Эндокринология : учебник. И.И. Дедов - 2009. - 432 с.: ил.
|
|
|
|
ГЛАВА 10 ПОЛИЭНДОКРИНОПАТИИ
Сочетание нескольких эндокринных заболеваний встречается достаточно часто. Так, при акромегалии часто развиваются новообразования щитовидной железы, при синдроме Кушинга - сахарный диабет. Заболевания сразу нескольких эндокринных желез встречаются при многих наследственных синдромах: синдром Тернера (гипогонадизм, сахарный диабет, аутоиммунный тиреоидит), Клайнфелтера (гипогонадизм, сахарный диабет), Дауна (гипогонадизм, сахарный диабет, аутоиммунный тиреоидит). К классическим и наиболее типичным полиэндокринопатиям относят аутоиммунные полигландулярные синдромы и синдромы множественных эндокринных неоплазий.
10.1. АУТОИММУННЫЕ ПОЛИГЛАНДУЛЯРНЫЕ СИНДРОМЫ
Аутоиммунные полигландулярные синдромы (АПС) - это первичное аутоиммунное поражение двух и более периферических эндокринных желез, приводящее к их недостаточности, часто сочетающееся с различными органоспецифическими неэндокринными заболеваниями аутоиммунного генеза. На основании клинических и иммуногенетических особенностей выделяют аутоиммунный полигландулярный синдром l типа (АПС-l) и аутоиммунный полигландулярный синдром 2 типа (АПС-2) (табл. 10.1 и 10.2).
Табл. 10.1. Аутоиммунные полигландулярные синдромы
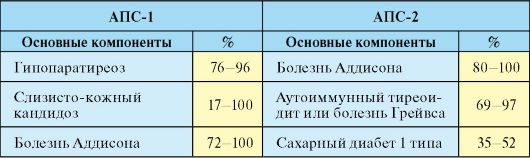 Окончание табл. 10.1
Окончание табл. 10.1
 10.1.1. Аутоиммунный полигландулярный синдром 1 типа
10.1.1. Аутоиммунный полигландулярный синдром 1 типа
Аутоиммунный полигландулярный синдром l-го типа (АПС-l, кандидополиэндокринный синдром, APECED - autoimmune polyendocrinopathy, candidiasis, ectodermal-dystrophy; MEDAC - multiple endocrine deficiency autoimmune candidiasis) - редкое заболевание, для которого характерна классическая триада, описанная Дж. Уайткером: слизисто-кожный кандидоз, гипопаратиреоз, первичная хроническая надпочечниковая недостаточность (болезнь Аддисона) (табл. 10.3). Классической триаде могут сопутствовать первичный гипогонадизм, значительно реже первичный гипотиреоз, сахарный диабет 1 типа (табл. 10.1 и 10.2).
Этиология
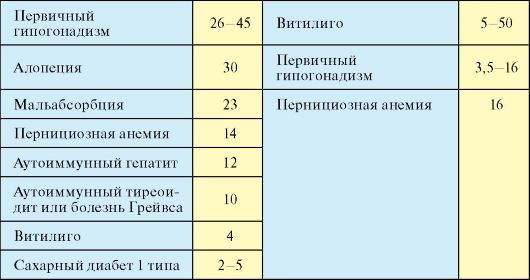
АПС-l является моногенным аутосомно-рецессивным заболеванием. Ген, различные мутации в котором приводят развитию АПС-l, расположен на хромосоме 21 (21q22.3). Этот ген, получивший название AIRE (autoimmune regulator - аутоиммунный регулятор), кодирует белок AIRE, который, наиболее вероятно, является регулятором транскрипции.
Патогенез
В основе патогенеза лежит аутоиммунная деструкция эндокринных желез. При АПС-l с высокой частотой определяются антитела к ферментам надпочечникового стероидогенеза P450scc (20,22-десмолаза),
Р450с17 (17α-гидроксилаза) и Р450с21 (21α-гидроксилаза), антитела против панкреатических β-клеток (к глутаматациддекарбоксилазе и L-аминоациддекарбоксилазе) и других пораженных тканей.
Табл. 10.2. Сравнительная характеристика аутоиммунных полигландулярных синдромов
 Эпидемиология
Эпидемиология
АПС-l, являясь казуистически редкой патологией, чаще встречается в финской популяции, среди иранских евреев и сардинцев, что, вероятно, связано с длительной генетической изолированностью этих народов. Частота новых случаев в Финляндии составляет 1 на 25000 населения.
Клинические проявления
АПС-l дебютирует в детском возрасте, несколько чаще встречается у мужчин. В большинстве случаев его первым проявлением является слизисто-кожный кандидоз, развивающийся в первые 10 лет жизни, при этом наблюдается поражение слизистых оболочек полости рта, гениталий, а также кожи, ногтевых валиков, ногтей. На фоне слизисто-кожного кандидоза у большинства пациентов развивается гипо-
паратиреоз (см. п. 8.5). В среднем через 2 года после начала гипопаратиреоза развивается первичная хроническая надпочечниковая недостаточность (см. п. 4.5), которая обычно протекает в латентной форме, без выраженной гиперпигментации кожи и слизистых. У 10-20 % женщин с АПС-l встречается первичный гипогонадизм, развивающийся в результате аутоиммунной деструкции яичников (аутоиммунный оофорит); клинически он проявляется первичной или вторичной аменореей. Нередко на первый план в клинической картине выходят аутоиммунные неэндокринные заболевания (аутоиммунный гепатит с исходом в цирроз печени).
Диагностика
♦ Характерное сочетание нескольких эндокринных заболеваний.
♦ При установленном диагнозе АПС-1 - периодическое обследование пациентов с целью раннего выявления других компонентов синдрома (гипогонадизм, сахарный диабет и т.д).
Дифференциальная диагностика
Изолированные спорадические эндокринопатии, являющиеся компонентами синдрома.
Лечение
Заместительная терапия недостаточности нескольких эндокринных желез. При назначении заместительной терапии глюкокортикоидами следует иметь в виду, что передозировка может способствовать декомпенсации гипопаратиреоза и спровоцировать гипокальциемию.
Прогноз
Определяется сочетанием аутоиммунных заболеваний у отдельных пациентов.
10.1.2. Аутоиммунный полигландулярный синдром 2 типа
АПС-2 обозначаются различные варианты сочетаний аутоиммунной патологии надпочечников (болезнь Аддисона), щитовидной железы (аутоиммунный тиреоидит (АИТ) или болезнь Грейвса) и сахарного диабета 1 типа (СД-1) (табл. 10.1. и 10.2). Кроме того, им могут сопутствовать другие аутоиммунные эндокринные (оофорит) и неэндокринные (витилиго, пернициозная анемия) заболевания. Наиболее типичными и частыми вариантами АПС-2 являются синдром Шмидта (сочетание первичного гипокортицизма и гипотиреоза в исходе АИТ) и синдром Карпентера (сочетание СД-1 и АИТ).
Этиология
В настоящее время достоверных данных о каких-либо иммуногенетических, серологических и морфологических различиях между аутоиммунными эндокринопатиями в изолированной форме и теми же заболеваниями в рамках АПС-2 не получено. Многие из заболеваний, встречаясь в рамках АПС-2, ассоциированы с гаплотипами HLA-B8, -R3, -DR4, -DR5. Чаще АПС-2 встречается спорадически, однако описано немало случаев семейных форм, при которых АПС-2 развился у разных членов семьи в нескольких поколениях. При этом может наблюдаться разное сочетание заболеваний, встречающихся в рамках АПС-2, у разных членов семьи.
Патогенез
Аутоиммунная деструкция нескольких эндокринных желез с развитием их недостаточности.
Эпидемиология
АПС-2 примерно в 8 раз чаще встречается у женщин, манифестирует в среднем в возрасте между 20 и 50 годами, при этом интервал между клиническим дебютом его отдельных компонентов может составить более 20 лет (в среднем 7 лет). У 40-50 % больных с исходно изолированным первичным гипокортицизмом (см. п. 4.5) рано или поздно развивается другая аутоиммунная эндокринопатия.
Клинические проявления
Сочетание клинических проявлений нескольких эндокринных заболеваний.
Диагностика
♦ Принципы не отличаются от диагностики изолированных эндокринопатий.
♦ Периодическое обследование пациентов с аутоиммунными эндокринопатиями на предмет развития другого заболевания - компонента АПС-2 (определение уровня ТТГ с целью ранней диагностики гипотиреоза у пациентов с СД-1 и болезнью Аддисона).
Дифференциальная диагностика
1. Изолированные аутоиммунные эндокринопатии.
2. Типичной ошибкой является интерпретация умеренного повышения уровня ТТГ в фазе декомпенсации надпочечниковой недостаточности как проявления первичного гипотиреоза. Тест необходимо повторить после достижения компенсации надпочечниковой недостаточности.
3. При тяжелом тиреотоксикозе при болезни Грейвса у пациента могут быть явления относительной надпочечниковой недостаточности (легкая гиперпигментация, гипотония и т.д.), которую необходимо дифференцировать от истинной (проба с 1-24АКТГ).
Лечение
Комбинированная заместительная терапия недостаточности нескольких эндокринных желез.
Прогноз
Определяется сочетанием аутоиммунных заболеваний у отдельных пациентов.
10.2. СИНДРОМЫ МНОЖЕСТВЕННЫХ ЭНДОКРИННЫХ НЕОПЛАЗИЙ
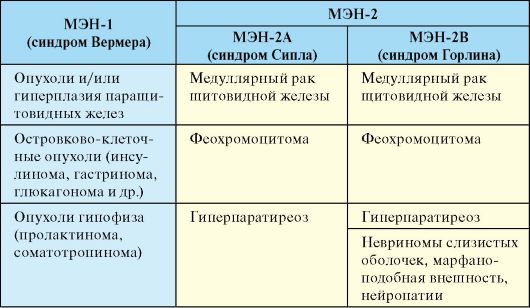
Синдромы множественных эндокринных неоплазий (МЭН) - группа аутосомно-доминантно наследуемых синдромов, характеризующихся устойчивым сочетанием развития опухолей желез внутренней секреции, имеющих одинаковое эмбриональное происхождение (табл. 10.3).
Табл. 10.3. Синдромы множественных эндокринных неоплазий
 10.2.1. Синдром множественных эндокринных неоплазий 1-го типа
10.2.1. Синдром множественных эндокринных неоплазий 1-го типа
МЭН-1 (синдром Вермера) - аутосомно-доминантно наследуемое сочетание опухолей и/или гиперплазии паращитовидных желез с островково-клеточными опухолями (инсулинома, гастринома и др.) и аденомами гипофиза (табл. 10.4). Набор компонентов синдрома варьирует не только между семьями, но и внутри одной семьи, несколько членов которой имеют синдром МЭН-1. В большинстве случаев встречается сочетание двух эндокринных опухолей. На момент постановки диагноза все 3 компонента МЭН-1 имеют место только в трети случаев.
Табл. 10.4. Синдром МЭН-1
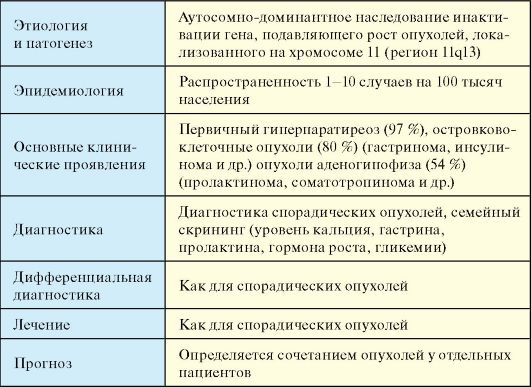 Этиология и патогенез
Этиология и патогенез
Инактивации гена, подавляющего рост опухолей, локализованного на хромосоме 11 (регион 11q13). Развитие опухоли происходит при инактивации обоих аллелей инактивирующего гена. Потери гетерозиготности региона 11q13 были обнаружены в подавляющем боль-
шинстве семейных случаев, а также при спорадически встречающемся синдроме МЭН-1.
Эпидемиология
Является редкой эндокринной патологией: распространенность 1-10 случаев на 100 тысяч населения.
Клинические проявления
1. Первичный гиперпаратиреоз (97 %) (см. п. 8.3) при МЭН по своим проявлениям не отличается от спорадических форм (см. п. 8.3). Особенностью является высокая частота рецидивов после субтотальной паратиреоидэктомии. Обнаружение гиперплазии четырех паращитовидных желез является поводом для целенаправленного поиска МЭН-1 и МЭН-2. На долю МЭН приходится 10-15 % всех случаев первичного гиперпаратиреоза.
2. Островково-клеточные опухоли (80 %) (см. гл. 9); чаще всего выявляются гастриномы (25-60 % всех гастрином обнаруживается при МЭН-1) и инсулиномы. Значительно реже встречаются ВИПомы, глюкагономы и функционально неактивные эндокринные опухоли или карциноиды.
3. Опухоли аденогипофиза (54 %) (см. гл. 2) обнаруживаются у половины пациентов. Наиболее часто встречаются пролактиномы, реже соматотропиномы или гормонально-неактивные опухоли, крайне редко - кортикотропиномы.
Диагностика
♦ Строится на принципах, описанных для спорадических опухолей.
♦ Семейному скринингу при МЭН-1 (уровень кальция, гастрина, пролактина, гормона роста, гликемии) с интервалом раз в 2 года подлежат все родственники больного первой и второй степени родства от 15 до 65 лет.
Дифференциальная диагностика
Строится на принципах, описанных для спорадических опухолей. Лечение
Строится на принципах, описанных для спорадических опухолей. При сочетании гиперпаратиреоза с гастриномой вначале добиваются полного подавления желудочной секреции с помощью блокаторов протонной помпой, затем производят субтотальную или тотальную паратиреоидэктомию, после этого производят операцию по поводу
гастриномы. При сочетании других опухолей вопрос о последовательности операций решается индивидуально.
Прогноз
Определяется набором опухолей у отдельных пациентов.
10.2.2. Синдром множественных эндокринных неоплазий 2-го типа
МЭН-2А (синдром Сиппла) - аутосомно-доминантно наследуемое сочетание медуллярного рака щитовидной железы, феохромоцитомы и гиперпаратиреоза. При синдроме МЭН-2В к указанным компонентам добавляются невриномы слизистых оболочек, патология мышц и скелета (марфаноподобная внешность) и нейропатии (табл. 10.5).
Табл. 10.5. Синдром МЭН-2
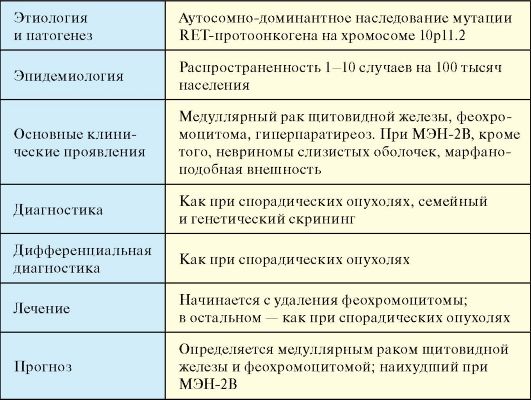 Этиология и патогенез
Этиология и патогенез
Развитие синдрома МЭН-2 связано с мутациями RET-протоонкогена, который локализован на хромосоме 10 (регион 10р11.2) и кодирует
поверхностный мембранный гликопротеид, относящийся к семейству рецептора тирозинкиназы. Мутации RET-протоонкогена выявлены у 90 % пациентов с синдромом МЭН-2. Тип мутации в значительной степени взаимосвязан с различными вариантами фенотипа синдрома МЭН-2. При МЭН-2А и семейной форме медуллярного рака щитовидной железы (МРЩЖ) выявляются миссенс-мутации, приводящие к замене аминокислот в экзонах экстрацеллюлярного домена кодируемого белка. При МЭН-2В практически всегда выявляется мутация в 918-м кодоне 16-го экзона. Мутации RET-протоонкогена приводят к его активации, что в свою очередь обусловливает активацию клеточного роста, которая может закончиться опухолевой трансформацией.
Эпидемиология
Является редкой эндокринной патологией: распространенность 1-10 случаев на 100 тысяч населения.
Клинические проявления
1. Медуллярный рак щитовидной железы (МРЩЖ) - опухоль парафолликулярных клеток щитовидной железы, основным продуктом секреции которой является кальцитонин (п. 3.11). Клиническая картина МРЩЖ бедна, и симптоматика либо вообще отсутствует, либо (в 1/3 случаев) в поздних стадиях проявляется диареей и/или карциноидным синдромом (см. п. 9.7), поскольку МРЩЖ обладает способностью к продукции вазоактивных пептидов (простагландины, серотонин, гистамин). Очень редко МРЩЖ продуцирует АКТГ с развитием синдрома Кушинга (см п. 4.4). Чаще всего диагноз МРЩЖ устанавливается при обследовании по поводу узлового зоба
(см. п. 3.8).
2. Феохромоцитома (см. п. 4.8) выявляется у половины больных, чаще уже после манифестации МРЩЖ. В типичном случае опухоль двусторонняя (70 %). Частота экстраадреналовых и злокачественных вариантов ниже, чем при спорадических феохромоцитомах.
3. Первичный гиперпаратиреоз (см. п. 8.3).
4. При синдроме МЭН-2В, кроме того, развивается патология опорно-двигательного аппарата (марфаноидная внешность, искривления позвоночника и грудной клетки, конская стопа, вывихи головок бедер, арахнодактилия), невриномы слизистых оболочек
(видны на губах, щеках, языке, но могут поражать весь ЖКТ; представляют собой бело-розовые безболезненные узелки размером 1-3 мм), удлиненное лицо с чертами прогнатизма, значительное утолщение губ.
Диагностика
1. Диагностика отдельных компонентов МЭН-2, которая стоится на принципах, описанных для отдельных заболеваний (см. п. 3.11,
4.8, 8.3).
2. Семейный скрининг:
♦ пентагастриновый тест (или тест с глюконатом кальция) (гл. 3.11), определение уровня метанефринов (гл. 4.8) и кальция проводится ежегодно родственникам первой и второй степени родства в возрасте от 6 до 50 лет;
♦ генетический скрининг с целью ранней диагностики МРЩЖ: По наличию или отсутствию мутации RET-протоонкогена пациенты подразделяются на 3 группы:
- обнаружение мутации RET-протоонкогена в лейкоцитах периферической крови указывает на то, что в данном случае МРЩЖ имеет место в рамках МЭН-2 или является семейным;
- отсутствие мутации RET-протоонкогена в периферических лейкоцитах и ее наличие в самой опухоли свидетельствует о соматической мутации и, таким образом, о спорадической форме
МРЩЖ;
- отсутствие мутации в самой опухоли и лейкоцитах не позволяет сделать никакого заключения.
Родственники больных 1-й группы имеют наибольший потенциальный риск, и в этой ситуации ставится вопрос о профилактической экстирпации щитовидной железы. При отсутствии у пациентов 2-й группы признаков феохромоцитомы и гиперпаратиреоза нет необходимости в обследовании родственников. Обследование родственников больных 3-й группы обязательно.
Дифференциальная диагностика
Строится на принципах, описанных для спорадических опухолей.
Лечение
Лечение при синдроме начинается с удаления феохромоцитомы. При любом варианте МРЩЖ обязательна экстирпация щитовидной железы с систематическим удалением шейных лимфатических узлов
и клетчатки. При радикально проведенной операции необходимо пожизненное наблюдение за пациентом, включающее определение уровня кальцитонина и проведение пентагастринового теста или теста с глюконатом кальция.
Прогноз
Прогноз при синдроме МЭН-2 значительно хуже, чем при других вариантах МЭН, поскольку он определяется такими заболеваниями, как медуллярный рак щитовидной железы и феохромоцитома. Пятилетняя выживаемость при МРЩЖ колеблется от 40 до 80 %. Наихудший прогноз при МРЩЖ в рамках МЭН-2В.