норме. Препараты принимаются за 10-20 минут до основных приемов пищи, обычно 3 раза в день.
III. Препараты, снижающие всасывание глюкозы в кишечнике.
К этой группе относится акарбоза и гуаровая смола. Механизм действия акарбозы заключается в обратимой блокаде α-гликозидаз тонкой кишки, в результате которой замедляются процессы последовательного ферментирования и всасывания углеводов, снижается скорость резорбции и поступления глюкозы в печень и снижается уровень постпрандиальной гликемии. Начальная доза акарбозы составляет 50 мг 3 раза в день, в дальнейшем доза может быть увеличена до 100 мг 3 раза в сутки; препарат принимается непосредственно перед едой или во время еды. Основным побочным эффектом акарбозы является кишечная диспепсия (диареи, метеоризм), которая связана с поступлением невсосавшихся углеводов в толстую кишку. Сахароснижающий эффект акарбозы весьма умерен (табл. 7.10).
В клинической практике таблетированные сахароснижающие препаратов эффективно комбинируется друг с другом и с препаратами инсулина, поскольку у большинства пациентов одновременно определяется как тощаковая, так и постпрандиальная гипергликемия. Существуют многочисленные фиксированные комбинации препаратов в одной таблетке. Наиболее часто в одной таблетке комбинируют метформин с различными ПСМ, а также метформин с тиазолидиндионами.
Табл. 7.10. Механизм действия и потенциальная эффективность таблетированных сахароснижающих препаратов
 IV. Инсулины и аналоги инсулинов
IV. Инсулины и аналоги инсулинов
На определенном этапе препараты инсулинов начинают получать до 30-40 % пациентов с СД-2. Показания для инсулинотерапии при СД-2 приведены в начале п. 7.4. Наиболее частый вариант перевода пациентов с СД-2 на инсулинотерапию заключается в назначении инсулина пролонгированного действия (инсулин НПХ, гларгин или детемир) в комбинации с принимаемыми таблетированными сахароснижающими препаратами. В ситуации, когда уровень гликемии натощак не удается контролировать назначением метформина или последний противопоказан, пациенту назначается вечерняя (на ночь) инъекция инсулина. При невозможности контролировать при помощи таблетированных препаратов как тощаковую, так и постпрандиальную гликемию, пациент переводится на моноинсулинотерапию. Обычно, при СД-2 инсулинотерапия ведется по так называемой «традиционной» схеме, которая подразумевает назначение фиксированных доз инсулина пролонгированного и короткого действия. В этом плане
удобны стандартные смеси инсулинов, содержащие в одном флаконе инсулин короткого (ультракороткого) и пролонгированного действия. Выбор традиционной инсулинотерапии определяется тем, что при СД-2 она зачастую назначается пожилым пациентам, обучение которых самостоятельному изменению дозы инсулина затруднено. Кроме того, интенсивная инсулинотерапия, целью которой является поддержание компенсации углеводного обмена на уровне, приближающемся к нормогликемии, несет повышенный риск гипогликемий. Если для молодых пациентов легкие гипогликемии не представляют серьезной опасности, у пожилых пациентов со сниженным порогом ощущения гипогликемии они могут иметь весьма неблагоприятные последствия со стороны сердечно-сосудистой системы. Молодым пациентам с СД-2, а также пациентам перспективным в плане возможности эффективного обучения, может быть назначен интенсивный вариант инсулинотерапии.
Прогноз
Основной причиной инвалидизации и смерти пациентов с СД-2 являются поздние осложнения (см. п. 7.8), чаще всего диабетическая макроангиопатия. Риск развития отдельных поздних осложнений определяется комплексом факторов, которые обсуждаются в соответствующих главах. Универсальным фактором риска их развития является хроническая гипергликемия. Так, снижение уровня HbA1c у пациентов с СД-2 на 1 % приводит к уменьшению общей смертности примерно на 20 %, на 2 % и 3 % - соответственно примерно на 40 %
и 60 %.
7.7. ОСТРЫЕ ОСЛОЖНЕНИЯ САХАРНОГО ДИАБЕТА
7.7.1. Диабетический кетоацидоз
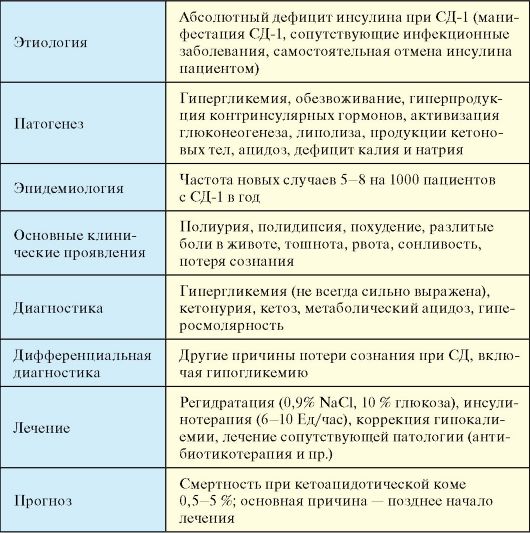
Диабетический кетоацидоз (ДКА) - декомпенсация СД-1, обусловленная абсолютным дефицитом инсулина, при отсутствии своевременного лечения заканчивающаяся кетоацидотической комой (КК) и смертью.
Этиология
Причиной ДКА является абсолютный дефицит инсулина. Той или иной выраженности ДКА определяется у большинства пациентов на момент манифестации СД-1 (10-20 % всех случаев ДКА).
У пациента с установленным диагнозом СД-1 ДКА может развиться при прекращении введения инсулина, зачастую самим пациентом (13 % случаев ДКА), на фоне сопутствующих заболеваний, в первую очередь, инфекционных, при отсутствии увеличения дозы инсулина
(30-40 %).
Табл. 7.11. Диабетический кетоацидоз
 До
20 % случаев развития ДКА у молодых пациентов с СД-1 связаны с
психологическими проблемами и/или нарушениями пищевого поведения (боязнь
прибавки веса, боязнь гипогликемий, подростковые проблемы). Достаточно
частой причиной ДКА в ряде стран является
До
20 % случаев развития ДКА у молодых пациентов с СД-1 связаны с
психологическими проблемами и/или нарушениями пищевого поведения (боязнь
прибавки веса, боязнь гипогликемий, подростковые проблемы). Достаточно
частой причиной ДКА в ряде стран является
отмена инсулина самим пациентом из-за дороговизны препаратов для некоторых слоев населения (табл. 7.11).
Патогенез
В основе патогенеза ДКА лежит абсолютный дефицит инсулина в сочетании с повышением продукции контринсулярных гормонов, таких как глюкагон, катехоламины и кортизол. В результате происходит значительное усиление продукция глюкозы печенью и нарушение ее утилизации периферическими тканями, нарастание гипергликемии и нарушение осмолярности внеклеточного пространства. Дефицит инсулина в сочетании с относительным избытком контринсулярных гормонов при ДКА приводит к высвобождению в циркуляцию свободных жирных кислот (липолиз) и их несдерживаемому окислению в печени до кетоновых тел (β-гидроксибутират, ацетоацетат, ацетон), в результате чего развивается гиперкетонемия, а в дальнейшем метаболический ацидоз. В результате выраженной глюкозурии развивается осмотический диурез, обезвоживание, потеря натрия, калия и других электролитов (рис. 7.9).
Эпидемиология
Частота новых случаев ДКА составляет 5-8 на 1000 пациентов с СД-1 в год и напрямую зависит от уровня организации медицинской помощи больным СД. Ежегодно в США происходит около 100 000 госпитализаций по поводу ДКА, при этом с учетом затраты на одного пациента за госпитализацию 13 тыс. долларов, ежегодно на стационарное лечение ДКА тратится более 1 миллиарда долларов в год. В РФ в 2005 г. ДКА зафиксирован у 4,31 % детей, 4,75 % подростков и 0,33 % взрослых пациентов с СД-1.
Клинические проявления
Развитие ДКА в зависимости от вызвавшей его причины может занимать от нескольких недель до суток. В большинстве случаев ДКА предшествуют симптомы декомпенсации диабета, но иногда они могут не успеть развиться. Клинические симптомы ДКА включают полиурию, полидипсию, похудение, разлитые боли в животе («диабетический псевдоперитонит»), дегидратацию, выраженную слабость, запах ацетона изо рта (или фруктовый запах), постепенное помутнение сознания. Истинная кома при ДКА в последнее время в силу ранней диагностики развивается относительно редко. При физикальном исследовании выявляются признаки обезвоживания: снижение
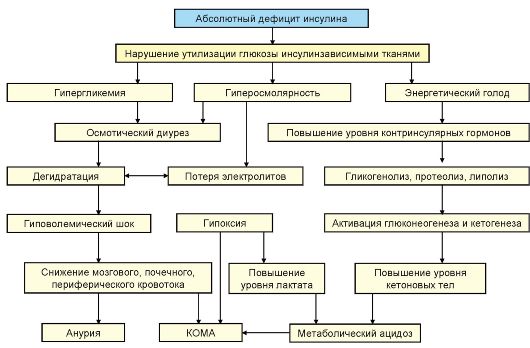 Рис. 7.9. Патогенез кетоацидотической комы
Рис. 7.9. Патогенез кетоацидотической комы
тургора кожи и плотности глазных яблок, тахикардия, гипотония. В далеко зашедших случаях развивается дыхание Куссмауля. Более чем у 25 % пациентов с ДКА развивается рвота, которая по цвету может напоминать кофейную гущу.
Диагностика
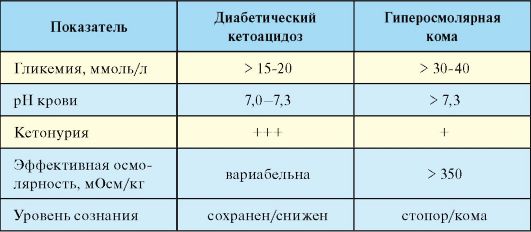
Базируется на данных клинической картины, указаниях на наличие у пациента СД-1, а также данных лабораторного исследования. Для ДКА характерна гипергликемия (в ряде случаев незначительная), кетонурия, метаболический ацидоз, гиперосмолярность (табл. 7.12).
Табл. 7.12. Лабораторная диагностика острых осложнений сахарного диабета
 При
обследовании пациентов с острой декомпенсацией СД необходимо
определение уровня гликемии, креатинина и мочевины, электролитов, на
основании чего производится расчет эффективной осмолярности. Кроме того,
необходима оценка кислотно-основного состояния. Эффективная осмолярность (ЭО) рассчитывается по следующей формуле: 2 * [Na+ (мЭкв/л) + глюкоза (ммоль/л)]. В норме ЭО составляет 285 - 295 мОсм/л.
При
обследовании пациентов с острой декомпенсацией СД необходимо
определение уровня гликемии, креатинина и мочевины, электролитов, на
основании чего производится расчет эффективной осмолярности. Кроме того,
необходима оценка кислотно-основного состояния. Эффективная осмолярность (ЭО) рассчитывается по следующей формуле: 2 * [Na+ (мЭкв/л) + глюкоза (ммоль/л)]. В норме ЭО составляет 285 - 295 мОсм/л.
У большинства пациентов с ДКА определяется лейкоцитоз, выраженность которого пропорциональна уровню кетоновых тел в крови. Уровень натрия, как правило, снижен вследствие осмотического оттока жидкости из интрацеллюлярных пространств в экстрацеллюлярные в ответ на гипергликемию. Реже уровень натрия может быть снижен ложноположительно как следствие выраженной гипер-
триглицеридемии. Уровень калия сыворотки исходно может быть повышен вследствие его перемещения из экстрацеллюлярных пространств.
Дифференциальная диагностика
Другие причины потери сознания у пациентов с СД. Дифференциальная диагностика с гиперосмолярной комой, как правило, не вызывает затруднений (развивается у пожилых пациентов с СД-2) и не имеет большого клинического значения, т.к. принципы лечения обоих состояний сходны. При невозможности оперативно выяснить причину потери сознания пациента с СД ему показано введение глюкозы, т.к. гипогликемические состояния встречаются значительно чаще, а быстрая положительная динамика на фоне введения глюкозы сама по себе позволяет выяснить причину потери сознаний.
Лечение
Лечение ДКА подразумевает регидратацию, коррекцию гипергликемии, электролитных расстройств, а также лечение заболеваний, вызвавших декомпенсацию диабета. Лечение наиболее оптимально проводить в реанимационном отделении специализированного лечебного учреждения. У взрослых пациентов без тяжелой сопутствующей сердечной патологии еще на догоспитальном этапе в качестве первоочередной меры с целью регидратации рекомендуется введение изотонического раствора (0,9 % NaCl) ориентировочно со скоростью литр в час (около 15-20 мл на килограмм веса в час). Полное возмещение дефицита жидкости, которое при ДКА составляет 100-200 мл на кг веса, должно быть достигнуто в пределах первых суток лечения. При сопутствующей сердечной или почечной недостаточности этот период времени должен быть увеличен. Для детей рекомендуемый объем изотонического раствора для регидратационной терапии составляет 10-20 мл на кг массы тела в час, при этом за первые 4 часа он не должен превысить 50 мл на кг веса. Полную регидратацию рекомендуется достигнуть примерно через 48 часов. После того как на фоне параллельно проводимой инсулинотерапии уровень гликемии снизится примерно до 14 ммоль/л, переходят на переливание 10 % раствора глюкозы, которым и продолжается регидратация.
В настоящее время принята концепция «малых доз» инсулина при лечении ДКА. Используется только инсулин короткого действия. Наиболее оптимально использование внутривенного введения инсу-
лина. Внутримышечное введение инсулина, которое менее эффективно, возможно только при умеренной тяжести ДКА, при стабильной гемодинамике и при невозможности проведения внутривенной терапии. В последнем случае инъекции делаются в прямую мышцу живота, при этом на инсулиновый шприц надевается игла для внутримышечных инъекций (для надежного внутримышечного попадания), и по этой игле инсулин набирается из флакона в шприц.
Возможно несколько вариантов внутривенного введения инсулина. Во-первых, инсулин может вводиться «в резинку» инфузионной системы, при этом необходимое количество инсулина набирается в инсулиновый шприц, после чего в него добирается 1 мл изотонического раствора. Вплоть до достижения уровнем гликемии 14 ммоль/л ежечасно пациенту вводится по 6-10 Ед инсулина короткого действия; в дальнейшем (параллельно со сменой регидратационного раствора с изотонического на 10 % глюкозу) в зависимости от ежечасно определяемых показателей гликемии доза инсулина снижается до 4-8 Ед в час. Рекомендованная скорость снижения уровня гликемии не должна превышать 5 ммоль/л в час. Другой вариант внутривенной инсулинотерапии подразумевает использование перфузора. Для приготовления раствора для перфузора исходят из соотношения: к 50 Ед инсулина короткого действия добавляется 2 мл 20 % раствора альбумина человека, после чего добавляется 50 мг 0,9 % изотонического раствора. В случае, если выбран внутримышечный путь введения инсулина, исходно вводится 20 Ед инсулина короткого действия, после чего ежечасно по 6 Ед, а после достижения уровнем гликемии 14 ммоль/л доза снижается до 4 Ед в час. После полной стабилизации гемодинамики и компенсации кислотно-основных нарушений пациент переводится на подкожные инъекции инсулина.
Как указывалось, несмотря на значительный дефицит калия в организме (общая потеря 3-6 ммоль/кг), при ДКА его уровень до начала инсулинотерапии может быть несколько повышен. Тем не менее, начало переливания раствора хлорида калия рекомендуется проводить одновременно с началом инсулинотерапии, если уровень калия плазмы меньше 5,5 ммоль/л. Успешная коррекция дефицита калия происходит только на фоне нормализации рН. При низком рН поступление калия внутрь клетки значительно снижено, в связи с этим, по возможности дозу переливаемого хлорида калия желательно адаптировать к конкретному показателю рН (табл. 7.13).
Табл. 7.13. Схема коррекции дефицита калия
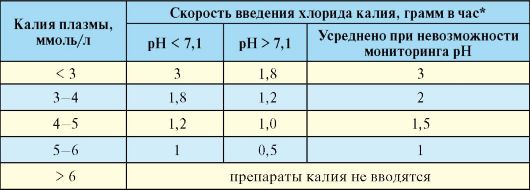 * Для расчета используют следующие данные:
* Для расчета используют следующие данные:
1 г KCl = 13,4 ммоль; 1 ммоль KCl = 0,075 г. В 4% раствор КС1: в 100 мл - 4 г КС1, в 25 мл - 1 г КС1, в 10 мл 0,4 г КС1.
Причиной декомпенсации диабета зачастую являются инфекционные заболевания (пиелонефрит, инфицированная язва при синдроме диабетической стопы, пневмония, синусит и проч.). Существует правило, согласно которому при ДКА антибиотикотерапия назначается практически всем пациентам с субфебрилитетом или лихорадкой даже при отсутствии видимого очага инфекции, поскольку собственно для ДКА повышение температуры тела не характерно.
Прогноз
Смертность при ДКА составляет 0,5-5 %, при этом большинство случаев обусловлено поздним и неквалифицированным оказанием медицинской помощи. Смертность наиболее высока (до 50 %) среди пациентов пожилого возраста.
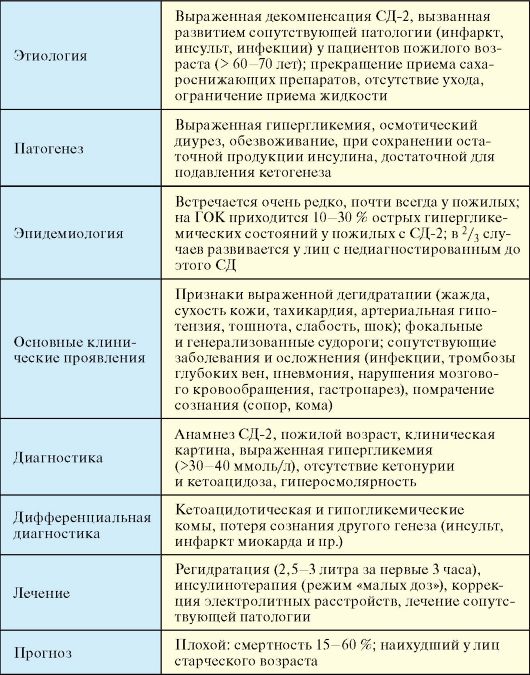
7.7.2. Гиперосмолярная кома
Гиперосмолярная кома (ГОК) - редкое острое осложнение СД-2, развивающееся вследствие выраженной дегидратации и гипергликемии на фоне отсутствия абсолютного дефицита инсулина, сопровождающееся высокой летальностью (табл. 7.14).
Этиология
ГОК, как правило, развивается у пожилых пациентов с СД-2. Такие пациенты чаще всего одиноки, живут без ухода, пренебрегают своим состоянием и самоконтролем и принимают недостаточно жидкости. Часто к декомпенсации приводят инфекции (синдром диабетической стопы, пневмонии, острый пиелонефрит), нарушения мозгового
кровообращения и другие состояния, в результате которых пациенты плохо передвигаются, не принимают сахароснижающие препараты и жидкость.
Табл. 7.14. Гиперосмолярная кома (ГОК)
 Патогенез
Патогенез
Нарастающая гипергликемия и осмотический диурез обусловливают выраженную дегидратацию, которая по указанным выше причинам не восполняется извне. Результатом гипергликемии и дегидратации является гиперосмолярность плазмы. Неотъемлемым компонентом патогенеза ГОК является относительный дефицит инсулина и избыток контринсулярных гормонов, тем не менее, сохраняющейся при СД-2 остаточной секреции инсулина оказывается достаточно для подавления липолиза и кетогенеза, вследствие чего не происходит развития кетоацидоза.
В ряде случаев может определяться умеренной выраженности ацидоз как результат гиперлактатемии на фоне тканевой гипоперфузии. При выраженной гипергликемии для сохранения осмотического баланса в цереброспинальной жидкости увеличивается содержание натрия, поступающего из клеток головного мозга, куда в обмен попадает калий. Нарушается трансмембранный потенциал нервных клеток. Развивается прогрессирующее помрачение сознания в сочетании с судорожным синдромом (рис. 7.10).
Эпидемиология
На ГОК приходится 10-30 % острых гипергликемических состояний у взрослых и пожилых пациентов с СД-2. Примерно в 2/3 случаев ГОК развивается у лиц с недиагностированным до этого СД.
Клинические проявления
Особенностями клинической картины гиперосмолярной комы являются:
- комплекс признаков и осложнений дегидратации и гипоперфузии: жажда, сухость слизистых, тахикардия, артериальная гипотензия, тошнота, слабость, шок;
- фокальные и генерализованные судороги;
- лихорадка, тошнота и рвота (40-65 % случаев);
- из сопутствующих заболеваний и осложнений часто встречаются тромбозы глубоких вен, пневмония, нарушения мозгового кровообращения, гастропарез.
Диагностика
Базируется на данных клинической картины, возрасте пациента и анамнезе СД-2, выраженной гипергликемии при отсутствии кетонурии и кетоацидоза. Типичные лабораторные признаки ГОК представлены в табл. 7.12.
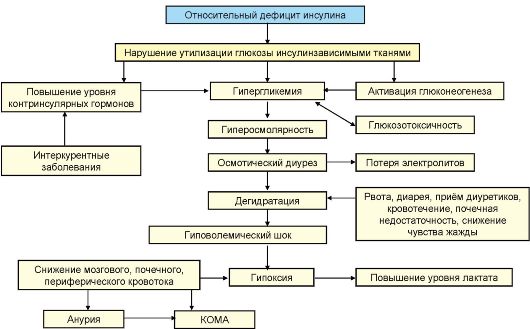 Рис. 7.10. Патогенез гиперосмолярной комы
Рис. 7.10. Патогенез гиперосмолярной комы
Дифференциальная диагностика
Другие острые состояния, развивающиеся у пациентов с СД, чаще всего с сопутствующей патологией, приведшей к выраженной декомпенсации СД.
Лечение
Лечение и мониторинг при ГОК, за исключением некоторых особенностей, не отличаются от таковых, описанных для кетоацидотической диабетической комы (п. 7.7.1):
• больший объем начальной регидратации 1,5-2 л за 1-й час; 1 л - за 2-й и 3-й час, далее по 500 мл/ч изотонического раствора хлорида натрия;
• потребность во введении калийсодержащих растворов, как правило, больше, чем при кетоацидотической коме;
• инсулинотерапия аналогична таковой при КК, но потребность в инсулине меньше и уровень гликемии необходимо снижать не быстрее, чем на 5 ммоль/л в час во избежание развития отека мозга;
• введения гипотонического раствора (NaCl 0,45 %) лучше избегать (только при выраженной гипернатриемии: > 155 ммоль/л и/или эффективной осмолярности > 320 мОсм/л);
• во введении бикарбоната нет необходимости (только в специализированных реанимационных отделениях при ацидозе с рН < 7,1).
Прогноз
Летальность при ГОК высока и составляет 15-60 %. Наихудший прогноз у пожилых пациентов с тяжелой сопутствующей патологией, которая, зачастую, и является причиной декомпенсации СД и развития ГОК.
7.7.3. Гипогликемия
Гипогликемия - снижение уровня глюкозы в сыворотке крови (<2,2- 2,8 ммоль/л), сопровождающее клинический синдром, характеризующийся признаками активации симпатической нервной системы и/или дисфункцией центральной нервной системы. Гипогликемия как лабораторный феномен не тождественен понятию «гипогликемическая симптоматика», поскольку лабораторные данные и клиническая картина не всегда совпадают.
Этиология
- Передозировка препаратов инсулина и его аналогов, а также препаратов сульфонилмочевины;
- недостаточный прием пищи на фоне неизменной сахароснижающей терапии;
- прием алкогольных напитков;
- физические нагрузки на фоне неизменной сахароснижающей терапии и/или без дополнительного приема углеводов;
- развитие поздних осложнений СД (автономная нейропатия с гастропарезом, почечная недостаточность) и ряда других заболеваний (надпочечниковая недостаточность, гипотиреоз, печеночная недостаточность, злокачественные опухоли) при неизменной сахароснижающей терапии (продолжение приема и кумуляция ТСП на фоне почечной недостаточности, сохранение прежней дозы инсулина);
- нарушение техники введения инсулина (внутримышечная инъекция вместо подкожной);
- артифициальная гипогликемия (сознательная передозировка сахароснижающих препаратов самим пациентом);
- органический гиперинсулинизм - инсулинома (см. п. 10.3).
Патогенез
Патогенез гипогликемии заключается в нарушении баланса между поступлением глюкозы в кровь, ее утилизацией, уровнем инсулина и контринсулярных гормонов. В норме при уровне гликемии в пределах 4,2-4,7 ммоль/л продукция и высвобождение инсулина из β-клеток подавлены. Снижение уровня гликемии менее 3,9 ммоль/л сопровождается стимуляцией продукции контринсулярных гормонов (глюкагон, кортизол, гормон роста, адреналин). Нейрогликопеническая симптоматика развивается при снижении уровня гликемии менее 2,5-2,8 ммоль/л. При передозировке инсулином и/или препаратами сульфонилмочевины гипогликемия развивается вследствие прямого гипогликемизирующего действия экзогенного или эндогенного гормона. В случае передозировки препаратами сульфонилмочевины гипогликемическая симптоматика может многократно рецидивировать после купирования приступа вследствие того, что длительность действия ряда препаратов может достигать суток и более. ТСП, которые не оказывают стимулирующего влияния на продукцию инсулина (метформин, тиазолидиндионы), сами по себе гипогликемии вызвать не могут, но при их добавлении к препаратам сульфонилмочевины или инсулину прием последних в прежней дозе может стать причиной гипогликемии вследствие кумуляции сахароснижающего эффекта комбинированной терапии (табл. 7.15).
Табл. 7.15. Гипогликемия
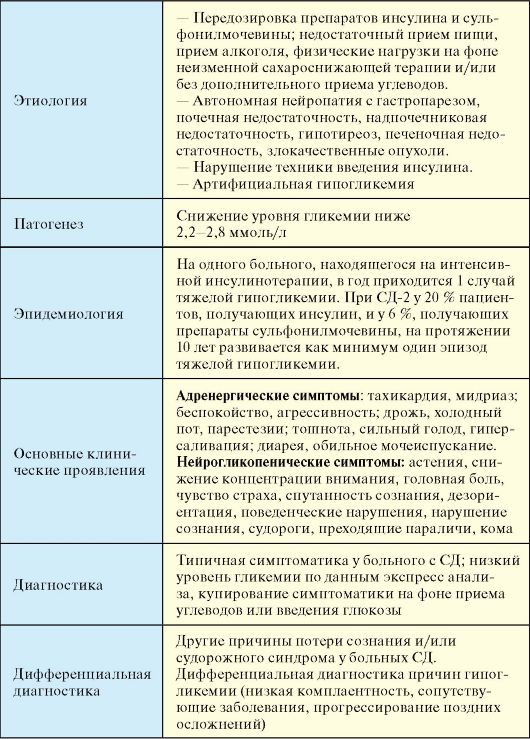 Окончание табл. 7.15
Окончание табл. 7.15
 При приеме алкоголя происходит подавление глюконеогенеза в печени, который является важнейшим фактором, противодействующим гипогликемии. Физические нагрузки способствуют
инсулиннезависимой утилизации глюкозы, благодаря чему на фоне
неизменной сахароснижающей терапии и/или при отсутствии дополнительного
приема углеводов могут явиться причиной гипогликемии.
При приеме алкоголя происходит подавление глюконеогенеза в печени, который является важнейшим фактором, противодействующим гипогликемии. Физические нагрузки способствуют
инсулиннезависимой утилизации глюкозы, благодаря чему на фоне
неизменной сахароснижающей терапии и/или при отсутствии дополнительного
приема углеводов могут явиться причиной гипогликемии.
Эпидемиология
Легкие, быстро купирующиеся гипогликемии у пациентов с СД-1, получающих интенсивную инсулинотерапию, могут развиваться несколько раз в неделю, и относительно безвредны. На одного больного, находящегося на интенсивной инсулинотерапии, в год приходится 1 случай тяжелой гипогликемии. В большинстве случаев гипогликемии развиваются в ночное время. При СД-2 у 20 % пациентов, получающих инсулин, и у 6 %, получающих препараты сульфонилмочевины, на протяжении 10 лет развивается как минимум один эпизод тяжелой гипогликемии.
Клинические проявления
Выделяют две основные группы симптомов: адренергические, связанные с активацией симпатической нервной системы и выбросом адреналина надпочечниками, и нейрогликопенические, связанные с нарушением функционирования центральной нервной системы на фоне дефицита ее основного энергетического субстрата. К адренергическим симптомам относятся: тахикардия, мидриаз; беспокойство, агрессивность; дрожь, холодный пот, парестезии; тошнота, сильный голод, гиперсаливация; диарея, обильное мочеиспускание. К нейрогликопеническим симптомам относят астению,
снижение концентрации внимания, головную боль, чувство страха, спутанность сознания, дезориентацию, галлюцинации; речевые, зрительные, поведенческие нарушения, амнезию, нарушение сознания, судороги, преходящие параличи, кому. Четкой зависимости выраженности и последовательности развития симптомов по мере утяжеления гипогликемии может не быть. Могут возникать только адренергические или только нейрогликопенические симптомы. В отдельных случаях, несмотря на восстановление нормогликемии и продолжающуюся терапию, пациенты могут пребывать в ступорозном или даже коматозном состоянии на протяжении нескольких часов и даже дней. Длительная гипогликемия или ее частые эпизоды могут привести к необратимым изменениям в ЦНС (прежде всего в коре больших полушарий), проявления которых значительно варьируют от делириозных и галлюцинаторно-параноидных эпизодов до типичных эпилептических припадков, неизбежным исходом которых является стойкое слабоумие.
Гипергликемия субъективно переносится пациентами легче, чем эпизоды даже легкой гипогликемии. Поэтому многие пациенты из-за боязни гипогликемии считают необходимым поддержание гликемии на относительно высоком уровне, который фактически соответствует декомпенсации заболевания. Преодоление этого стереотипа требует порой немалых усилий врачей и обучающего персонала.
Диагностика
Клиническая картина гипогликемии у пациента с СД в сочетании с лабораторным (как правило, при помощи глюкометра) выявлением низкого уровня глюкозы крови.
Дифференциальная диагностика
Другие причины, приводящие к потере сознания. Если причина потери сознания больного СД неизвестна и невозможно проведение экспресс-анализа уровня гликемии, ему показано введение глюкозы. Нередко возникает необходимость выяснения причины развития частых гипогликемий у пациентов с СД. Наиболее часто они являются следствием неадекватной сахароснижающей терапии и низкого уровня знаний пациента о своем заболевании. Следует помнить о том, что к снижению потребности в сахароснижающей терапии вплоть до ее полной отмены («исчезнувший СД») могут приводить ряд заболеваний (надпочечниковая недостаточность, гипотиреоз, почечная и печеночная недостаточность), в том числе злокачественные опухоли.
Лечение
Для лечения легкой гипогликемии, при которой пациент в сознании и может сам оказать себе помощь, обычно достаточно принять пищу или жидкость, содержащую углеводы в количестве 1-2 хлебных единиц (10-20 г глюкозы). Такое количество содержится, например, в 200 мл сладкого фруктового сока. Напитки более эффективно купируют гипогликемию, поскольку в жидком виде глюкоза значительно скорее всасывается. Если симптоматика продолжает нарастать, несмотря на продолжающийся прием углеводов, необходимо внутривенное введение глюкозы или внутримышечное глюкагона. Аналогичным образом лечится и тяжелая гипогликемия, протекающая с потерей сознания. В этом случае пациенту вводится около 50 мл 40 % раствора глюкозы внутривенно. Введение глюкозы необходимо продолжать вплоть до купирования приступа и нормализации гликемии, хотя большей дозы - до 100 мл и более, как правило, не требуется. Глюкагон вводится (как правило, приготовленным в заводских условиях наполненным шприцем) внутримышечно или подкожно. Через несколько минут уровень гликемии благодаря индукции глюкагоном гликогенолиза нормализуется. Однако это происходит не всегда: при высоком уровне инсулина в крови глюкагон неэффективен. Период полувыведения глюкагона короче, чем инсулина. При алкоголизме и болезнях печени синтез гликогена нарушен, и введение глюкагона может оказаться неэффективным. Побочным эффектом введения глюкагона может быть рвота, создающая опасность аспирации. Близким пациента желательно владеть техникой инъекции глюкагона.
Прогноз
Легкие гипогликемии у обученных пациентов на фоне хорошей компенсации заболевания безопасны. Частые гипогликемии являются признаком плохой компенсации СД; в большинстве случаев у таких пациентов в остальное время суток определяется более или менее выраженная гипергликемия и высокий уровень гликированного гемоглобина. У пожилых пациентов с поздними осложнениями СД гипогликемии могут провоцировать такие сосудистые осложнения, как инфаркт миокарда, инсульт, кровоизлияние в сетчатку. Гипогликемическая кома длительностью до 30 мин при адекватном лечении и быстром возвращении сознания, как правило, не имеет каких-либо осложнений и последствий.
7.8. ПОЗДНИЕ ОСЛОЖНЕНИЯ САХАРНОГО ДИАБЕТА
Поздние осложнения развиваются при обоих типах СД. Клинически выделяют пять основных поздних осложнений СД: макроангиопатию, нефропатию, ретинопатию, нейропатию и синдром диабетической стопы. Неспецифичность поздних осложнений для отдельных типов СД определяется тем, что их основным патогенетическим звеном является хроническая гипергликемия. В связи с этим на момент манифестации СД-1 поздние осложнения у пациентов практически никогда не встречаются, развиваясь через годы и десятилетия в зависимости от эффективности проводимой терапии. Наибольшее клиническое значение при СД-1, как правило, приобретает диабетическая микроангиопатия (нефропатия, ретинопатия) и нейропатия (синдром диабетической стопы). При СД-2, напротив, поздние осложнения часто выявляются уже на момент установления диагноза. Во-первых, это связано с тем, что СД-2 манифестирует задолго до установления диагноза. Во-вторых, атеросклероз, клинически проявляющийся макроангиопатией, имеет много общих с СД звеньев патогенеза. При СД-2 наибольшее клиническое значение, как правило, приобретает диабетическая макроангиопатия, которая на момент постановки диагноза выявляется у подавляющего большинства пациентов. В каждом конкретном случае набор и выраженность отдельных поздних осложнений варьируют от их парадоксального полного отсутствия, несмотря на значительную длительность заболевания вплоть до сочетания всех возможных вариантов в тяжелой форме.
Поздние осложнения являются основной причиной смерти пациентов с СД, а принимая во внимание его распространенность - важнейшей медико-социальной проблемой здравоохранения большинства стран. В связи с этим основной целью лечения и наблюдения пациентов с СД является профилактика (первичная, вторичная, третичная) его поздних осложнений.
7.8.1. Диабетическая макроангиопатия
Диабетическая макроангиопатия - собирательное понятие, объединяющее атеросклеротическое поражение крупных артерий при СД,
клинически проявляющееся ишемической болезнью сердца (ИБС), облитерирующим атеросклерозом сосудов головного мозга, нижних конечностей, внутренних органов и артериальной гипертензией (табл. 7.16).
Табл. 7.16. Диабетическая макроангиопатия
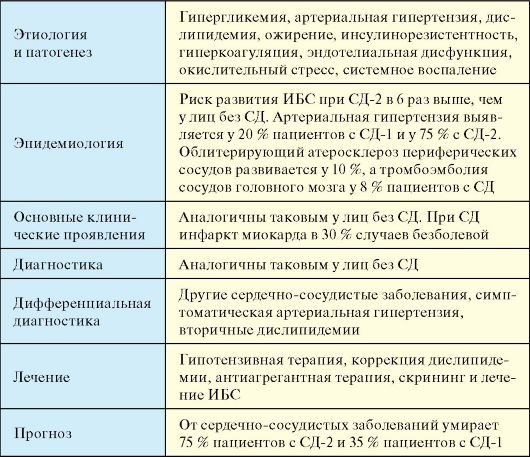 Этиология и патогенез
Этиология и патогенез
Вероятно, аналогичны этиологии и патогенезу атеросклероза у лиц без СД. Атеросклеротические бляшки не отличаются по микроскопическому строению у лиц с СД и без него. Тем не менее при СД на первый план могут выступать дополнительные факторы риска, или же СД усугубляет известные неспецифические факторы. К таковым при СД следует отнести:
1. Гипергликемию. Является фактором риска развития атеросклероза. Увеличение уровня HbA1c на 1 % у пациентов с СД-2 увеличива-
ет риск развития инфаркта миокарда на 15 %. Механизм атерогенного действия гипергликемии не вполне ясен, возможно, он связан с гликозированием конечных продуктов метаболизма ЛПНП и коллагена сосудистой стенки.
2. Артериальную гипертензию (АГ). В патогенезе большое значение придается почечному компоненту (диабетическая нефропатия). АГ при СД-2 - не менее значимый фактор риска инфаркта и инсульта, чем гипергликемия.
3. Дислипидемию. Гиперинсулинемия, являющаяся неотъемлемым компонентом инсулинорезистентности при СД-2, обусловливает снижение уровня ЛПВП, повышение уровня триглицеридов и снижение плотности, т.е. усиление атерогенности ЛПНП.
4. Ожирение, которым страдает большинство пациентов с СД-2, является независимым фактором риска атеросклероза, инфаркта миокарда и инсульта (см. п. 11.2).
5. Инсулинорезистентность. Гиперинсулинемия и высокий уровень инсулин-проинсулин-подобных молекул повышает риск развития атеросклероза, что, возможно, связано с эндотелиальной дисфункцией.
6. Нарушение коагуляции крови. При СД определяется повышение уровня фибриногена, активатора ингибитора тромбоцитов и фактора Виллебранда, в результате чего формируется протромботическое состояние свертывающейся системы крови.
7. Эндотелиальную дисфункцию, характеризующуюся повышением экспрессии активатора ингибитора плазминогена и молекул клеточной адгезии.
8. Окислительный стресс, приводящий к повышению концентрации окисленных ЛПНП и F2-изопростанов.
9. Системное воспаление, при котором происходит повышение экспрессии фибриногена и С-реактивного белка.
Наиболее значимыми факторами риска развития ИБС при СД-2 являются повышенный уровень ЛПНП, пониженный ЛПВП, артериальная гипертензия, гипергликемия и курение. Одним из отличий атеросклеротического процесса при СД является более распространенный и дистальный характер окклюзионного поражения, т.е. В процесс чаще вовлекаются относительно более мелкие артерии, что затрудняет хирургическое лечение и ухудшает прогноз.
Эпидемиология
Риск развития ИБС у лиц с СД-2 в 6 раз выше, чем у лиц без диабета, при этом он одинаков для мужчин и женщин. Артериальная гипертензия выявляется у 20 % пациентов с СД-1 и у 75 % с СД-2. В общем, у больных СД она встречается в 2 раза чаще, чем у лиц без него. Облитерирующий атеросклероз периферических сосудов развивается у 10 % больных с СД. Тромбоэмболия сосудов головного мозга развивается у 8 % пациентов с СД (в 2-4 раза чаще, чем у лиц без СД).
Клинические проявления
В основном не отличаются от таковых у лиц без СД. В клинической картине СД-2 макрососудистые осложнения (инфаркт миокарда, инсульт, окклюзионное поражение сосудов ног) зачастую выступают на первый план, и именно при их развитии у пациента нередко впервые обнаруживается гипергликемия. Возможно, вследствие сопутствующей автономной нейропатии до 30 % инфарктов миокарда у лиц с СД протекают без типичного ангинозного приступа (безболевой инфаркт).
Диагностика
Принципы диагностики осложнений атеросклероза (ИБС, нарушение мозгового кровообращения, окклюзионное поражение артерий ног) не отличаются от таковых для лиц без СД. Измерение артериального давления (АД) должно проводиться на каждом визите пациента с СД к врачу, а определение показателей липидного спектра крови (общий холестерин, триглицериды, ЛПНП, ЛПВП) при СД необходимо проводить не реже, чем раз в год.
Дифференциальная диагностика
Другие сердечно-сосудистые заболевания, симптоматическая артериальная гипертензия, вторичные дислипидемии.
Лечение
♦ Контроль артериального давления. Должный уровень систолического АД при СД составляет менее 130 ммHg, а диастолического 80 ммНg (табл. 7.3). Большинству пациентов для достижения этой цели необходимо назначение нескольких гипотензивных препаратов. Препаратами выбора гипотензивной терапии при СД являются ингибиторы АПФ и блокаторы рецепторов ангиотензина, которые при необходимости дополняются тиазидными диуретиками. Препаратами выбора для пациентов с СД, перенесших инфаркт миокарда, являются β-адреноблокаторы.
♦ Коррекция дислипидемии. Целевые уровни показателей липидного спектра представлены в табл. 7.3. Препаратами выбора гиполипидемической терапии являются ингибиторы 3-гидрокси-3-метилглу- тарил-КоА-редуктазы (статины).
♦ Антиагрегантная терапия. Терапия аспирином (75-100 мг/сут) показана пациентам с СД старше 40 лет при повышенном риске развития сердечно-сосудистой патологии (отягощенный семейный анамнез, артериальная гипертензия, курение, дислипидемия, микроальбуминурия), а также всем пациентам с клиническими проявлениями атеросклероза в качестве вторичной профилактики.
♦ Скрининг и лечение ИБС. Нагрузочные тесты для исключения ИБС показаны пациентам с симптомами сердечно-сосудистых заболеваний, а также при выявлении патологии при ЭКГ.
Прогноз
От сердечно-сосудистых заболеваний умирает 75 % пациентов с СД-2 и 35 % пациентов с СД-1. Примерно 50 % больных СД-2 умирают от осложнений ИБС, 15 % от тромбоэмболии сосудов головного мозга. Смертность от инфаркта миокарда у лиц с СД превышает 50 %.
7.8.2. Диабетическая ретинопатия
Диабетическая ретинопатия (ДР) - микроангиопатия сосудов сетчатки глаза, характеризующаяся развитием микроаневризм, кровоизлияний, экссудативных изменений и пролиферацией новообразованных сосудов, приводящая к частичной или полной потере зрения (табл. 7.17).
Этиология
Основным этиологическим фактором развития ДР является хроническая гипергликемия. Другие факторы (артериальная гипертензия, дислипидемия, курение, беременность и др.) имеют меньшее значение.
Патогенез
Основными звеньями патогенеза ДР являются:
• микроангиопатия сосудов сетчатки, приводящая к сужению просвета сосудов с развитием гипоперфузии;
• дегенерация сосудов с образованием микроаневризм;
• прогрессирующая гипоксия, стимулирующая пролиферацию сосудов и приводящая к жировой дистрофии и отложению солей кальция в сетчатке;
Табл. 7.17. Диабетическая ретинопатия
• 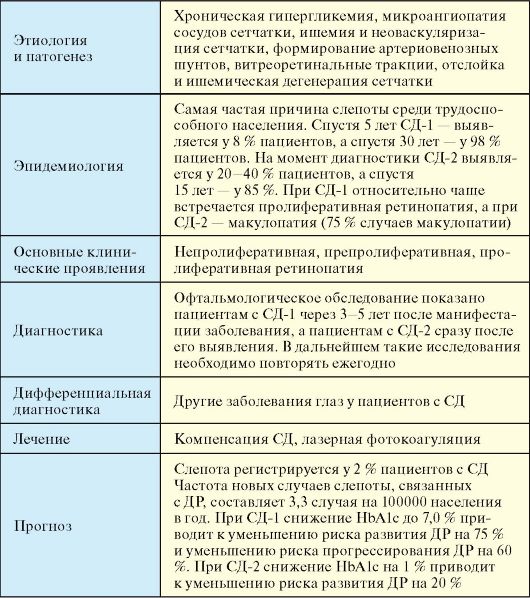 микроинфаркты с экссудацией, приводящие к образованию мягких «ватных пятен»;
микроинфаркты с экссудацией, приводящие к образованию мягких «ватных пятен»;
• отложение липидов с формированием плотных экссудатов;
• разрастание в сетчатке пролиферирующих сосудов с образованием шунтов и аневризм, приводящих к дилатации вен и усугублению гипоперфузии сетчатки;
• феномен обкрадывания с дальнейшим прогрессированием ишемизации, являющийся причиной образования инфильтратов и рубцов;
• отслоение сетчатки в результате ее ишемической дезинтеграции и образования витреоретинальных тракций;
• кровоизлияния в стекловидное тело в результате геморрагических инфарктов, массивной сосудистой инвазии и разрыва аневризм;
• пролиферация сосудов радужной оболочки (диабетический рубеоз), приводящая к развитию вторичной глаукомы;
• макулопатия с отеком сетчатки.
Эпидемиология
ДР является самой частой причиной слепоты среди трудоспособного населения развитых стран, а риск развития слепоты у пациентов с СД в 10-20 раз выше, чем в общей популяции. На момент диагностики СД-1 ДР не обнаруживается практически ни у кого из пациентов, спустя 5 лет заболевание выявляется у 8 % пациентов, а при тридцатилетнем стаже диабета - у 98 % пациентов. На момент диагностики СД-2 ДР выявляется у 20-40 % пациентов, а среди пациентов с пятнадцатилетнем стажем СД-2 - у 85 %. При СД-1 относительно чаще встречается пролиферативная ретинопатия, а при СД-2 - макулопатия (75 % случаев макулопатии).
Клинические проявления
Согласно общепринятой классификации, выделяют 3 стадии ДР
(табл. 7.18).
Диагностика
Полное офтальмологическое обследование, включающее прямую офтальмоскопию с фотографированием сетчатки, показано пациентам с СД-1 через 3-5 лет после манифестации заболевания, а пациентам с СД-2 - сразу после его выявления. В дальнейшем такие исследования необходимо повторять ежегодно.
Табл. 7.18. Классификация диабетической ретинопатии
 Дифференциальная диагностика
Дифференциальная диагностика
Другие заболевания глаз у пациентов с СД.
Лечение
Базовым принципом лечения диабетической ретинопатии, как и других поздних осложнений, является оптимальная компенсация СД. Наиболее эффективным методом лечения диабетической ретинопатии и предупреждения слепоты является лазерная фотокоагуляция. Целью
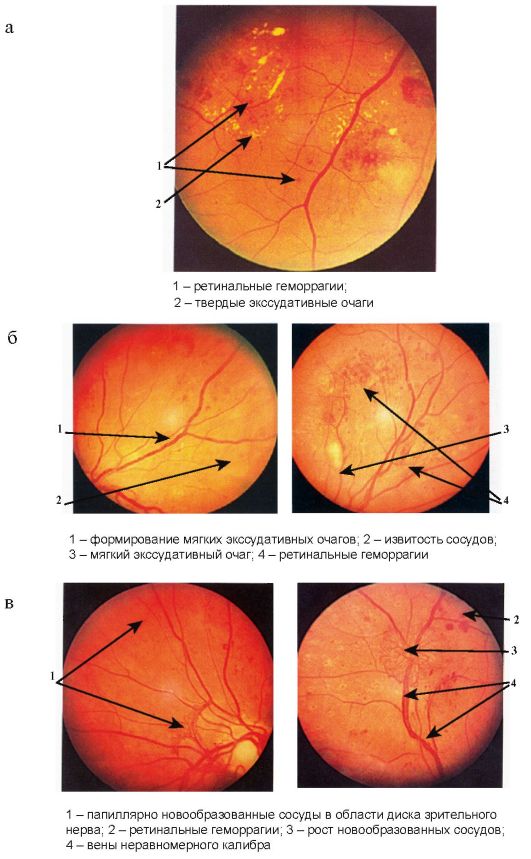 Рис. 7.11. Диабетическая ретинопатия:
Рис. 7.11. Диабетическая ретинопатия:
а) непролиферативная; б) препролиферативная; в) пролиферативная
лазерной фотокоагуляции является прекращение функционирования новообразованных сосудов, которые представляют основную угрозу развития таких тяжелых осложнений, как гемофтальм, тракционная отслойка сетчатки, рубеоз радужки и вторичная глаукома.
Прогноз
Слепота регистрируется у 2 % пациентов с СД (3-4 % пациентов с СД- 1 и 1,5-2 % пациентов с СД-2). Примерная частота новых случаев слепоты, связанных с ДР, составляет 3,3 случая на 100000 населения в год. При СД-1 снижение HbA1c до 7,0 % приводит к уменьшению риска развития ДР на 75 % и уменьшению риска прогрессирования ДР на 60 %. При СД-2 снижение HbA1c на 1 % приводит к уменьшению риска развития ДР на 20 %.
7.8.3. Диабетическая нефропатия
Диабетическая нефропатия (ДНФ) определяется как альбуминурия (более 300 мг альбумина в сутки или протеинурия более 0,5 г белка в сутки) и/или снижение фильтрационной функции почек у лиц с СД при отсутствии мочевых инфекций, сердечной недостаточности или других заболеваний почек. Микроальбуминурия определяется как экскреция альбумина 30-300 мг/сут или 20-200 мкг/мин.
Этиология и патогенез
Основными факторами риска ДНФ являются длительность СД, хроническая гипергликемия, артериальная гипертензия, дислипидемия, заболевания почек у родителей. При ДНФ в первую очередь поражается клубочковый аппарат почки.
1. Одним из возможным механизмов, по которому гипергликемия способствует развитию поражения клубочков, является аккумуляция сорбитола за счет активизации полиолового пути метаболизма глюкозы, а также ряда конечных продуктов гликирования.
2. Гемодинамические нарушения, а именно внутриклубочковая артериальная гипертензия (повышение кровяного давления внутри клубочков почки) является важнейшим компонентом патогенеза
ДНФ.
Причиной внутриклубочковой гипертензии является нарушение тонуса артериол: расширение приносящей и сужение выносящей.
Табл. 7.19. Диабетическая нефропатия
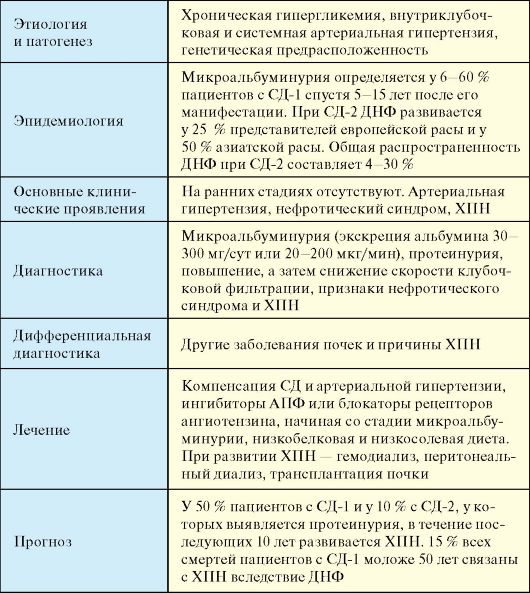 Это,
в свою очередь, происходит под воздействием ряда гуморальных факторов,
таких как ангиотензин-2 и эндотелин, а также вследствие нарушения
электролитных свойств базальной мембраны клубочков. Кроме того,
внутриклубочковой гипертензии способствует системная гипертензия,
которая определяется у большинства пациентов с ДНФ. Вследствие
внутриклубочковой гипертензии происходит повреждение базальных мембран и
фильтрационных пор,
Это,
в свою очередь, происходит под воздействием ряда гуморальных факторов,
таких как ангиотензин-2 и эндотелин, а также вследствие нарушения
электролитных свойств базальной мембраны клубочков. Кроме того,
внутриклубочковой гипертензии способствует системная гипертензия,
которая определяется у большинства пациентов с ДНФ. Вследствие
внутриклубочковой гипертензии происходит повреждение базальных мембран и
фильтрационных пор,
через которые начинают проникать следовые (микроальбуминурия), а затем значимые количества альбумина (протеинурия). Утолщение базальных мембран вызывает изменение их электролитных свойств, что само по себе приводит к попаданию большего количества альбумина в ультрафильтрат даже при отсутствии изменения размера фильтрационных пор.
3. Генетическая предрасположенность. У родственников пациентов с ДНФ с повышенной частотой встречается артериальная гипертензия. Существуют данные о связи ДНФ с полиморфизмом гена АПФ. Микроскопически при ДНФ выявляется утолщение базальных мембран клубочков, экспансия мезангия, а также фиброзные изменения приносящих и выносящих артериол. На конечной стадии, которая клинически соответствует хронической почечной недостаточности (ХПН), определяется очаговый (Киммельстиля-Уилсона), а затем диффузный гломерулосклероз.
Эпидемиология
Микроальбуминурия определяется у 6-60 % пациентов с СД-1 спустя 5-15 лет после его манифестации. ДНФ определяется у 35 % с СД-1, чаще у мужчин и у лиц, у которых СД-1 развился в возрасте моложе 15 лет. При СД-2 ДНФ развивается у 25 % представителей европейской расы и у 50 % азиатской расы. Общая распространенность ДНФ при СД-2 составляет 4-30 %.
Клинические проявления
Относительно ранним клиническим проявлением, которое косвенно связано с ДНФ, является артериальная гипертензия. Другие клинически явные проявления относятся к поздним. К ним можно отнести проявления нефротического синдрома и хронической почечной недостаточности.
Диагностика
Скрининг на ДНФ у лиц с СД подразумевает ежегодное тестирование на микроальбуминурию при СД-1 спустя 5 лет после манифестации заболевания, а при СД-2 - сразу после его выявления. Кроме того, необходимо как минимум ежегодное определение уровня креатинина для расчета скорости клубочковой фильтрации (СКФ). СКФ может быть рассчитана при помощи различных формул, например, по формуле Кокрофта-Голта:
 Для мужчин: а = 1,23 (норма СКФ 100 - 150 мл/мин) Для женщин: а = 1,05 (норма СКФ 85 - 130 мл/мин)
Для мужчин: а = 1,23 (норма СКФ 100 - 150 мл/мин) Для женщин: а = 1,05 (норма СКФ 85 - 130 мл/мин)
На начальных стадиях ДНФ может быть выявлено повышение СКФ, которая постепенно падает по мере развития ХПН. Микроальбуминурия начинает определяться через 5-15 лет после манифестации СД-1; при СД-2 в 8-10 % случаев она обнаруживается сразу после его выявления, вероятно, вследствие длительного бессимптомного течения заболевания до постановки диагноза. Пик развития явной протеинурии или альбуминурии при СД-1 приходится между 15 и 20 годами после его начала. Протеинурия свидетельствует о необратимости ДНФ, которая рано или поздно приведет к ХПН. Уремия в среднем развивается через 7-10 лет после появления явной протеинурии. Следует заметить, что СКФ не коррелирует с протеинурией.
Дифференциальная диагностика
Другие причины протеинурии и почечной недостаточности у лиц с СД. В большинстве случаев ДНФ сочетается с артериальной гипертензией, диабетической ретинопатией или нейропатией, при отсутствии которых дифференциальная диагностика должна быть особо тщательной. В 10 % случаев при с СД-1 и в30 % случаев при СД-2 протеинурия не связана с ДНФ.
Лечение
♦ Основными условиями первичной и вторичной профилактики
ДНФ являются компенсация СД и поддержание нормального системного артериального давления. Кроме того, первичная профилактика ДНФ подразумевает уменьшение потребления белковой пищи - менее 35 % суточного калоража.
♦ На стадиях микроальбуминурии и протеинурии пациентам показано назначение ингибиторов АПФ или блокаторов рецепторов ангиотензина. При сопутствующей артериальной гипертензии они назначаются в гипотензивных дозах, при необходимости в комбинации с другими гипотензивными препаратами. При нормальном артериальном давлении эти препараты назначаются в дозах, не приводящих к развитию гипотонии. Как ингибиторы АПФ (при СД-1 и СД-2), так и блокаторы рецепторов ангиотензина (при СД-2) способствуют предотвращению перехода микроальбуминурии в протеинурию. В ряде случаев на фоне указанной терапии в сочетании с компенсацией диабета по другим параметрам микроальбуминурия ликвидируется. Кроме того, начиная со стадии микроальбуминурии необходимо
сокращение потребление белков менее 10 % суточного калоража (или менее 0,8 грамм на кг веса) и соли менее 3 грамм в день.
♦ На стадии ХПН, как правило, требуется коррекция сахароснижающей терапии. Большинство пациентов с СД-2 необходимо перевести на инсулинотерапию, поскольку кумуляция ТСП несет риск развития тяжелой гипогликемии. У большинства пациентов с СД-1 происходит снижение потребности в инсулине, поскольку почка является одним из основных мест его метаболизма. При повышении уровня креатинина сыворотки до 500 мкмоль/л и более необходимо ставить вопрос о подготовке пациента к экстракорпоральному (гемодиализ, перитонеальный диализ) или хирургическому (трансплантация почки) методу лечения. Трансплантация почки показана при уровне креатинина до 600-700 мкмоль/л и снижении скорости клубочковой фильтрации менее 25 мл/мин, гемодиализ - 1000-1200 мкмоль/л и менее 10 мл/мин соответственно.
Прогноз
У 50 % пациентов с СД-1 и 10 % с СД-2, у которых выявляется протеинурия, в течение последующих 10 лет развивается ХПН. 15 % всех смертей пациентов с СД-1 моложе 50 лет связано с ХПН вследствие ДНФ.
7.8.4. Диабетическая нейропатия
Диабетическая нейропатия (ДНЕ) представляет собой сочетание синдромов поражения нервной системы, которые могут быть классифицированы в зависимости от преимущественного вовлечения в процесс ее различных отделов (сенсомоторная, автономная), а также распространенности и тяжести поражения (табл. 7.20).
I. Сенсомоторная нейропатия:
- симметричная;
- фокальная (мононейропатия) или полифокальная (краниальная, проксимальная моторная, мононейропатия конечностей и туловища).
II. Автономная (вегетативная) нейропатия:
- кардиоваскулярная (ортостатическая гипотензия, синдром сердечной денервации);
- гастроинтестинальная (атония желудка, дискинезия желчных путей, диабетическая энтеропатия);
- урогенитальная (с нарушением функций мочевого пузыря и половой функции);
- нарушение у пациента способности распознавать гипогликемию;
- нарушение функции зрачка;
- нарушение функций потовых желез (дистальный ангидроз, гипергидроз при еде).
Табл. 7.20. Диабетическая нейропатия
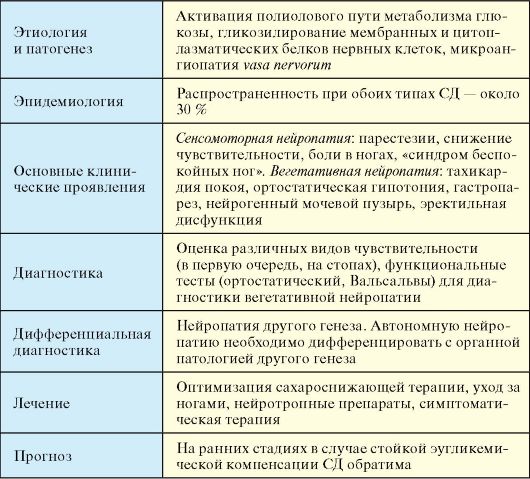 Этиология и патогенез
Этиология и патогенез
Основной причиной ДНЕ является гипергликемия. Предполагается несколько механизмов ее патогенеза:
• активизация полиолового пути метаболизма глюкозы, в результате чего в нервных клетках происходит накопление сорбитола, фруктозы и снижение содержания миоинозитола и глутатиона. Это, в свою очередь, приводит к активизации свободнорадикальных процессов и снижению уровня оксида азота;
• неэнзиматическое гликозилирование мембранных и цитоплазматических белков нервных клеток;
• микроангиопатия vasa nervorum, которая приводит к замедлению капиллярного кровотока и гипоксии нервов.
Эпидемиология
Распространенность ДНЕ при обоих типах СД составляет около 30 %. При СД-1 спустя 5 лет от начала заболевания она начинает выявляться у 10 % пациентов. Частота новых случаев ДНЕ при СД-2 составляет около 6 % пациентов в год. Наиболее частым вариантом является дистальная симметричная сенсомоторная ДНЕ.
Клинические проявления
Сенсомоторная ДНЕ проявляется комплексом двигательных и чувствительных нарушений. Частым симптомом дистальной формы ДНЕ являются парестезии, которые проявляются ощущением «ползания мурашек», онемением. Пациенты часто жалуются на зябкость ног, хотя они остаются теплыми на ощупь, что является признаком, позволяющим отличить полинейропатию от ишемических изменений, когда ноги на ощупь холодные. Ранним проявлением сенсорной нейропатии является нарушение вибрационной чувствительности. Характерным является синдром «беспокойных ног», представляющий собой сочетание ночных парестезии и повышенной чувствительности. Боли в ногах чаще беспокоят ночью, при этом иногда пациент не может выносить прикосновения одеяла. В типичном случае боли в противоположность таковым при облитерирующих заболеваниях артерий могут уменьшаться при ходьбе. Спустя годы боль может спонтанно прекратиться вследствие гибели мелких нервных волокон, отвечающих за болевую чувствительность. Гипоэстезия проявляется выпадением чувствительности по типу «чулок» и «перчаток». Нарушение глубокой, проприоцептивной чувствительности приводит к нарушению координации и затруднению передвижений (сенсорная атаксия). Пациент жалуется на «чужие ноги», ощущение «стояния на вате». Нарушение трофической иннервации приводит к дегенеративным изменениям кожи, костей и сухожилий. Нарушение болевой чувствительности приводит к частым, не замечаемым пациентом микротравмам стоп, которые легко инфицируются. Нарушение координации и ходьбы приводит к нефизиологическому перераспределению нагрузки на суставы стопы. В результате нарушаются анатомические взаимоотношения в опорно-двигательном аппарате ноги.
Деформируется свод стопы, развиваются отечность, фрактуры, хронические гнойные процессы (см. п. 7.8.5).
Выделяют несколько форм автономной ДНЕ. Причина кардиоваскулярной формы - нарушение иннервации сердечно-легочного комплекса и крупных сосудов. Блуждающий нерв является наиболее длинным нервом, в связи с чем поражается раньше других. В результате преобладания симпатических влияний развивается тахикардия покоя. Неадекватная реакция на ортостаз проявляется ортостатической гипотензией и синкопальными состояниями. Вегетативная денервация легочно-сердечного комплекса приводит к отсутствию вариабельности сердечного ритма. С автономной нейропатией связывают повышенную распространенность среди больных СД безболевых инфарктов миокарда.
Симптомами гастроинтестинальной формы ДНЕ являются гастропарез с замедленным или, наоборот, быстрым опорожнением желудка, что может создать сложности в подборе инсулинотерапии, поскольку время и объем всасывания углеводов неопределенно варьируют; атония пищевода, рефлюкс-эзофагит, дисфагия; водянистая диарея. Для урогенитальной формы ДНЕ характерны атония мочеточников и мочевого пузыря, приводящая к склонности к мочевым инфекциям; эректильная дисфункция (около 50 % больных СД); ретроградная эякуляция.
Другие возможные проявления вегетативной ДНЕ - нарушение способности распознавать гипогликемию, нарушение функции зрачка, нарушение функции потовых желез (ангидроз), диабетическая амиотрофия.
Диагностика
Неврологическое обследование пациентов с СД необходимо проводить ежегодно. Как минимум оно подразумевает проведение тестов, направленных на выявление дистальной сенсомоторной нейропатии. Для этого используется оценка вибрационной чувствительности при помощи градуированного камертона, тактильной чувствительности при помощи монофиламента, а также температурной и болевой чувствительности. По показаниям изучается состояние вегетативной нервной системы: для диагностики недостаточности парасимпатической иннервации сердца используют ряд функциональных проб, таких как измерение ЧСС при глубоком дыхании с оценкой вариабельности
сердечного ритма и пробу Вальсальвы; для диагностики недостаточности симпатической иннервации сердца используют ортостатическую пробу.
Дифференциальная диагностика
Нейропатии другого генеза (алкогольная, уремическая, при В12-дефицитной анемии и др.). Диагноз дисфункции того или иного органа в результате вегетативной нейропатии устанавливается только после исключения органной патологии.
Лечение
1. Оптимизация сахароснижающей терапии.
2. Уход за ногами (см. п. 7.8.5).
3. Эффективность нейротропных препаратов (α-липоевая кислота) подтверждается не во всех исследованиях.
4. Симптоматическая терапия (обезболивание, силденафил при эректильной дисфункции, флудрокортизон при ортостатической гипотонии и др.).
Прогноз
На начальных стадиях ДНЕ может быть обратимой на фоне стойкой компенсации СД. ДНЕ определяется у 80 % пациентов с язвенным поражением и является основным фактором риска ампутации ног
при СД.
7.8.5. Синдром диабетической стопы
Синдром диабетической стопы (СДС) - патологическое состояние стопы при СД, возникающее на фоне поражения периферических нервов, кожи и мягких тканей, костей и суставов и проявляющееся острыми и хроническими язвами, костно-суставными поражениями и гнойнонекротическими процессами (табл. 7.21).
Этиология и патогенез
Патогенез СДС многокомпонентен и представлен сочетанием нейропатических и перфузионных нарушений с выраженной склонностью к инфицированию. Исходя из преобладания в патогенезе того или иного из перечисленных факторов, выделяют 3 основные формы
СДС:
Табл. 7.21. Синдром диабетической стопы
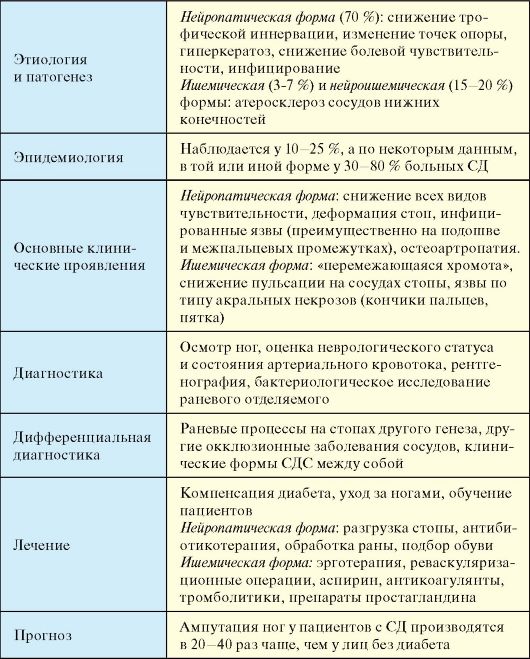 I. Нейропатическая форма (60-70 %):
I. Нейропатическая форма (60-70 %):
- без остеоартропатии;
- с диабетической остеоартропатией.
II. Нейроишемическая (смешанная) форма (15-20 %).
III. Ишемическая форма (3-7 %).
Нейропатическая форма СДС. При диабетической нейропатии в первую очередь поражаются дистальные отделы наиболее длинных нервов. Длительный дефицит трофической импульсации приводит к гипотрофии кожи, костей, связок, сухожилий и мышц. Результатом гипотрофии соединительных структур является деформация стопы с нефизиологичным перераспределением опорной нагрузки и ее чрезмерным увеличением на отдельные участки. В этих местах, например в области проекции головок плюсневых костей, отмечаются утолщение кожи и формирование гиперкератозов. Постоянное давление на эти участки приводит к воспалительному аутолизу подлежащих мягких тканей, что создает предпосылки для формирования язвенного дефекта. В результате атрофии и нарушения потоотделения кожа становится сухой, легко трескается. Из-за снижения болевой чувствительности пациент часто не обращает внимания на происходящие изменения. Он не может своевременно обнаружить неудобство обуви, что приводит к образованию потертостей и мозолей, не замечает внедрения инородных тел, мелких ранок в местах растрескивания. Ситуацию усугубляет нарушение глубокой чувствительности, проявляющееся в нарушении походки, неправильной установке ноги. Наиболее часто язвенный дефект инфицируется стафилококками, стрептококками, бактериями кишечной группы; нередко присоединяется анаэробная флора. Нейропатическая остеоартропатия является результатом выраженных дистрофических изменений в костносуставном аппарате стопы (остеопороз, остеолиз, гиперостоз).
Ишемическая форма СДС является следствием атеросклероза артерий нижних конечностей, приводящего к нарушению магистрального кровотока, т.е. является одним из вариантов диабетической макроангиопатии.
Эпидемиология
СДС наблюдается у 10-25 %, а по некоторым данным, в той или иной форме у 30-80 % больных СД. В США ежегодные расходы на лечение больных с СД c СДС составляют 1 млрд. долларов.
Клинические проявления
При нейропатической форме СДС выделяют два наиболее частых вида поражения: нейропатическая язва и остеоартропатия (с развитием
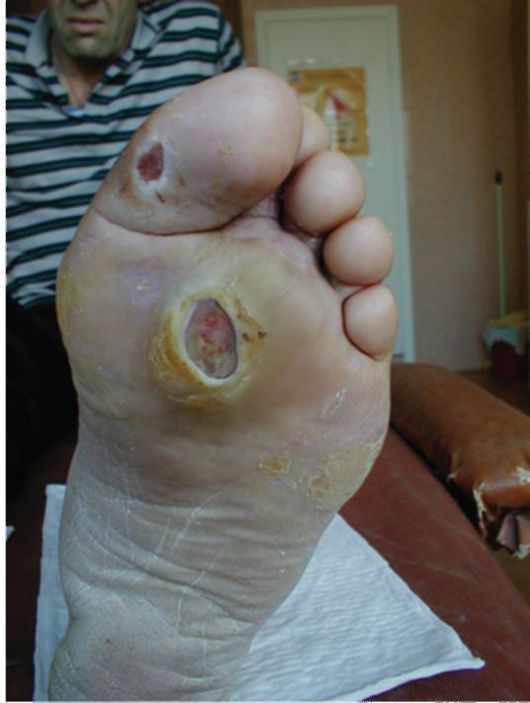 Рис. 7.12. Нейропатическая язва при синдроме диабетической стопы
Рис. 7.12. Нейропатическая язва при синдроме диабетической стопы
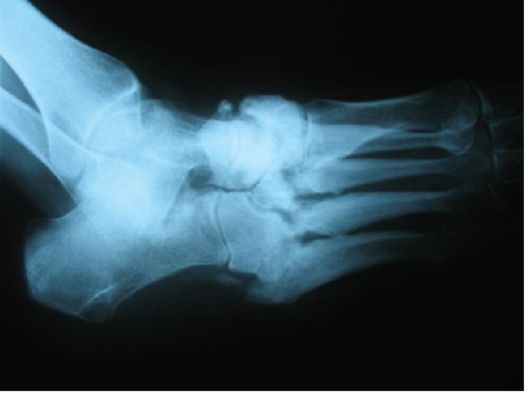 Рис. 7.13. Сустав Шарко при синдроме диабетической стопы
Рис. 7.13. Сустав Шарко при синдроме диабетической стопы
сустава Шарко). Нейропатические язвы, как правило, локализуются в области подошвы и межпальцевых промежутков, т.е. на участках стопы, испытывающих наибольшее давление (рис. 7.12).
Деструктивные изменения костно-связочного аппарата стопы могут прогрессировать на протяжении многих месяцев и привести к выраженной костной деформации - диабетической остео-артропатии и формированию сустава Шарко, при этом стопу образно сравнивают с «мешком с костями»
(рис. 7.13).
При ишемической форме СДС
кожа на стопах холодная, бледная или цианотичная; реже имеет розовато-красный оттенок изза расширения поверхностных капилляров в ответ на ишемию. Язвенные дефекты возникают по типу акральных некрозов - на кончиках пальцев, краевой поверхности пяток (рис. 7.14).
Пульс на артериях стопы, подколенных и бедренных артериях ослаблен или не пальпируется.
В типичных случаях пациенты предъявляют жалобы на «перемежающуюся хромоту». Тяжесть ишемического поражения конечности определяется тремя основными факторами: тяжестью стеноза, развитием коллатерального кровотока, состоянием свертывающей системы крови.
Диагностика
Осмотр ног больного СД должен производиться каждый раз во время визита к врачу, не реже раза в полгода. Диагностика СДС включает:
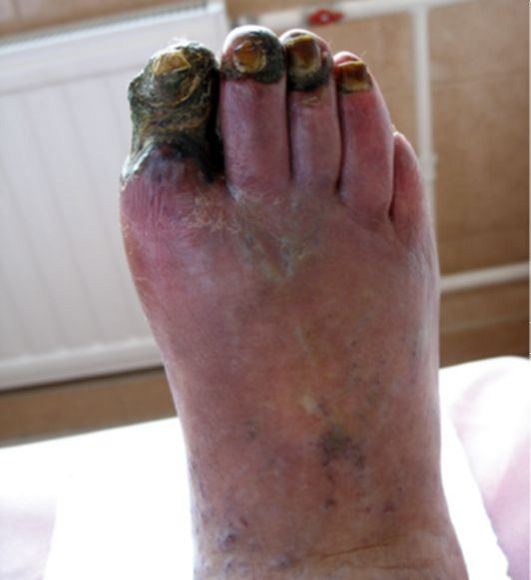 Рис. 7.14. Акральные некрозы при ишемической форме синдрома диабетической стопы
Рис. 7.14. Акральные некрозы при ишемической форме синдрома диабетической стопы
• осмотр ног;
• оценку неврологического статуса - различных видов чувствительности, сухожильных рефлексов, электромиографию;
• оценку состояния артериального кровотока - ангиографию, доплерометрию, доплерографию;
• рентгенографию стоп и голеностопных суставов;
• бактериологическое исследование раневого отделяемого.
Дифференциальная диагностика
Проводится с раневыми процессами на стопах другого генеза, а также другими окклюзионными заболеваниями сосудов нижних конечностей и патологией суставов стопы. Кроме того, необходимо дифференцировать клинические формы СДС (табл. 7.22).
Лечение
Лечение нейропатически-инфицированной формы СДС включает комплекс следующих мероприятий:
- оптимизацию компенсации СД, как правило, увеличение дозы инсулина, а при СД-2 - перевод на него;
- системную антибиотикотерапию;
- полную разгрузку стопы (это может в течение нескольких недель привести к заживлению язв, существующих годами);
- местную обработку раны с удалением участков гиперкератоза;
- уход за ногами, правильный подбор и ношение специальной обуви. Своевременно проводенная консервативная терапия позволяет
избежать оперативного вмешательства в 95 % случаев.
Табл. 7.22. Дифференциальная диагностика клинических форм СДС
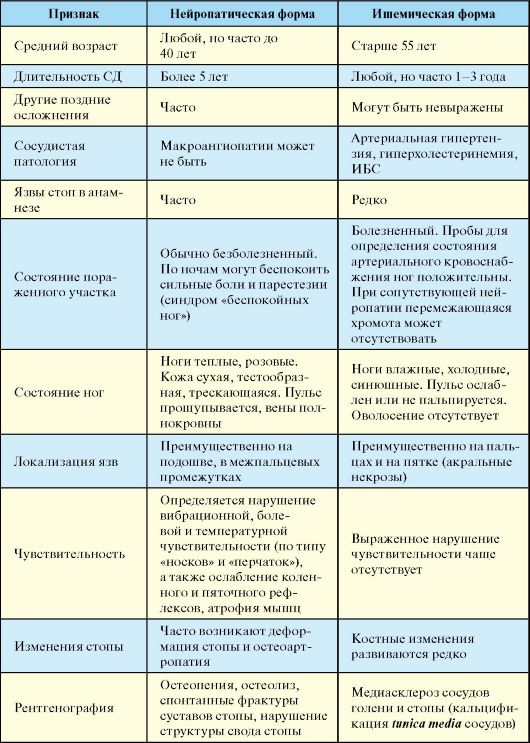 Лечение ишемической формы СДС включает:
Лечение ишемической формы СДС включает:
• оптимизацию компенсации СД, как правило, увеличение дозы инсулина, а при СД-2 - перевод на него;
• при отсутствии язвенно-некротических поражений эрготерапию (1-2-часовая ходьба в день, способствующая развитию коллатерального кровотока);
• реваскуляризационные операции на пораженных сосудах;
• консервативную терапию: антикоагулянты, аспирин (до 100 мг/сут), при необходимости - фибринолитики, препараты простагландина E1 и простациклина.
При развитии обширного гнойно-некротического поражения при всех вариантах СДС ставится вопрос об ампутации.
Прогноз
От 50 до 70 % от общего количества выполненных ампутаций ног приходится на долю больных СД. Ампутация ног у пациентов с СД производятся в 20-40 раз чаще, чем у лиц без диабета.
7.9. САХАРНЫЙ ДИАБЕТ И БЕРЕМЕННОСТЬ
Гестационный сахарный диабет (ГСД) - это нарушение толерантности к глюкозе, впервые выявленное во время беременности (табл. 7.23). Это определение не исключает того, что патология углеводного обмена могла предшествовать наступлению беременности. ГСД следует отличать от ситуаций, когда у женщины с диагностированным ранее диабетом (в силу возраста, чаще СД-1) наступает беременность.
Этиология и патогенез
При ГСД сходны с таковыми при СД-2. Высокий уровень овариальных и плацентарных стероидов, а также увеличение образования кортизола корой надпочечников приводят при беременности к развитию физиологической инсулинорезистентности. Развитие ГСД связывают с тем, что инсулинорезистентность, закономерно развивающаяся при беременности, и, следовательно, повышенная потребность в инсулине у предрасположенных лиц превышает функциональную способность β-клеток ПЖЖ. После родов с возвращением гормональных и метаболических взаимоотношений к исходному уровню он, как правило, проходит.
Табл. 7.23. Гестационный сахарный диабет
 ГСД
обычно развивается в середине 2 триместр, между 4 и 8 месяцами
беременности. Подавляющее большинство пациенток имеет избыток массы тела
и отягощенный по СД-2 анамнез. Факторы риска развития ГСД, а также
группы женщин с низким риском развития ГСД приведены в табл. 7.24.
ГСД
обычно развивается в середине 2 триместр, между 4 и 8 месяцами
беременности. Подавляющее большинство пациенток имеет избыток массы тела
и отягощенный по СД-2 анамнез. Факторы риска развития ГСД, а также
группы женщин с низким риском развития ГСД приведены в табл. 7.24.
Табл. 7.24. Факторы риска развития гестационного сахарного диабета
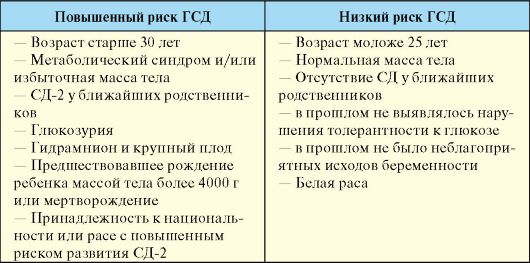 Гипергликемия
матери приводит к гипергликемии в системе кровообращения ребенка.
Глюкоза легко проникает через плаценту и непрерывно переходит к плоду из
крови матери. Также происходят активный транспорт аминокислот и перенос
кетоновых тел плоду. В отличие от этого инсулин, глюкагон и свободные
жирные кислоты матери в кровь плода не попадают. В первые 9-12 недель
беременности ПЖЖ плода еще не вырабатывает собственный инсулин. Это
время соответствует той фазе органогенеза плода, когда при постоянной
гипергликемии у матери могут формироваться различные пороки развития
(сердца, позвоночника, спинного мозга, ЖКТ). С 12-й недели беременности
ПЖЖ плода начинает синтезировать инсулин, и в ответ на гипергликемию
развиваются реактивная гипертрофия и гиперплазия β-клеток фетальной ПЖЖ.
Вследствие гиперинсулинемии развиваются макросомия плода, а также
угнетение синтеза лецитина, что объясняет высокую частоту развития
респираторного дистресссиндрома у новорожденных. В результате
гиперплазии β-клеток и гиперинсулинемии появляется склонность к тяжелым и
длительным гипогликемиям.
Гипергликемия
матери приводит к гипергликемии в системе кровообращения ребенка.
Глюкоза легко проникает через плаценту и непрерывно переходит к плоду из
крови матери. Также происходят активный транспорт аминокислот и перенос
кетоновых тел плоду. В отличие от этого инсулин, глюкагон и свободные
жирные кислоты матери в кровь плода не попадают. В первые 9-12 недель
беременности ПЖЖ плода еще не вырабатывает собственный инсулин. Это
время соответствует той фазе органогенеза плода, когда при постоянной
гипергликемии у матери могут формироваться различные пороки развития
(сердца, позвоночника, спинного мозга, ЖКТ). С 12-й недели беременности
ПЖЖ плода начинает синтезировать инсулин, и в ответ на гипергликемию
развиваются реактивная гипертрофия и гиперплазия β-клеток фетальной ПЖЖ.
Вследствие гиперинсулинемии развиваются макросомия плода, а также
угнетение синтеза лецитина, что объясняет высокую частоту развития
респираторного дистресссиндрома у новорожденных. В результате
гиперплазии β-клеток и гиперинсулинемии появляется склонность к тяжелым и
длительным гипогликемиям.
Эпидемиология
СД страдают 0,3 % всех женщин репродуктивного возраста, 0,2-0,3 % беременных уже исходно больны СД, а в 1-14 % беременностей развивается ГСД или манифестирует истинный СД. Распространенность ГСД варьирует в разных популяциях, так, в США он выявляется примерно у 4 % беременных (135 тыс. случаев в год).
Клинические проявления
При ГСД отсутствуют. Могут быть неспецифические симптомы декомпенсации СД.
Диагностика
Определение уровня глюкозы крови натощак показано всем беременным женщинам в рамках биохимического анализа крови. Женщинам, которые относятся к группе риска (табл. 7.24), показано проведение орального глюкозотолерантного теста (ОГТТ). Описано много вариантов его проведения у беременных. Наиболее простой из них подразумевает следующие правила:
• 3 дня до обследования женщина находится на обычном питании и придерживается обычной для себя физической активности;
• тест проводится утром натощак, после ночного голодания не менее 8 часов;
• после взятия пробы крови натощак женщина в течение 5 минут выпивает раствор, состоящий из 75 грамм сухой глюкозы, растворенной в 250-300 мл воды; повторное определение уровня гликемии проводится через 2 часа.
• диагноз ГСД устанавливается по следующим критериям:
- глюкоза цельной крови (венозной, капиллярной) натощак > 6,1 ммоль/л или
- глюкоза плазмы венозной крови ≥ 7 ммоль/л или
- глюкоза цельной капиллярной крови или плазмы венозной крови через 2 часа после нагрузки 75 г глюкозы ≥ 7,8 ммоль/л.
• если у женщины, которая относится к группе риска, результаты исследования соответствуют норме, тест проводится повторно на 24-28 неделе беременности.
Дифференциальная диагностика
ГСД и истинный СД; глюкозурия беременных.
Лечение
Риск для матери и плода, а также подходы к лечению СД и особенности контроля за ним при ГСД и при истинном СД одинаковы. Поздние осложнения СД во время беременности могут значительно прогрессировать, однако при качественной компенсации СД показаний для прерывания беременности нет. Женщина, страдающая СД (как правило, речь идет о СД-1), должна планировать беременность в молодом возрасте, когда риск развития осложнений наиболее низок. Если планируется беременность, то рекомендуется отменять конт-
рацепцию спустя несколько месяцев после достижения оптимальной компенсации. Противопоказаниями к планированию беременности являются тяжелая нефропатия с прогрессирующей почечной недостаточностью, тяжелая ИБС, тяжелая пролиферативная ретинопатия, не поддающаяся коррекции, кетоацидоз на ранних сроках беременности (кетоновые тела являются тератогенными факторами).
Целью лечения ГСД и истинного СД во время беременности является достижение следующих лабораторных показателей:
• гликемия натощак < 5-5,8 ммоль/л;
• гликемия через 1 ч после еды < 7,8 ммоль/л;
• гликемия через 2 ч после еды < 6,7 ммоль/л;
• среднее значение дневного гликемического профиля < 5,5 ммоль/л;
• уровень HbA1c при ежемесячном контроле, как у здоровых (4-6 %).
При СД-1, как и вне беременности, женщина должна получать интенсивную инсулинотерапию, однако уровень гликемии во время беременности рекомендуется оценивать 7-8 раз в сутки. При невозможности достижения нормогликемической компенсации на фоне обычных инъекций, необходимо рассмотреть вопрос о переводе пациентки на инсулинотерапию при помощи дозатора инсулина.
На первым этапе лечения ГСД назначается диетотерапия, заключающаяся в ограничении суточного калоража примерно до 25 кКал/кг фактического веса, в первую очередь за счет легкоусваиваемых углеводов и жиров животного происхождения, а также расширение физических нагрузок. Если на фоне диетотерапии не удается достичь поставленных целей лечения, пациентке необходимо назначить интенсивную инсулинотерапию. Любые таблетированные сахароснижающие препараты (ТСП) при беременности противопоказаны. На инсулинотерапию оказывается необходимым переводить около 15 % женщин.
Прогноз
При неудовлетворительной компенсации ГСД и СД во время беременности вероятность развития различной патологии у плода составляет 30 % (риск в 12 раз выше, чем в общей популяции). Более чем у 50 % женщин, у которых во время беременности выявлялся ГСД, в течение последующих 15 лет манифестирует СД-2.