Эндокринология : учебник. И.И. Дедов - 2009. - 432 с.: ил.
|
|
|
|
ГЛАВА 2 ГИПОТАЛАМО-ГИПОФИЗАРНЫЕ ЗАБОЛЕВАНИЯ
2.1. АНАТОМИЯ И ФИЗИОЛОГИЯ ГИПОТАЛАМО-ГИПОФИЗАРНОЙ СИСТЕМЫ
Гипофиз - железа внутренней секреции, расположенная в гипофизарной ямке турецкого седла клиновидной кости (рис. 2.1). Масса гипофиза составляет 0,5-0,7 г, размеры 1,3 * 0,6 * 1,0 см, но они могут меняться в зависимости от возраста и пола (у женщин он больше, чем у мужчин). В гипофизе различают две доли: переднюю (аденогипофиз) и заднюю (нейрогипофиз). Аденогипофиз состоит из трех
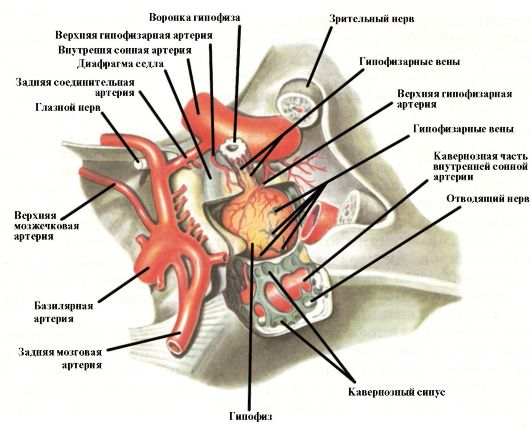 Рис. 2.1. Гипофиз (вид сверху)
Рис. 2.1. Гипофиз (вид сверху)
типов клеток: ацидофильных, базофильных, составляющих группу хромофилов, и хромофобов. Ацидофильные (эозинофильные) клетки вырабатывают гормон роста (ГР; соматотрофы) и пролактин (лактотрофы), базофильные клетки - тиреотропный гормон (ТТГ; тиреотрофы), адренокортикотропный гормон (АКТГ, кортикотрофы), а также гонадотропины (гонадотрофы): фолликулостимулирующий (ФСГ) и лютеинизирующий (ЛГ) гормоны. Хромофобные клетки рассматривают как источник, из которого дифференцируются хромофилы.
В нейрогипофизе оканчиваются волокна гипоталамо-гипофизарного тракта, идущие от супраоптического и паравентрикулярных ядер гипоталамуса. Аксоны нейросекреторных клеток заканчиваются аксовазальными синапсами, по которым поступают секретируемые в ядрах гипоталамуса вазопрессин (антидиуретический гормон) и окситоцин.
Аденогипофиз является ключевым регулятором эндокринной системы. Секретируемые им гормоны (ЛГ, ФСГ, ТТГ, АКТГ) регулируют функцию периферических эндокринных желез: щитовидной, коры надпочечников, гонад. Другие гормоны (ГР, пролактин) оказывают прямое действие на органы и ткани-мишени.
Гипоталамус расположен на основании мозга и ограничен спереди перекрестом зрительных нервов, сзади мамиллярными телами, по бокам - зрительными нервами. Сверху в гипоталамическую область внедряется III желудочек мозга. Масса гипоталамуса взрослого человека составляет около 4 г. Проводящие пути тесно связывают гипоталамус с соседними структурами головного мозга. Взаимосвязь гипофиза и гипоталамуса осуществляется через портальную систему. Портальная система гипофиза включает первичную капиллярную сеть, которая контактирует с терминалями аксонов аркуатного, вентромедиального и паравентрикулярного ядер гипоталамуса. Капилляры первичного сплетения собираются в портальные вены, идущие вдоль гипофизарной ножки в переднюю долю гипофиза, где они распадаются на вторичную капиллярную сеть. Синусоиды вторичной капиллярной сети собираются в выносящие вены, по которым кровь, обогащенная гормонами передней доли гипофиза, поступает в системный кровоток. Известные в настоящее время гормоны гипоталамуса подразделяют на гормоны, усиливающие (рилизинг-гормоны, либерины) и тормозящие (статины) выделение соответствующих тропных гормонов, при этом их роль не сводится к схеме один либерин (статин) - один гормон гипофиза. Так, тиролиберин может сти-
мулировать продукцию ТТГ и пролактина; гонадолиберин является общим рилизинг-гормоном для ЛГ и ФСГ; соматостатин подавляет секрецию ГР и АКТГ.
Пролактин - белковый гормон, основной физиологической функцией которого является обеспечение лактации. Стимулирующее влияние на секрецию пролактина оказывает процесс кормления грудью. Основным ингибитором секреции пролактина является дофамин, синтезирующийся в гипоталамусе.
Гормон роста (ГР, соматотропин) - полипептидный гормон эффекты которого на органы и ткани реализуются инсулиноподобным ростовым фактором-1 (ИРФ-1), синтезирующимися в печени под влиянием ГР. Основным эффектом ГР у детей и подростков является стимуляция продольного роста костей (преимущественно длинных трубчатых и в меньшей степени губчатых). Кроме того, ГР стимулирует синтез белка и задержку азота, оказывает липолитическое и антинатрийуретическое действие. Введение физиологических доз ГР дает кратковременный инсулиноподобный (снижение гликемии), а затем контринсулярный эффект. Синтез и секреция ГР контролируются двумя гипоталамическими нейропептидами - рилизинг-гормоном ГР (соматолиберин, ГР-РГ) и соматостатином. В течение дня уровень ГР в плазме сохраняется низким; пик содержания ГР отмечается после приема пищи, и его уровень прогрессивно увеличивается во время сна. У растущих детей интегральная суточная продукция ГР существенно выше, чем у взрослых.
Лютеинизирующий гормон (ЛГ) в яичниках стимулирует овуляцию и синтез андрогенов клетками теки, а в яичках является регулятором продукции тестостерона клетками Лейдига.
Фолликулостимулирующий гормоны (ФСГ) в яичниках стимулирует рост клеток гранулезы и секрецию эстрогенов; в яичках - вместе с тестостероном стимулирует сперматогенез (см. п. 5.1. и 6.1).
Адренокортикотропный гормон (АКТГ, кортикотропин) является стимулятором продукции кортизола и андрогенов в коре надпочечников (см. п. 4.1).
Основной функцией тиреотропного гормона (ТТГ) является стимуляция синтеза и секреции гормонов щитовидной железы, а также трофическое воздействие на тиреоциты (см. п. 3.1).
Самостоятельной и во многом автономной системой является нейрогипофиз, состоящий, как указывалось, из аксонов супраоптического и паравентрикулярного ядер гипоталамуса.
Вазопрессин (аргинин-вазопрессин, антидиуретический гормон, АДГ) является белком, состоящим из 9 аминокислот. Рецепторы АДГ находятся в дистальных извитых канальцах нефрона; их активация приводит к усилению реабсорбции воды. В физиологических условиях секреция АДГ регулируется осморецепторами гипоталамуса: гиперосмолярность плазмы приводит к стимуляции секреции АДГ. Другими непрямыми стимуляторами секреции АДГ являются гиповолемия и артериальная гипотензия.
Окситоцин так же, как и вазопрессин, состоит из 9 аминокислот, но отличается от него двумя аминокислотными остатками. Окситоцин, воздействуя на мускулатуру матки, увеличивает силу ее сокращений, обеспечивая таким образом родовую деятельность и послеродовое сокращение матки. Стимулируя сокращение миоэпителиальных клеток альвеол молочных желез, окситоцин способствует поступлению молока в млечные протоки. Физиологическими стимуляторами секреции окситоцина являются растяжение половых путей женщины и кормление грудью.
2.2. МЕТОДЫ ОБСЛЕДОВАНИЯ ПАЦИЕНТОВ С ГИПОТАЛАМО-ГИПОФИЗАРНОЙ ПАТОЛОГИЕЙ
2.2.1. Физикальные методы
Клиническая картина гипоталамо-гипофизарной патологии отличается значительным разнообразием, поэтому какие-либо специфические физикальные методы для обследования пациентов с патологией гипофиза отсутствуют. Тем не менее, вопреки бытующим представлениям, именно данные клинической картины имеют наибольший удельный вес для постановки диагноза.
2.2.2. Лабораторные методы
Для диагностики нарушения продукции гипофизарных гормонов чаще всего используется определение базального уровня гормонов, реже различные функциональные пробы (табл. 2.1). Исследование уровня гипоталамических гормонов клинического значения в настоящее время не имеет.
Табл. 2.1. Лабораторная диагностика гипоталамо-гипофизарных заболеваний
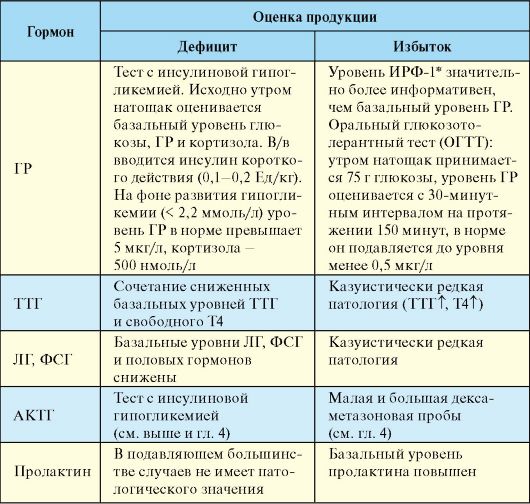 *ИРФ-1 - инсулиноподобный ростовой фактор.
*ИРФ-1 - инсулиноподобный ростовой фактор.
2.2.3. Инструментальные методы
К инструментальным методам, которые используются для визуализации гипоталамо-гипофизарной области, относятся рентгенокраниография, компьютерная томография (КТ) и магнитно-резонансная томография (МРТ). Среди дополнительных методов следует указать на определение полей зрения (периметрия), которое показано пациентам с макроаденомами гипофиза, а также после перенесенных нейрохирургических вмешательств, которые могут осложниться спаечным процессом с нарушением зрительной функции.
Рентгенодиагностика интраселлярных опухолей гипофиза основывается на определении размеров турецкого седла. В норме размеры турецкого седла составляют: сагиттальный - 12-15 мм, вертикальный - 8-9 мм (рис. 2.2).
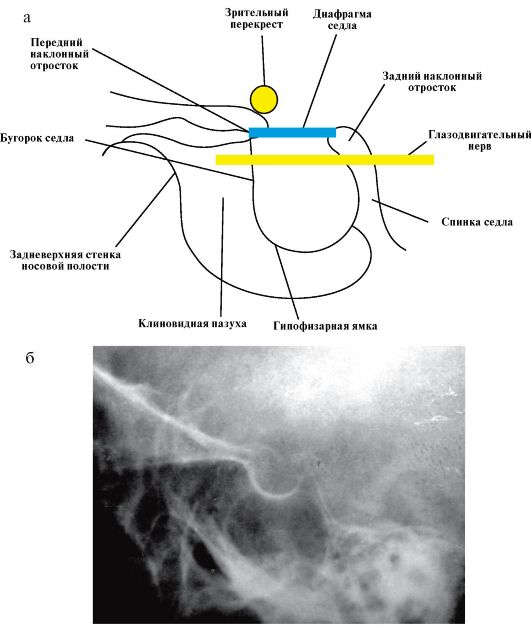 Рис. 2.2. Рентгенокраниография:
Рис. 2.2. Рентгенокраниография:
а - рентгенографические ориентиры турецкого седла; б - рентгенограмма турецкого седла в норме
Большие аденомы гипофиза вызывают значительное увеличение размеров, расширение входа в турецкое седло, истончение и укорочение клиновидных отростков, углубление дна, выпрямление, разрушение спинки седла, двухконтурность (нечеткое очертание одного или двух контуров) седла, как правило, не представляют диагностических трудностей (рис. 2.3). При микроаденомах гипофиза, которые не выходят за пределы турецкого седла, какие-либо изменения на рентгенограммах чаще всего отсутствуют.
Более информативным и относительно недорогим методом визуализации гипофиза является КТ. Тем не менее факторами, существенно ограничивающими его использование, являются плоскостной характер изображение, помехи от костных структур, невозможность дифференцировать небольшие патологические образования, рентгенологическая плотность которых близка к плотности цереброспинальной жидкости или нормальной мозговой ткани.
Методом выбора визуализации гипофиза и гипоталамуса, а также методом выбора топической диагностики новообразований гипоталамо-гипофизарной области является МРТ (рис. 2.4). Она позволяет различить малейшие изменения структуры гипофиза, наличие кистозного компонента опухоли, кисты, кровоизлияния и проч. Костная ткань и различные обызвествления на МРТ не дифференцируются.
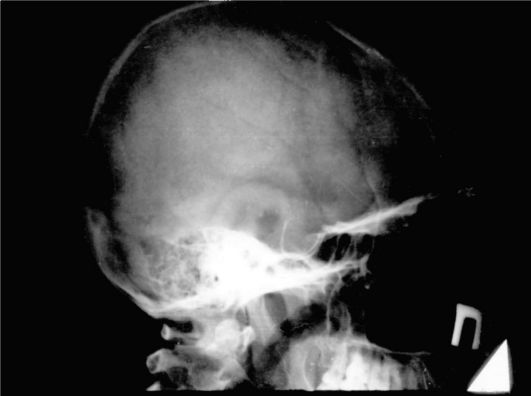 Рис. 2.3. Рентгенограмма черепа при макроаденоме гипофиза
Рис. 2.3. Рентгенограмма черепа при макроаденоме гипофиза
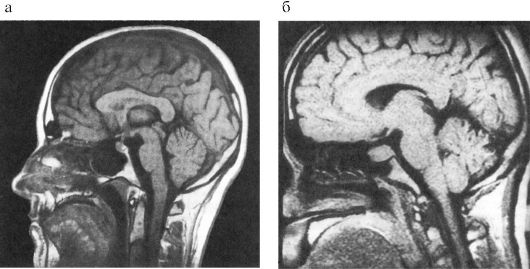 Рис. 2.4. Магнитно-резонансная томография гипоталамо-гипофизарной области:
Рис. 2.4. Магнитно-резонансная томография гипоталамо-гипофизарной области:
а - норма (сагиттальная проекция);
б - макроаденома гипофиза (сагиттальная проекция)
Использование контрастных веществ значительно увеличивает диагностические возможности МРТ. Важным преимуществом МРТ является отсутствие лучевой нагрузки и, таким образом, возможность многократного динамического обследования пациента.
2.3. ГОРМОНАЛЬНО-НЕАКТИВНЫЕ ОБЪЕМНЫЕ ОБРАЗОВАНИЯ И ИНФИЛЬТРАТИВНЫЕ ПРОЦЕССЫ ГИПОТАЛАМО-ГИПОФИЗАРНОЙ ОБЛАСТИ
К этой группе патологических изменений гипоталамо-гипофизарной области относятся гормонально-неактивные аденомы, краниофарингиома, редко встречающиеся инфильтративные заболевания, такие как гистиоцитоз Х, саркоидоз, а также такие объемные образования, как кисты, гемангиомы, ганглионевриномы, гамартомы и др.
Гормонально-неактивные аденомы гипофиза (ГНАГ) представляют собой аденомы гипофиза, протекающие без клинических проявлений гиперсекреции гипофизарных гормонов. Объединение всех этих отличающихся по этиологии процессов продиктовано тем, что они вызывают сходные клинические проявления и синдромы (табл. 2.2).
Этиология
В настоящее время развитие как ГНАГ, так и других аденом гипофиза связывается с моноклональными соматическими мутациями. В качестве факторов инициации клеточной трансформации предполагается влияние гормонов гипоталамуса и нейротрансмиттеров. Многие ГНАГ, которые клинически себя не проявляют гиперпродукцией какого-либо гормона, на самом деле могут продуцировать гликопротеидные гормоны (гонадотропины, α-субъединицу гликопротеидных гормонов), которые выявляются при иммуногистохимическом исследовании удаленной опухоли. Характер роста ГНАГ варьирует от весьма медленного, замершего на стадии микроаденомы, вплоть до бурного, с быстрым прогрессированием гипофизарной недостаточности и неврологической симптоматикой.
Табл. 2.2. Гормонально-неактивные объемные образования и инфильтративные процессы гипоталамо-гипофизарной области
 Окончание табл. 2.2
Окончание табл. 2.2
 Краниофарингиома -
гипоталамическая опухоль, происходящая из остатков кармана Ратке
(эпителиальное выпячивание задней стенки глотки зародыша, являющееся
зачатком аденогипофиза). Развитие опухоли связано с нарушением
эмбриональной дифференцировки клеток кармана Ратке. Опухоль может
локализоваться в гипоталамусе, III желудочке, турецком седле и чаще
имеет кистозное строение. Краниофарингиомы гормонально неактивны, в
основе клинических проявлений опухоли лежит механическое сдавление
окружающих структур головного мозга.
Краниофарингиома -
гипоталамическая опухоль, происходящая из остатков кармана Ратке
(эпителиальное выпячивание задней стенки глотки зародыша, являющееся
зачатком аденогипофиза). Развитие опухоли связано с нарушением
эмбриональной дифференцировки клеток кармана Ратке. Опухоль может
локализоваться в гипоталамусе, III желудочке, турецком седле и чаще
имеет кистозное строение. Краниофарингиомы гормонально неактивны, в
основе клинических проявлений опухоли лежит механическое сдавление
окружающих структур головного мозга.
Среди опухолей гипоталамической области, помимо краниофарингиомы, встречаются глиомы, гемангиомы, дисгерминомы, гамартомы, ганглионевриномы, эпендимомы, медуллобластомы, липомы, нейробластомы, лимфомы, плазмоцитомы, коллоидные и дермоидные кисты, саркомы. Вовлечение в патологический процесс гипоталамуса возможно при диссеминированном специфическом или неспецифическом инфекционном процессе, а также при диссеминации системных заболеваний.
Патогенез
Патогенез ГНАГ и инфильтративной патологии гипоталамо-гипофизарной области определяется скоростью и распространенностью
деструктивного процесса, а также возрастом, в котором развивается то или иное заболевание. При этом в результате роста опухоли и деструкции гипоталамо-гипофизарных структур могут развиваться несколько типичных синдромов, выраженность и набор которых у отдельных пациентов значительно варьирует.
♦ Аденогипофизарная недостаточность (см. п. 2.6.), выраженность которой может варьировать, начиная от выпадения какой-то одной функции (дефицит гормона роста или гонадотропинов) вплоть до пангипопитуитаризма.
♦ Несахарный диабет (см. п. 2.7.). При низком повреждении ножки гипофиза сохраняется секреция вазопрессина аксонами срединного возвышения, и несахарный диабет не развивается. При деструктивном процессе в области гипоталамуса или высоком повреждении ножки гипофиза продукция вазопрессина снижается.
♦ Гиперпролактинемия (см. п. 2.4.). Наиболее частым гормональным феноменом при ГНАГ является различной выраженности гиперпролактинемия. Причиной гиперпролактинемии при сдавлении ножки гипофиза опухолью или при инфильтративном процессе является прекращение поступления дофамина, который подавляет продукцию пролактина. Наряду с гиперпролактинемией полное сдавление или деструкция ножки гипофиза сопровождается дефицитом всех остальных гормонов аденогипофиза (ЛГ, ФСГ, ГР, ТТГ, АКТГ). Указанный феномен известен под названием синдром изолированного гипофиза.
♦ Хиазмальный синдром (битемпоральная гемианопсия), обусловленный сдавлением крупной аденомой гипофиза перекреста зрительных нервов (рис. 2.5).
Эпидемиология
На долю ГНАГ приходится 25 % всех аденом гипофиза (самая частая аденома гипофиза); среди опухолей с супраселлярной локализацией 70 % всех аденом являются ГНАГ. По аутопсийным данным распространенность микроинсиденталом гипофиза достигает 10-25 %. Частота новых случаев клинически значимых ГНАГ ориентировочно составляет 6 случаев на 1 млн. населения в год.
Краниофарингиома является редким заболеванием, но самой частой супраселлярной опухолью у детей (5-10 % опухолей головного мозга у детей).
Гемангиомы с поражением гипоталамуса выявляются во время перинатального периода и до 2 лет.
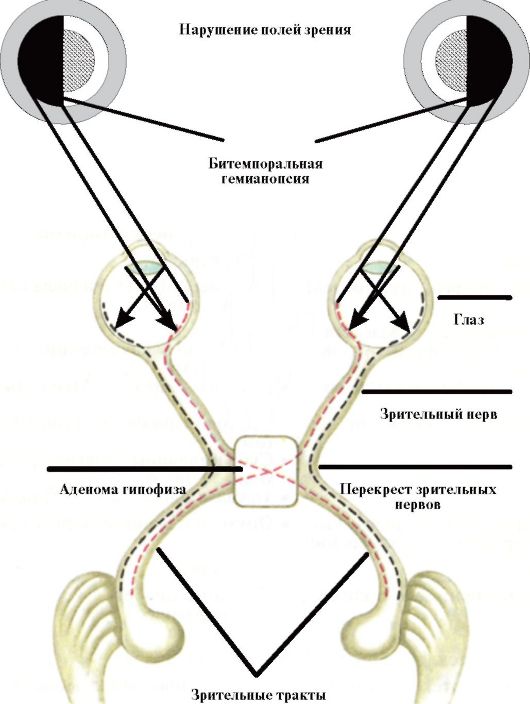 Рис. 2.5. Формирование хиазмального синдрома (битемпоральная гемианопсия) при экстраселлярном росте аденомы гипофиза
Рис. 2.5. Формирование хиазмального синдрома (битемпоральная гемианопсия) при экстраселлярном росте аденомы гипофиза
Дисгерминома и гамартрома обычно диагностируются в возрасте от 2 до 25 лет. В возрасте от 10 до 25 лет могут появиться дермоидные опухоли, липомы, нейробластомы.
Время манифестации глиомы достаточно обширно - от периода новорожденности до 50 лет.
Клинические проявления
Клиническая симптоматика определяется набором и выраженностью упомянутых выше синдромов.
♦ Аденогипофизарная недостаточность. Наиболее частым и ранним симптомом у женщин являются нарушения менструального цикла вплоть до аменореи. Это заставляет женщин обращаться к гинекологу, что позволяет устанавливать диагноз на относительно более ранних стадиях опухолевого процесса, чем у мужчин, которые не склонны обращаться за медицинской помощью при появлении эректильной дисфункции. Реже на первый план выступают другие проявления гипофизарной недостаточности: прогрессирующая общая слабость и гипотония как проявления вторичного гипокортицизма и гипотиреоза. В ряде случаев развивается развернутая клиническая картина пангипопитуитаризма (см. п. 2.6). При развитии ГНАГ, краниофарингиомы или другого деструктивного процесса гипоталамо-гипофизарной области в детском возрасте, как правило, происходит задержка полового и физического развития (задержка роста, отсутствие пубертата).
♦ Несахарный диабет. Клинически проявляется выраженной полиурией (без глюкозурии) и полидипсией (см. п. 2.7). Нарушение водного обмена может иметь трехфазный характер: вначале остро развивается полиурия, затем примерно на 7-10 день у молодых пациентов следует фаза нормального водного обмена и, наконец, стойкое развитие несахарного диабета. Эта триада объясняется тем, что вначале остро утрачивается поступление АДГ в заднюю долю гипофиза, затем происходит ее аутолиз с выделением гормонов в кровоток и, наконец, наступает полное прекращение поступления в кровь АДГ. Степень полиурии тем ниже, чем ниже уровень кортизола. По мере снижения продукции АКТГ (в рамках прогрессирующего гипопитуитаризма) уменьшается и выраженность полиурии (синдром Ханна). Это связано с антагонистическим взаимодействием кортизола и АДГ.
♦ Гиперпролактинемия. Как правило, протекает бессимптомно, но ряд проявлений (аменорея) могут быть обусловлены как повышением продукции пролактина, так и гипогонадотропным гипогонадизмом.
♦ Неврологическая симптоматика: хиазмальный синдром (при ГНАГ в 75 % случаев - битемпоральная гемианопсия, в 15 % случаев - квадрианопсия), паралич черепно-мозговых нервов, головная боль, тошнота, рвота.
Диагностика
У пациентов с соответствующей симптоматикой проводится МРТ гипофиза. Обычно к моменту установления диагноза ГАНГ имеют значительные размеры и выраженные нейроофтальмологические
проявления. При кранофарингиоме в 80 % случаев при рентгенографии в опухоли выявляются кальцинаты. При гормональном исследовании выявляется той или иной выраженности дефицит тропных гормонов гипофиза (см. табл. 2.1), гиперпролактинемия (как правило, легкая или умеренная).
Дифференциальная диагностика
При гиперпролактинемии необходима дифференциальная диагностика ГНАГ с пролактиномой, что имеет принципиальное клиническое значение, поскольку в последнем случае пациенту показано лишь консервативное лечение дофаминомиметиками. Быстрый рост опухоли и небольшое (до 200 мкг/л) увеличение уровня пролактина более характерны для гормонально-неактивной аденомы. В качестве маркера ГНАГ предлагается исследование уровней хромогранинов и секретогранина, а также β-ХГЧ.
Краниофарингиому необходимо дифференцировать от других заболеваний, протекающих с задержкой полового и физического развития и гипопитуитаризмом, а также от других опухолей гипофиза и головного мозга. Опухолевые процессы гипоталамо-гипофизарной области нередко приходится дифференцировать от системных и генетических поражений.
Лечение
Хирургическое лечение (удаление опухоли) при ГНАГ показано при макроаденомах супраселлярной локализации, а также при наличии неврологической симптоматики (хиазмальный синдром) и гипофизарной недостаточности, при этом в большинстве случаев возможен транссфеноидальный доступ. Кроме того, хирургическое лечение показано в большинстве случаев краниофарингиом. Большинство ГНАГ и краниофарингиом радиорезистентны. При парциальном и тотальном гипопитуитаризме показана заместительная гормонотерапия (см. п. 2.6). При случайно выявленных ГНАГ (инциденталомах) и отсутствии данных за гормональную активность показано динамическое наблюдение: при образованиях менее 1 см контрольная МРТ проводится в сроки через 1,2 и 5 лет, при образованиях более 1 см - через 6 месяцев, а далее через 1, 2 и 5 лет.
Прогноз
После операций по поводу крупных ГНАГ и краниофарингиом зрительная функция восстанавливается в большинстве случаев. Полное восстановление функции аденогипофиза после операций по поводу
гормонально неактивных макроаденом и крупных краниофарингиом происходит только в 10 % случаев, в связи с чем большая часть пациентов после операции продолжает получать заместительную терапию в том или ином объеме. Для краниофарингиомы характерны рецидивы после удаления первичной опухоли. Большинство случайно выявленных гормонально неактивных микроаденом (инциденталом) характеризуется постоянством размеров или очень медленным ростом и весьма редко приобретает клиническое значение с развитием гипофизарной недостаточности, гиперпролактинемии или неврологической симптоматики.
2.4. ГИПЕРПРОЛАКТИНЕМИЧЕСКИЙ ГИПОГОНАДИЗМ
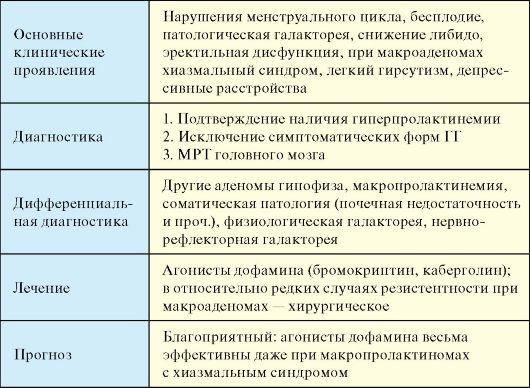
Гиперпролактинемический гипогонадизм (ГГ) - клинический синдром, обусловленный избытком пролактина, включающий той или иной степени гипогонадизм и патологическое отделяемое из молочных желез (необязательный признак) (табл. 2.3).
Табл. 2.3. Гиперпролактинемический гипогонадизм (ГГ)
 Окончание табл. 2.3
Окончание табл. 2.3
 Понятия гиперпролактииемия и
ГГ не являются синонимами. Гиперпролактинемия (повышение сывороточного
уровня пролактина) подразделяется на физиологическую (при беременности,
лактации, у новорожденных), бессимптомную (биохимическую) и
патологическую.
Понятия гиперпролактииемия и
ГГ не являются синонимами. Гиперпролактинемия (повышение сывороточного
уровня пролактина) подразделяется на физиологическую (при беременности,
лактации, у новорожденных), бессимптомную (биохимическую) и
патологическую.
Этиология
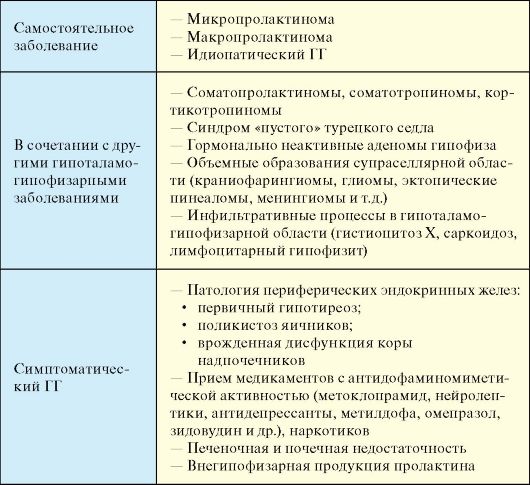
Этиологическая классификация ГГ представлена в табл. 2.4. Он может быть самостоятельным заболеванием, а также результатом другой гипоталамо-гипофизарной патологией или носить симптоматический характер.
Ранее существовала концепция, рассматривающая идиопатические формы ГГ (аденома отсутствует), микропролактиномы и макропролактиномы как стадии единого процесса, при котором снижение или отсутствие ингибирующего влияния гипоталамуса на секрецию пролактина приводит сначала к гиперплазии пролактотрофов, а затем к формированию микро- и макроаденом гипофиза.
Табл. 2.4. Этиология гиперпролактинемического гипогонадизма
 В
настоящее время доминирует гипотеза первично гипофизарного поражения
(аденомы), которая возникает вследствие соматической мутации; конкретная
мутация до настоящего времени не идентифицирована. Этиология
идиопатического ГГ, при котором отсутствуют аденома гипофиза и другие
видимые причины повышения уровня пролактина, неизвестна.
В
настоящее время доминирует гипотеза первично гипофизарного поражения
(аденомы), которая возникает вследствие соматической мутации; конкретная
мутация до настоящего времени не идентифицирована. Этиология
идиопатического ГГ, при котором отсутствуют аденома гипофиза и другие
видимые причины повышения уровня пролактина, неизвестна.
Причиной развития гиперпролактинемии и ГГ в сочетании с другими гипоталамо-гипофизарными заболеваниями, как правило, является нарушение анатомических взаимоотношений гипофиза и гипоталамуса, в результате которого происходит сдавление ножки гипофиза, приводящее к нарушению поступления в него дофамина, ингибирующего продукцию пролактина. Любые факторы, приводящие к сни-
жению продукции дофамина, начиная от приема ряда медикаментов, заканчивая многими соматическими и эндокринными заболеваниями могут обусловить развитие симптоматического ГГ и гиперпролактинемии.
Пролактин может циркулировать в различных молекулярных формах, при этом пролактин с большей, чем в норме (23 кДа) молекулярной массой (макропролактин) не обладает биологической активностью и представляет собой либо комплекс пролактин-антитело к пролактину, либо димеры и тетрамеры пролактина.
Макропролактинемия не сопровождается явной симптоматикой, не требует лечения и обычно выявляется случайно (бессимптомная, биохимическая гиперпролактинемия).
Патогенез
В основе патогенеза ГГ лежит гиперсекреция пролактина, который блокирует циклическое выделение гонадолиберина, что приводит к снижению цикличности выделения ЛГ, ФСГ, гиполютеиновой дисфункции яичников, ановуляции, гипоэстрогении.
Ряд симптомов обусловлен экстрагенитальным действием пролактина: увеличение конверсии углеводов в жиры способствует ожирению; стимуляция продукции дегидроэпиандростерона надпочечниками ведет к развитию «мягкой» гиперандрогении, и, наконец, гиперпролактинемия способствует формированию синдрома остеопении, влияя на обмен витамина D, что усугубляется дефицитом эстрогенов. Пролактин вызывает лактацию и понижает либидо. Развивающийся на фоне гиперпролактинемии дефицит эстрогенов вызывает диспареунию и способствует ожирению.
Эпидемиология
Пролактинома является наиболее частой функционирующей аденомой гипофиза. Аутопсийные исследования обнаружили микропролактиномы у 10 % умерших. Микропролактиномы встречаются в несколько раз чаще, чем макропролактиномы, при этом в большей степени у женщин. Макропролактиномы встречаются с одинаковой частотой у лиц обоего пола. Суммарно патологическая и биохимическая гиперпролактинемия встречается в 1 случае на 500 человек взрослого населения. Гиперпролактинемия обнаруживается примерно у 8 % женщин с олигоменореей. Средний возраст женщин при дебюте заболевания - 25-30 лет, мужчин - 45-50 лет.
Клинические проявления
Клиническая картина значительно варьирует от бессимптомного течения даже в случае значительно повышения уровня пролактина, до развернутой симптоматики (аменорея, галакторея, бесплодие). Наиболее частыми симптомами ГГ являются:
1. Нарушения менструального цикла варьируют от опсоолигоменореи до аменореи, чаще всего вторичной.
2. Бесплодие (первичное и вторичное) является одной из основных жалоб при ГГ, а устранение бесплодия для многих женщин является основной целью лечения. При гинекологическом осмотре могут выявляться гипоплазия матки, отсутствие симптома «зрачка», симптома «натяжения» слизи. У заболевших в допубертатном периоде могут отмечаться гипоплазия клитора, малых половых губ.
3. Патологическая галакторея редко бывает первым симптомом ГГ (не более чем в 20 % случаев). Ее выраженность варьирует от обильной и спонтанной до единичных капель при сильном надавливании. Фиброзно-кистозная мастопатия и рак молочной железы у пациенток с ГГ встречаются не чаще, чем в среднем в популяции. Типична жировая инволюция молочной железы, не соответствующая возрасту.
4. Снижение либидо, аноргазмия, фригидность, сухость во влагалище имеют место у большинства пациенток, но эти жалобы редко предъявляются активно.
5. Эректильная дисфункция является основной жалобой у мужчин; может выявляться олигоспермия; гинекомастия и галакторея встречаются крайне редко.
6. Неврологическая симптоматика при макроаденомах (хиазмальный синдром, головные боли, паралич черепно-мозговых нервов).
7. Прочие возможные симптомы: умеренное ожирение (около 80 % пациенток), избыточный рост волос на лице, вокруг сосков и по белой линии живота (25 % пациенток), депрессивные расстройства.
Диагностика
1. Подтверждение наличия гиперпролактинемии при гормональном исследовании у пациента с соответствующей клинической симптоматикой. Однократное обнаружение в крови повышенного уровня пролактина еще не позволяет установить диагноз ГГ. Сам по себе уровень пролактина может косвенно свидетельствовать о генезе ГГ. Так, при уровне пролактина более 3000 мЕд/л, как правило, имеет место адено-
ма гипофиза. При идиопатическом и медикаментозном ГГ он существенно ниже.
2. Исключение симптоматических форм ГГ (определение функционального состояния щитовидной железы, исключение синдрома поликистозных яичников, печеночной и почечной недостаточности, нервно-рефлекторных и медикаментозных влияний).
3. МРТ головного мозга с целью визуализации аденомы или установления идиопатического характера ГГ.
Дифференциальная диагностика
♦ Гормонально неактивные аденомы гипофиза (см. п. 2.3) и синдром «пустого» турецкого седла (см. п. 2.8). При ГНАГ уровень пролактина, как правило, повышен лишь умеренно. Кроме того, такие аденомы не уменьшаются на фоне терапии дофаминомиметиками.
♦ Гормонально-активные опухоли гипофиза (соматотропинома, пролакто-соматотропинома).
♦ Симптоматическая гиперпролактинемия (первичный гипотиреоз, употребление наркотиков и т.д.).
♦ Макропролактинемию можно заподозрить по отсутствию специфической клинической картины и подтвердить с помощью определения макропролактина.
♦ Соматическая патология (почечная недостаточность и проч.).
♦ Физиологическая галакторея (может сохраняться до 2-3 лет после рождения ребенка и окончания грудного вскармливания).
♦ Нервно-рефлекторная галакторея и гиперпролактинемия (астеноневротическое расстройство с элементами канцерофобии, при котором пациентки постоянно проверяют наличие отделяемого из молочных желез и этой самопальпацией рефлекторно поддерживают галакторею).
Лечение
1. Медикаментозная терапия агонистами дофамина показана при микро- и макропролактиномах, а также при идиопатическом ГГ. Стимулируя гипофизарные дофаминергические рецепторы, они блокирут синтез и выделение пролактина, уменьшают частоту митозов в пролактотрофах, ингибируют рост пролактинсекретирующих аденом гипофиза. Нормализация секреции пролактина у большинства больных приводит к восстановлению циклической активности
гипоталамуса, повышению продукции гонадотропных гормонов, восстановлению двухфазного менструального цикла и фертильности. Кроме того, происходит значительное уменьшение размера истинных пролактином, в связи с этим медикаментозная терапия эффективна и при макропролактиномах даже с хиазмальным синдромом и другой неврологической симптоматикой. При лечении бесплодия дофаминомиметики (бромокриптин или каберголин) отменяют после наступления беременности:
- бромокриптин является первым препаратом из этой группы и его отличает неселективность воздействия на рецепторы дофамина и короткий период полувыведения. Его применяют, начиная с 1,25 мг 1-3 раза в день во время еды с дальнейшим увеличением дозы до 2,5 мг 2-4 раза в день. Однократная доза бромокриптина ингибирует секрецию пролактина в среднем на 12 ч. Побочные явления (тошнота, ортостатическая гипотензия, запор) обычно кратковременны и исчезают при уменьшении дозы.
- каберголин является селективным агонистом дофаминовых D2-рецепторов; вызывает значительно более длительное и эффективное подавление продукции пролактина, в связи с чем, может приниматься 1-2 раза в неделю в дозе 0,25-2 мг (при необходимости до 4,0 мг и более).
2. Хирургическое лечение показано при макропролактиномах, резистентных к дофаминомиметикам. Зачастую, такие аденомы в итоге, по данным иммуногистохимического исследования, оказываются не истинными пролактиномами, а ГНАГ или смешанными опухолями.
Прогноз
При истинных микро- и макропролактиномах терапия дофаминомиметиками эффективна в подавляющем большинстве случаев, как в плане уменьшения размеров опухоли, так и в плане нормализации уровня пролактина и фертильности. Длительные (более 5 лет) ремиссии после прекращения лечения наблюдаются у 5-10 % больных. После наступления менопаузы самопроизвольная ремиссия гипепролактинемии наступает у 1/3 пациенток. Нередко ремиссия развивается после родов.
2.5. АКРОМЕГАЛИЯ И ГИГАНТИЗМ
Акромегалия и гигантизм - тяжелые, хронические нейроэндокринные заболевания, возникающие вследствие избыточной продукции гормона роста (ГР) аденомой гипофиза (соматотропиномой). Эти два заболевания являются возрастными вариациями одного и того же патологического процесса, конкретные клинические проявления которого определяются степенью завершенности остеогенеза.
Этиология
При акромегалии аденомы гипофиза, секретирующие ГР, выявляются в 99 % случаев, при этом, как правило, речь идет о макроаденоме. Иммуногистохимически, помимо чистых соматотропных аденом (около 45 %), выделяют смешанные пролактосоматропиномы (около 30 %). Остальные 25 % аденом, кроме того, продуцируют другие аденогипофизарные гормоны: ТТГ, α-субъединицу,
ЛГ, ФСГ.
Табл. 2.5. Акромегалия
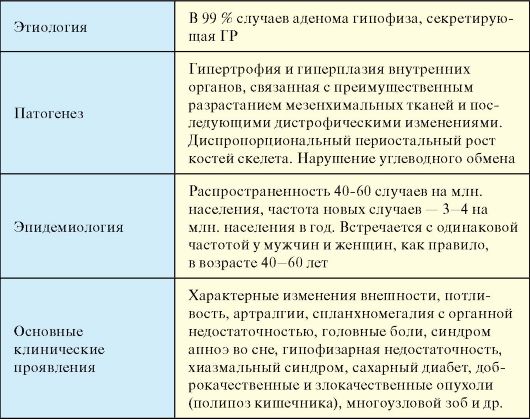 Окончание табл. 2.5
Окончание табл. 2.5
 По
своему происхождению соматотропиномы являются моноклональными
опухолями, развивающимися в результате соматической мутации
соматотрофов. В 40 % соматотропином может быть выявлена мутация
Gsp-белка, обеспечивающего димеризацию α- и β-субъединиц G-белков,
результатом которой является активация рецепторов соматолиберина
(рилизинг-гормон гормона роста, ГР-РГ). Такие опухоли чаще являются
микроаденомами. Соматотропинома может быть составной частью синдрома
множественных эндокринных неоплазий 1-го типа (МЭН-1) (см. п. 9.2.1).
По
своему происхождению соматотропиномы являются моноклональными
опухолями, развивающимися в результате соматической мутации
соматотрофов. В 40 % соматотропином может быть выявлена мутация
Gsp-белка, обеспечивающего димеризацию α- и β-субъединиц G-белков,
результатом которой является активация рецепторов соматолиберина
(рилизинг-гормон гормона роста, ГР-РГ). Такие опухоли чаще являются
микроаденомами. Соматотропинома может быть составной частью синдрома
множественных эндокринных неоплазий 1-го типа (МЭН-1) (см. п. 9.2.1).
Патогенез
Изменения в органах при акромегалии сводятся к их истинной гипертрофии и гиперплазии (спланхномегалии), что связано с преимущественным разрастанием мезенхимальных тканей. Разрастается паренхима и строма всех внутренних органов: легких, сердца, печени, поджелудочной железы, кишечника, селезенки. С прогрессированием заболевания в связи с пролиферацией соединительной ткани во всех органах происходят склеротические изменения, сопровождающиеся прогрессирующим развитием их недостаточности. Параллельно отмечается повышение риска возникновения доброкачественных и злокачественных новообразований во всех тканях и органах, включая эндокринные. У детей и подростков с продолжающимся ростом хроническая гиперпродукция гормона роста проявляется гигантизмом, характеризующимся чрезмерным, превышающим физиологические границы сравнительно пропорциональным эпифизарным и периостальным ростом костей, увеличением мягких тканей и органов. У взрослых, поскольку после окостенения эпифизарных хрящей дальнейший рост невозможен,
развивается акромегалия (от akros - крайний, megas - большой). При этой патологии также отмечается ускоренный рост тела, но не в длину, а в ширину за счет мягких тканей, что проявляется диспропорциональным периостальным ростом костей скелета, увеличением массы внутренних органов и характерным нарушением обмена веществ.
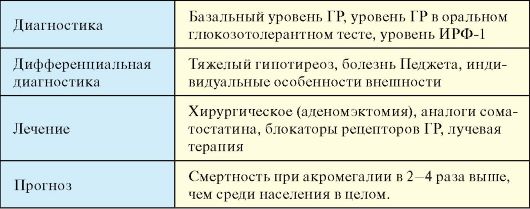
Эпидемиология
Распространенность акромегалии составляет около 40-60 случаев на миллион населения, частота новых случаев - 3-4 на миллион населения в год. Встречается с одинаковой частотой у мужчин и женщин, как правило, в возрасте 40-60 лет. Гигантизм является казуистически редкой патологией.
Клинические проявления
Акромегалия, как правило, характеризуется постепенным началом и торпидным течение с медленным нарастанием симптоматики и изменением внешности. Диагноз акромегалии в среднем устанавливается примерно через 7 лет после реального начала заболевания. Основными симптомами являются:
♦ Изменения внешности весьма характерны и в подавляющем большинстве случаев именно они позволяют заподозрить акромегалию. Характерно огрубение черт лица, связанное с увеличением надбровных дуг, скуловых костей, нижней челюсти. Отмечается гипертрофия мягких тканей лица: носа, губ, ушей. Увеличение нижней челюсти ведет к изменению прикуса (прогнатизм) за счет расхождения межзубных промежутков (диастема). Язык увеличен (макроглоссия), на нем часто видны отпечатки зубов. Изменение внешности развивается достаточно медленно, так что пациент сам его не замечает (рис. 2.6). Кроме того, происходит увеличение размеров кистей и стоп (пациенты часто указывают на увеличение размера обуви, порой значительное) (рис. 2.7). При гигантизме, в отличие от акромегалии, происходит увеличение линейного роста.
Выраженная гипертрофия хрящевой ткани суставов обусловливает артралгии. Увеличение количества и повышение функциональной активности потовых желез ведут к значительной потливости (при осмотре можно иногда увидеть ручейки пота, стекающие по телу больного). Активация и гипертрофия сальных желез, утолщение кожи приводят к ее характерному виду (плотная, утолщенная, с глубокими складками, более выраженными на волосистой части головы).
 Рис. 2.6. Динамика внешности пациента с акромегалией: а - 1972 г.; б - 1979 г.; в - 1991 г.
Рис. 2.6. Динамика внешности пациента с акромегалией: а - 1972 г.; б - 1979 г.; в - 1991 г.
♦ Спланхномегалия с последующим развитием органной недостаточности. Влияние ГР на мышцы и внутренние органы на начальных этапах заболевания малозаметно, а порой, особенно у спортсменов и лиц физического труда воспринимается позитивно, поскольку увеличиваются работоспособность и физическая активность, но по мере прогрессирования заболевания мышечные волокна дегенерируют, обусловливая нарастающую слабость, прогрессирующее снижение работоспособности. Некомпенсированная длительная гиперпродукция ГР ведет к развитию концентрической гипертрофии миокарда, которая сменяется гипертрофической миокардиодистрофией, а в запущенных случаях заболевания она переходит в дилатационную, что ведет к прогрессирующей сердечной недостаточности, являющаяся причиной гибели больных. У 30 % пациентов с акромегалией выявляется артериальная гипертензия.
♦ Головные боли, связанные с деструкцией турецкого седла, его диафрагмы и внутричерепной гипертензией.
♦ Синдром апноэ во сне развивается у 90 % больных с акромегалией. Это связано с разрастанием мягких тканей верхних дыхательных путей и поражением дыхательных центров.
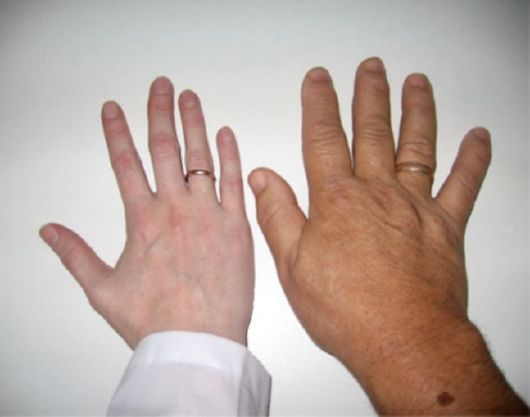 Рис. 2.7. Увеличение кисти, утолщение пальцев при акромегалии (рядом кисть здорового человека)
Рис. 2.7. Увеличение кисти, утолщение пальцев при акромегалии (рядом кисть здорового человека)
♦ Гипофизарная недостаточность связана с разрушением и сдавлением гипофиза опухолью. Репродуктивные расстройства (нарушения менструального цикла, эректильная дисфункция), помимо нарушения продукции гонадотропинов, часто связаны с гиперпролактинемией, которая в свою очередь может быть связана с сопутствующей гиперпродукцией пролактина опухолью (пролактосоматотропинома), либо со сдавлением ножки гипофиза.
♦ Хиазмальный синдром (см. п. 2.3).
♦ Симптоматический сахарный диабет (до 50 % пациентов).
♦ Развитие доброкачественных и злокачественных опухолей различной локализации вследствие хронической гиперподукции ростовых факторов (ИРФ-1 и др.). При акромегалии нередко выявляют узловой или диффузный зоб, аденоматозную гиперплазию надпочечников, фиброзно-кистозную мастопатию, миому матки, поликистоз яичников, полипоз кишечника. Полипы кишечника встречаются в 20-50 % случаев, кишечные аденокарциномы - в 7 % всех случаев акромегалии.
Диагностика
1. Повышение базального уровня ГР выявляют у большинства пациентов с развернутой клинической картиной акромегалии.
2. Оральный глюкозотолерантный тест подразумевает исследование уровня ГР исходно, а также в пробах крови через 30, 60, 90 и 120 минут после приема внутрь 75 г глюкозы. В норме при нагрузке глюкозой уровень ГР снижается. В активной фазе акромегалии уровень ГР не уменьшается ниже 2 нг/мл или выявляется парадоксальное повышение уровня ГР. Глюкозотолерантный тест показан в ситуации, когда у пациента с клиническими проявлениями акромегалии определяется лишь умеренное повышение базального уровня ГР, либо он в норме. Кроме того, тест используется для оценки эффективности лечения.
3. Весьма информативным исследованием является определение уровня ИРФ-1 (соматомедина С). У взрослых единственной причиной повышения уровня ИРФ-1 является акромегалия, а выявление нормального уровня ИРФ-1 практически исключает этот диагноз. ИРФ-1 в отличие от ГР имеет более длительный период полужизни в плазме и отражает уровень ГР на протяжении длительного времени.
4. МРТ гипофиза для визуализации аденомы.
5. Обследование на предмет возможных осложнений (полипоз кишечника, сахарный диабет, многоузловой зоб и др.).
Дифференциальная диагностика
Гигантизм дифференцируют от других форм высокорослости (конституционально высокий рост, синдром Клайнфелтера, первичный гипогонадизм различной этиологии). Акромегалию дифференцируют от тяжелого гипотиреоза, болезни Педжета, индивидуальных особенностей внешности.
Лечение
Целью лечения акромегалии являются ликвидация автономной гиперпродукции ГР, нормализация уровня ИРФ-1 в крови и отсутствие повышения плазменного уровня ГР в глюкозотолерантном тесте (75 г глюкозы) выше 1 нг/мл. Указанные критерии соответствуют ремиссии заболевания. Общий алгоритм лечения акромегалии представлен на рис. 2.8.
♦ Методом выбора при лечении больных с акромегалией является транссфеноидальное удаление аденомы гипофиза. При микроаденомах в 85 % случаев уровень ГР после операции возвращается к норме. В случае небольших инкапсулированных аденом оперативное лечение, как правило, приводит к стойкой ремиссии заболевания. При макроаденомах полное излечение после первой операции достигается в 30 % случаев. Наихудший прогноз имеют опухоли с экстраселлярным ростом.
♦ Аналоги соматостатина (октреотид, октреотид длительного действия, ланреотид) позволяют нормализовать уровни ГР и ИФР-1 у 50-70 % пациентов. Размеры аденомы гипофиза уменьшаются реже, только в 30-50 % случаев и обычно не намного.
♦ Блокаторы рецепторов ГР (пегвисомант) уменьшают синтез ИФР-1, конкурируя с эндогенным ГР за связывание с его рецептором. По предварительным данным при терапии пегвисомантом уровень ИРФ-1 снижается у 90 % пациентов. В силу отсутствия данных об отдаленных результатах лечения пока используется при неэффективности других методов.
♦ Лучевая терапия низко эффективна и может использоваться как вспомогательный метод лечения. Более перспективным методом является направленное облучение остаточной опухоли γ-частицами (гамма-нож).
Прогноз
Смертность при акромегалии в 2-4 раза выше, чем среди населения в целом, прежде всего из-за сердечно-сосудистых заболеваний. При
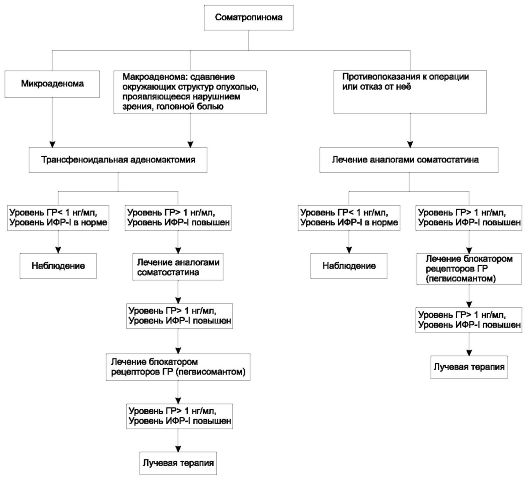 Рис. 2.8. Алгоритм лечения акромегалии
Рис. 2.8. Алгоритм лечения акромегалии
нормализации уровня ИФР-1 и устранении гиперсекреции СТГ смертность снижается до среднестатистического показателя.
2.6. ГИПОПИТУИТАРИЗМ
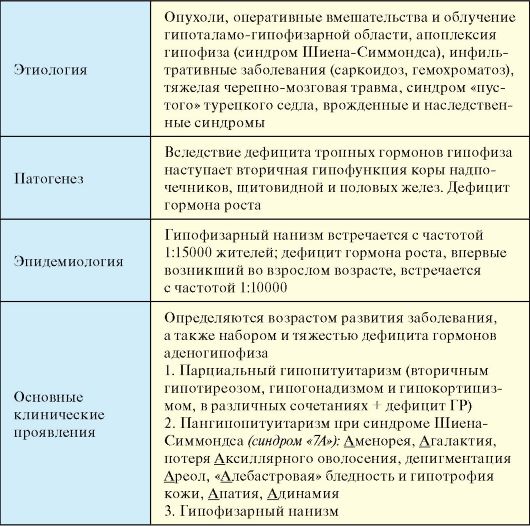
Гипоталамо-гипофизарная недостаточность (гипопитуитаризм) - клинический синдром, развивающийся в результате деструкции аденогипофиза с последующим снижением продукции тропных гормонов и нарушением деятельности периферических эндокринных желез (табл. 2.6).
Табл. 2.6. Гипоталамо-гипофизарная недостаточность
 Окончание табл. 2.6
Окончание табл. 2.6
 Выделяют пангипопитуитаризм - дефицит всех гормонов аденогипофиза и встречающийся значительно чаще парциальный гипопитуитаризм.
Выделяют пангипопитуитаризм - дефицит всех гормонов аденогипофиза и встречающийся значительно чаще парциальный гипопитуитаризм.
Этиология
1. Опухоли гипофиза, приводящие к его деструкции с выпадением продукции тропных гормонов.
2. Окологипофизарные опухоли (краниофарингиома, менингиома, метастазы различных опухолей и проч.).
3. Оперативные вмешательства в гипоталамо-гипофизарной области, облучение гипофиза.
4. Апоплексия гипофиза (септико-эмболический или ишемический инфаркт) или синдром Шиена-Симмондса (СШС). Классический СШС описан у женщин после родов, осложненных сепсисом, тромбоэмболиями и массивной кровопотерей. Гипертрофия передней доли гипофиза во время беременности, сменяющаяся ее инволюцией после родов, способствует тому, что все перечисленные осложнения ведут к нарушению кровообращения в гипофизе, ангиоспазмам, гипоксии и некрозу. В последние годы встречается редко. Синдром аналогичный СШС описан при кровопотерях другого генеза, в том числе и у мужчин.
5. Инфильтративные заболевания (саркоидоз, лимфоцитарный гипофизит, гемохроматоз, гистиоцитоз).
6. Тяжелая черепно-мозговая травма
7. Синдром «пустого» турецкого седла (см. п. 2.8). Как правило, легкий парциальный гипопитуитаризм, часто в сочетании с гиперпролактинемией, обнаруживается не более чем у 10 % пациентов.
8. Врожденные и наследственные синдромы:
- наследственный дефицит гормона роста и ряда тропных гормонов (мутация гена гормона роста, генов Pit-1 и Prop-1);
- дефекты развития гипоталамо-гипофизарной системы (голопрозэнцефалия, септооптическая дисплазия, врожденная аплазия и гипоплазия гипофиза и др.);
- идиопатический дефицит ГР и тропных гормонов гипофиза.
Патогенез
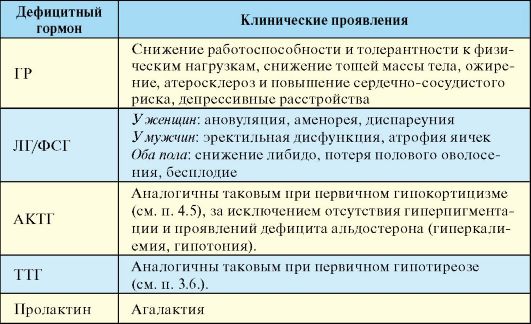
В основе патогенеза пангипопитуитаризма лежит дефицит тропных гормонов и гормона роста (ГР). В зависимости от локализации, обширности и интенсивности деструктивного процесса выпадение или снижение гормонообразования в гипофизе может быть равномерным и полным (пангипопитуитаризм) или частичным, при котором сохраняется продукция одного или нескольких гормонов. Хотя некротические процессы в гипофизе отмечены в 1,1-8,8 % всех аутопсий, частичная гормональная недостаточность развивается при поражении 60-70 % передней доли, а пангипопитуитаризм - при поражении 90 % и более. В результате наступает вторичная гипофункция коры надпочечников, щитовидной и половых желез. В редких случаях одновременного вовлечения в патологический процесс задней доли или ножки гипофиза возможно снижение уровня вазопрессина с развитием несахарного диабета. Одновременное снижение содержания АКТГ и кортикостероидов, антагонистичных вазопрессину в отношении водного обмена, может нивелировать, смягчать клинические проявления недостаточности вазопрессина. Выпадение продукции пролактина приводит к агалактии. При парциальном гипопитуитаризме наиболее часто страдают гонадотропная и соматотропная функции, значительно реже нарушается продукция АКТГ и ТТГ. У взрослых снижение продукции ГР с его универсальным влиянием на белковый синтез приводит к прогрессирующей атрофии гладкой и скелетной мускулатуры и внутренних органов.
Врожденный дефицит ГР в наиболее манифестной форме проявляется синдромом нанизма (от лат. nanos - карлик), который харак-
теризуется резким отставанием в росте и физическом развитии. Гипофизарный нанизм, не является однородным по этиологии и патогенезу состоянием: у большинства больных возникает патология регуляции и секреции других гипофизарных гормонов, как правило, имеются нарушения секреции ФСГ, ЛГ, ТТГ, что сопровождается различными сочетаниями эндокринных и обменных нарушений (пангипопитуитарный нанизм). Наследственные варианты недостаточности ГР, сочетающейся с дефицитом других тропных гормонов, чаще всего связаны с дефицитом фактора Ргор-1 или фактора Pit-1. Фактор Pit- 1 уже в ранних стадиях эмбриогенеза присутствует в соматотрофах, лактотрофах и тиротрофах, где играет важную роль в инициации экспресии генов, ответственных за синтез гормонов этими клетками аденогипофиза. Фактор Prop-1 (prophert, предвестник Pit1) определяет первоначальную закладку сомато-, пролакто- и тиреотрофов, дифференциация которых происходит при участии активатора транскрипции Pit-1. Мутации в указанных генах вызывают комбинированный дефицит ГР, пролактина и ТТГ. Большинство случаев гипофизарного нанизма приходятся на идиопатический дефицит ГР.
Эпидемиология
Точные данные о распространенности различных форм гипопитуитаризма отсутствуют. СШС в настоящее время следует рассматривать, как весьма редкое заболевание; в большинстве случаев он описывается у женщин в возрасте 20-40 лет. Гипофизарный нанизм встречается с частотой 1:15 000 жителей; разница в заболеваемости у мужчин и женщин отсутствует. Дефицит гормона роста, впервые возникший во взрослом возрасте, встречается с частотой 1:10 000.
Клинические проявления
1. Парциальный гипопитуитаризм клинически проявляется вторичным гипотиреозом, гипогонадизмом и гипокортицизмом в различных сочетаниях, а также весьма неспецифической симптоматикой дефицита ГР (табл. 2.7).
2. Пангипопитуитаризм при классическом течении синдрома Шиена-Симмондса (СШС) в большинстве случаев развивается медленно, в течение нескольких лет. Характерна неуклонно нарастающая потеря массы тела, при тяжелом течении достигающая 25-30 кг. Истощение обычно равномерное, мышцы атрофируются, внутренние органы уменьшаются в объеме. Характерны изменения кожных покровов: истончение и сухость придают коже вид папиросной бумаги,
Табл. 2.7. Клиническая картина гипопитуитаризма
 отмечаются
сморщивание, шелушение в сочетании с бледно-желтушной, восковидной
окраской. Исчезают волосы в подмышечных впадинах и на лобке,
депигментируются соски и кожа в области промежности. Характерны
аменорея, снижение либидо, эректильная дисфункция, постепенная атрофия
половых органов и молочных желез. При развитии заболевания после родов
характерны агалактия. В типичном случае обнаруживается синдром «7А» (Аменорея, Агалактия, потеря Аксиллярного оволосения, депигментация Ареол, «Алебастровая» бледность и гипотрофия кожи, Апатия, Адинамия).
Для поздних стадий характерна резкая общая слабость, апатия, адинамия,
вплоть до полной обездвиженности, гипотермия, ортостатический коллапс и
коматозное состояние, которые без лечения приводят к гибели больного. Острая аденогипофизарная недостаточность (гипофизарная кома) представляет собой сочетание острой надпочечниковой недостаточности и гипотиреоидной комы.
отмечаются
сморщивание, шелушение в сочетании с бледно-желтушной, восковидной
окраской. Исчезают волосы в подмышечных впадинах и на лобке,
депигментируются соски и кожа в области промежности. Характерны
аменорея, снижение либидо, эректильная дисфункция, постепенная атрофия
половых органов и молочных желез. При развитии заболевания после родов
характерны агалактия. В типичном случае обнаруживается синдром «7А» (Аменорея, Агалактия, потеря Аксиллярного оволосения, депигментация Ареол, «Алебастровая» бледность и гипотрофия кожи, Апатия, Адинамия).
Для поздних стадий характерна резкая общая слабость, апатия, адинамия,
вплоть до полной обездвиженности, гипотермия, ортостатический коллапс и
коматозное состояние, которые без лечения приводят к гибели больного. Острая аденогипофизарная недостаточность (гипофизарная кома) представляет собой сочетание острой надпочечниковой недостаточности и гипотиреоидной комы.
3. Гипофизарный нанизм проявляется резким отставанием в росте и физическом развитии. К людям карликового роста относят мужчин, имеющих рост ниже 130 см, и женщин - ниже 120 см. Наименьший описанный рост карлика составил 38 см. Дети с классической соматотропной недостаточностью чаще рождаются с нормальной массой
и длиной тела, и начинают заметно отставать в развитии с 2-4-летнего возраста. Для детей с органическим генезом дефицита ГР (краниофарингиома, черепно-мозговая травма и т.п.) характерны более поздние сроки проявления дефицита роста, после 5-6-летнего возраста. При идиопатическом гипофизарном нанизме на фоне отставания в росте отмечаются нормальные пропорции тела ребенка. У нелеченных взрослых отмечаются детские пропорции тела. Черты лица мелкие («кукольное лицо»), переносица западает. Кожа бледная, с желтоватым оттенком, сухая, иногда наблюдаются цианоз, мраморность кожи. У нелеченных больных рано появляются «старообразность», истончение и морщинистость кожи (геродерма), что связано с недостаточностью анаболического действия ГР и замедленной сменой клеточных генераций. Распределение подкожной жировой клетчатки колеблется от истощения до ожирения с преимущественно верхним, или «кушингоидным» (висцеральным) отложением. Волосы могут быть как нормальными, так и сухими, тонкими, ломкими. Вторичное оволосение чаще отсутствует. Мышечная система развита слабо. У мальчиков, как правило, имеется микропенис. Половое развитие задержано и наступает в сроки, когда костный возраст ребенка достигает пубертатного уровня. Значительная доля детей с дефицитом ГР имеет сопутствующий дефицит гонадотропинов.
Диагностика
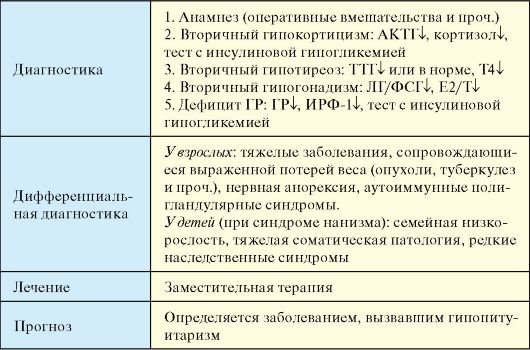
Для диагностики гипопитуитаризма у взрослых важнейшее значение имеют данные анамнеза (операции и облучение гипофиза, осложненные массивным кровотечением роды и т.д.). При гормональном исследовании определяется сочетание низких уровней гормонов периферических эндокринных желез (Т4, тестостерон, эстрадиол, кортизол) со сниженными или низкими уровнями тропных гормонов и ГР. В большинстве случаев необходимо подтверждение дефицита тропных гормонов и ГР в стимуляционных пробах (см. табл. 2.1). Всем пациентам показано проведение МРТ головного мозга.
Основными методами клинической диагностики гипофизарного нанизма являются антропометрия и сопоставление ее результатов с перцентильными таблицами. Для исключения различных скелетных дисплазий (ахондроплазия, гипохондроплазия) оцениваются пропорции тела. При рентгенографии кистей и лучезапястных суставов определяется костный (рентгенологический) возраст, при этом для гипофизарного нанизма характерна значительная задержка окос-
тенения. Дефицит ГР необходимо подтвердить пробой с инсулиновой гипогликемией (табл. 2.1). Весьма ценным исследованием в диагностике дефицита ГР является определение уровня ИРФ-1.
Дифференциальная диагностика
У взрослых гипоталамо-гипофизарную недостаточность необходимо дифференцировать с целым рядом заболеваний, приводящим к похудению (злокачественные опухоли, туберкулез, энтероколиты, спру и спруподобные синдромы, порфириновая болезнь и др.), в том числе от нервной анорексии (см. п. 11.4). Сочетание недостаточности нескольких эндокринных желез встречается в рамках аутоиммунных полигландулярных синдромов (см. п. 9.1).
Идиопатический гипофизарный нанизм дифференцируют от других форм низкорослости: при врожденном гипотиреозе, раннем половом созревании, врожденной дисфункции коры надпочечников, сахарном диабете (синдром Мориака, Нобекур), на фоне тяжелых соматических заболеваний, при генетических остеоартропатиях, а также с так называемой семейной низкорослостью (конституциональная задержка роста). В последнем случае, как правило, удается выявить аналогичные случаи низкорослости у одного из родителей.
Гипофизарный нанизм необходимо дифференцировать от ряда редких генетических синдромов, таких как прогерия (синдром Гетчинсона-Гилфорда), синдром Ларона (периферическая нечувствительность к ГР в результате дефекта гена его рецептора), синдром Рассела-Сильвера (внутриутробная задержка роста с асимметрией туловища), синдром Секкеля (птицеголовые карлики), синдром Прадера-Вилли (задержка роста с рождения, ожирение, крипторхизм, гипоспадия, олигофрения), синдром Лоуренса-Муна-Барде-Бидля (низкорослость, пигментная дегенерации сетчатки, атрофии дисков зрительных нервов, гипогонадизм, задержка умственного развития), ахондроплазия (задержка роста за счет диспропорционального укорочения конечностей).
Лечение
1. При возможности устранение причины заболевания (удаление опухоли гипофиза или гипоталамуса).
2. Заместительную гормонотерапию начинают с компенсации вторичного гипокортицизма препаратами кортикостероидов (см. п. 4.5). Назначение тиреоидных гормонов до компенсации гипокортицизма может привести к развитию острой надпочечниковой недостаточ-
ности. Недостаточность половых желез компенсируется с помощью эстрогенов, прогестинов и препаратов тестостерона. Гипотиреоз компенсируется препаратами левотироксина.
3. В основе патогенетической терапии гипофизарного нанизма лежит заместительная терапия препаратами генно-инженерного человеческого ГР (0,07-0,1 ЕД/кг массы тела ежедневно подкожно в 20.00-22.00 часа). Для лечения дефицита ГР у взрослых рекомендуемые дозы препарата составляют 0,125-0,25 ЕД/кг).
4. Лечение при гипопитуитарной коме аналогично таковому при острой надпочечниковой недостаточности (см. п. 4.5.3).
Прогноз
Определяется конкретным заболеванием, при котором развился гипопитуитаризм. При крупных аденомах гипофиза, по поводу которых предпринималось оперативное вмешательство, а также при инфильтративных заболеваниях процесс обычно необратим. При гипофизарном нанизме вовремя начатая заместительная терапия ГР позволяет достичь пациенту приемлемого для социальной адаптации роста.
2.7. НЕСАХАРНЫЙ ДИАБЕТ
Несахарный диабет (НД, diabetes insipidus) - клинический синдром, обусловленный снижением способности почек концентрировать мочу, связанный с дефицитом антидиуретического гормона (центральный НД) или с нарушением чувствительности почечных канальцев к его действию (почечный НД) (табл. 2.8). НД классифицируется по этиологии и патогенезу (табл. 2.9).
Этиология
В подавляющем большинстве случаев речь идет о центральном НД, который чаще всего связан с деструктивными процессами в области гипофиза (первично гипофизарные или метастатические опухоли, оперативные вмешательства и прочее) (табл. 2.9). Несколько реже НД развивается спонтанно и при визуализации гипофиза органической патологии не выявляется (идиопатический НД). В последнем случае у части пациентов выявлялись антитела к вазопрессин-продуцирующим клеткам аденогипофиза. В редких случаях НД является наследственным заболеванием; наиболее известен аутосомно-рецессивно наследуемый синдром Вольфрама (DIDMOAD), который может быть полным (имеются все проявления) и неполным (например, сочетание сахарного и несахарного диабета).
Табл. 2.8. Несахарный диабет
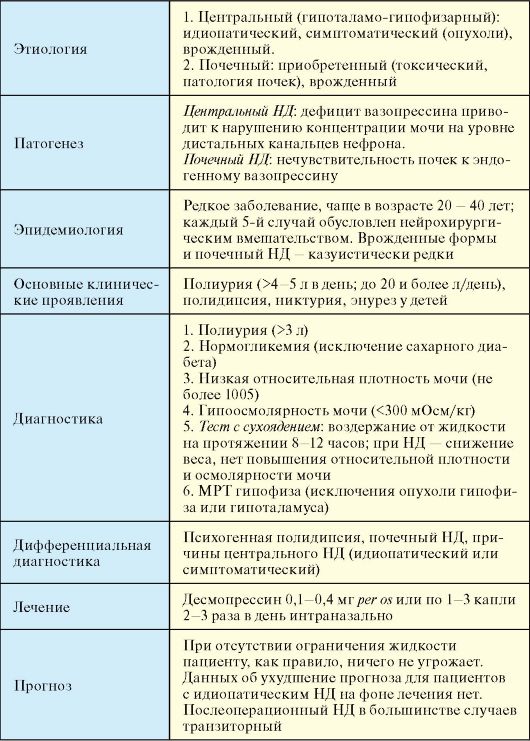 Табл. 2.9. Классификация и причины несахарного диабета
Табл. 2.9. Классификация и причины несахарного диабета
 Патогенез
Патогенез
При центральном НД дефицит вазопрессина приводит к нарушению концентрации мочи на уровне дистальных канальцев нефрона, в результате чего выделяется значительный объем мочи с низкой относительной плотностью. Стимуляция центра жажды приводит к полидипсии. Если пациенту с НД доступны неограниченные количества жидкости, его жизни долгое время ничего не угрожает. При воздержании от приема жидкости развивается гиперосмолярная дегидратация. Для того чтобы объем вторичной мочи не превысил 4 литров в сутки, достаточно 10 % нормально работающих вазопрессин-продуцирующих клеток нейрогипофиза. При длительно существующем центральном НД, при отсутствии лечения возможно развитие вторичной нечувствительности почек к экзогенно вводимому АДГ. Кроме того, постоянная перегрузка жидкостью может привести к опущению желудка, дискинезии желчных путей, синдрому раздраженного кишечника. НД, развившийся после нейрохирургического вмешательства, может быть как постоянным, так и транзиторным со спонтанной ремиссией в сроки от нескольких дней до нескольких лет. Течение НД, развившегося после черепно-мозговой травмы, непредсказуемо: спонтанные выздоровления описаны и через несколько лет после травмы.
Почечный НД является редким заболеванием, чаще наблюдается у детей и обусловлен либо анатомической неполноценностью нефрона, либо ферментативным или рецепторным дефектом, который препятствует реализации действия вазопрессина на проницаемость клеточной мембраны для воды. Возможно развитие нефрогенного НД при хронических заболеваниях почек и при медикаментозных тубулопатиях.
Эпидемиология
НД относительно редкое заболевание (0,5-0,7 % всех эндокринопатий), которое возникает с одинаковой частотой у лиц обоего пола, чаще в возрасте 20-40 лет; известны случаи заболевания в любом возрасте. Каждый 5-й случай НД обусловлен нейрохирургическим вмешательством. Врожденные формы, как и почечный НД - казуистически редкие заболевания, которые относительно чаще встречаются у детей, но иногда выявляются значительно позднее. Синдром DIDMOAD обычно диагностируют в детском возрасте, но известны случаи установления этого диагноза и в возрасте 20-30 лет.
Клинические проявления
Выраженность полиурии и полидипсии, зависит от степени недостаточности АДГ. При неполном дефиците АДГ клиническая симптоматика может быть не столь отчетлива. Количество выпиваемой жидкости колеблется от 3 до 18 л в день, но иногда при мучительной жажде, не покидающей больных ни днем, ни ночью, требуется 20-40 л воды. У детей учащенное ночное мочеиспускание (никтурия) может быть начальным признаком болезни. У маленьких детей вместо выраженной полиурии НД может проявляться диареей. Выделяемая моча обесцвечена, не содержит никаких патологических элементов, относительная плотность всех порций очень низкая (1000-1003). При идиопатическом НД начало заболевания обычно острое, внезапное, реже симптомы появляются постепенно и нарастают. Провоцировать манифестацию НД может беременность.
При длительно существующем нелеченном НД может быть обнаружено расширение мочевого пузыря, мочеточников и лоханок. В связи с хронической водной перегрузкой желудок нередко растягивается и опускается. При достаточном поступлении жидкости в организм сердечно-сосудистая система обычно не страдает (хотя имеется склонность к гипотензии), но по мере нарастания дегидратации в случаях, когда теряемая с мочой жидкость не восполняется (отсутствие воды, проведение дегидратационного теста с сухоядением и др.) возника-
ют симптомы обезвоживания: резкая общая слабость, тахикардия, гипотензия, коллапс. Даже при выраженной дегидратации, несмотря на уменьшение объема циркулирующей крови и снижение клубочковой фильтрации, полиурия сохраняется, концентрация мочи и ее осмолярность почти не возрастают. Если НД обусловлен интракраниальным образованием, отмечается неврологическая симптоматика и клинические проявления гипофизарной недостаточности.
Диагностика
1. Полиурия (не менее 3 литров в день).
2. Нормогликемия (исключение сахарного диабета).
3. Низкая относительная плотность мочи (при показателе более 1005 диагноз может быть надежно исключен).
4. Гипоосмолярность мочи (< 300 мОсм/кг).
5. Отсутствие патологии почек, гиперкальциемии и гипокалиемии.
6. Тест с сухоядением: воздержание от жидкости на протяжении 8-12 часов; в случае НД происходит снижение веса более чем на 5 % и не происходит повышения относительной плотности и осмолярности мочи (<300 мОсм/кг).
7. МРТ для исключения объемного образования гипоталамо-гипофизарной области.
Дифференциальная диагностика
♦ Психогенная полидипсия обусловлена чрезмерным приемом жидкости при невротических и психических расстройствах, а иногда при органической патологии головного мозга. Диурез при психогенной полидипсии может существенно превышать диурез при НД. Дифференциальную диагностику позволяет провести проба с сухоядением.
♦ Почечный НД позволяет исключить эффективность препаратов вазопрессина (купирование полиурии и полидипсии).
♦ Дифференциальная диагностика причин НД. В первую очередь необходимо исключить первичную или метастатическую опухоль гипоталамо-гипофизарной области. О последней в первую очередь нужно думать в случае развития НД в пожилом возрасте.
Лечение
Синтетический аналог вазопрессина - десмопрессин (адиуретин) используют в двух формах: в виде таблеток и спрея в нос.
Таблетированный десмопрессин назначается в дозе 0,1-0,4 мг 3 раза в день. Интраназально спрей назначают несколько раз в день. Лечение нефрогенного НД не разработано. Делаются попытки назначения высоких доз десмопрессина, тиазидных диуретиков (парадоксальный антидиуретический эффект), нестероидных противовоспалительных препаратов, препаратов лития и проч.
Прогноз
Послеоперационный НД, в большинстве случаев, оказывается транзиторным; идиопатический НД - наоборот, стойкий. Данные об ухудшении прогноза для пациентов с НД, получающих адекватную терапию, отсутствуют. Если НД развивается в рамках гипоталамогипофизарной недостаточности, прогноз определяется аденогипофизарной недостаточностью, а не НД.
2.8. СИНДРОМ «ПУСТОГО» ТУРЕЦКОГО СЕДЛА
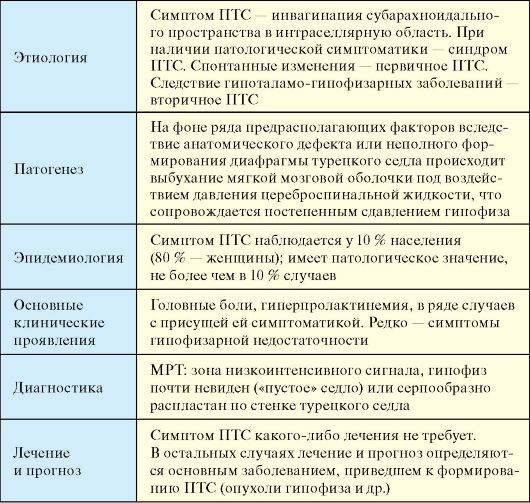
Синдром «пустого» турецкого седла (ПТС) - первичная или развившаяся после нейрохирургического вмешательства недостаточность диафрагмы турецкого седла, приводящая к внедрению в его полость мягкой мозговой оболочки, сдавлению и уменьшению гипофиза, что клинически может проявляться рядом локальных и обменно-эндокринных симптомов (табл. 2.10).
Этиология
Термином синдром «пустого» турецкого седла обозначается инвагинация субарахноидального пространства в интраселлярную область. В тех достаточно редких случаях, когда появляется патологическая симптоматика, говорят о синдроме «пустого» турецкого седла. Для обозначения спонтанных изменений используют термин первично «пустое» седло. Термином вторично «пустое» или «опустевшее» седло обозначаются случаи, когда изменения развиваются после гипоталамо-гипофизарных заболеваний или в результате их лечения, например после нейрохирургических вмешательств на гипофизе, при синдроме Шиена, на фоне медикаментозного лечения аденом гипофиза (пролактином - дофаминомиметиками, соматотропином - аналогами соматостатина; на фоне заместительной терапии первичного гипотиреоза, приведшего к развитию вторичной аденомы гипофиза и т.п.).
Табл. 2.10. «Пустое» турецкое седло
 Патогенез
Патогенез
В основе патогенеза первичного синдрома ПТС лежит недоразвитие диафрагмы турецкого седла, как правило, в сочетании с факторами, приводящими к его недостаточности, к которым можно отнести:
• повышение внутричерепного давления (легочная, сердечная недостаточность, артериальная гипертензия и т.д.);
• физиологическую или патологическую гиперплазию гипофиза или его стебля (многочисленные беременности, длительный прием оральных контрацептивов, длительная неадекватная заместительная терапия недостаточности периферических эндокринных желез);
• спонтанные некрозы опухолей, появление и изменение размера кист гипофиза.
В случаях первично «пустого» седла из-за анатомического дефекта не полностью сформированной диафрагмы турецкого седла создается возможность выбухания мягкой мозговой оболочки под воздействием давления цереброспинальной жидкости, что сопровождается постепенным уплощением гипофиза и увеличением размеров турецкого седла (рис. 2.9).
Полная форма ПТС встречается примерно в 75 % случаев, тогда как в 25 % случаев имеется лишь частичное заполнение полости турецкого седла цереброспинальной жидкостью. Патогенез эндокринно-обменных симптомов связан в основном со сдавлением не самого гипофиза, а его ножки.
Эпидемиология
Синдром «пустого» турецкого седла наблюдается у 10 % населения и в 9 случаях из 10 не сопровождается симптомами гипоталамогипофизарной дисфункции. В 80 % ПТС встречается у женщин, при этом 75 % пациентов страдают ожирением.
Клинические проявления
1. Нейроофтальмологические симптомы: наиболее часто (70 %) встречаются головная боль и головокружение, повышенная утомляемость, снижение работоспособности. Сосудистая компрессионная нейропатия зрительных нервов, хиазмальная симптоматика, ликво-
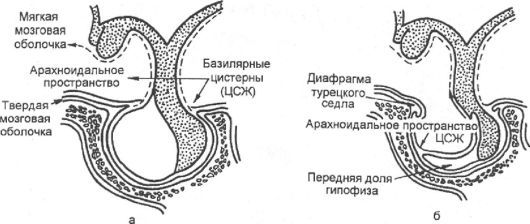 Рис. 2.9. Формирование «пустого» турецкого седла:
Рис. 2.9. Формирование «пустого» турецкого седла:
а - нормальные анатомические взаимоотношения; б - «пустое» турецкое седло; его расширение обусловлено выпячиванием арахноидального пространства через дефект диафрагмы седла
рея встречаются крайне редко, при этом они обусловлены не собственно «пустым» турецким седлом, а опухолью гипофиза или предшествовавшим оперативным вмешательством.
2. Эндокринно-обменные нарушения: среди всех возможных симптомов и лабораторных феноменов чаще всего встречается умеренная гиперпролактинемия (25 %). Не более чем в 10 % случаев встречается субклинически протекающая аденогипофизарная недостаточность, как правило, в виде умеренного дефицита гормона роста. Значительно реже обнаруживаются вторичный гипокортицизм, гипотиреоз, гипогонадизм; крайне редко - несахарный диабет.
Диагностика
ПТС, как правило, обнаруживается при МРТ, проводимой с целью топической диагностики опухолей гипофиза, чаще всего при обнаружении гиперпролактинемии. При этом в полости турецкого седла
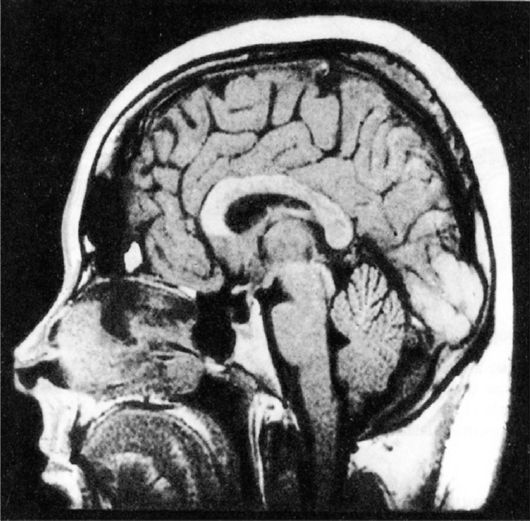 Рис. 2.10. МРТ
«пустого» турецкого седла. Виден распластанный по дну турецкого седла
гипофиз. Полость седла выполнена цереброспинальной жидкостью
Рис. 2.10. МРТ
«пустого» турецкого седла. Виден распластанный по дну турецкого седла
гипофиз. Полость седла выполнена цереброспинальной жидкостью
определяется зона низкоинтенсивного сигнала, что свидетельствует о наличии в интраселлярной области жидкостной структуры - цереброспинальной жидкости; гипофиз при этом деформирован, имеет форму серпа или полулуния, распластан по дну турецкого седла (рис. 2.10).
Лечение и прогноз
Определяются основным заболеванием (гиперпролактинемический гипогонадизм, вторичная микроаденома гипофиза при первичном гипотиреозе и др.). Собственно ПТС как МРТ-феномен лечения не требует.