Внутренние болезни: учебник. - 6-е изд., перераб. и доп. / В. И. Маколкин, С. И. Овчаренко, В. А. Сулимов. - 2012. - 768 с. : ил.
|
|
|
|
Глава 5. БОЛЕЗНИ СИСТЕМЫ КРОВИ
ГЕМОБЛАСТОЗЫ
Гемобластозы - опухолевые заболевания кроветворной ткани. Их подразделяют на две большие группы - лейкозы и гематосаркомы.
Лейкозы - опухоли из кроветворной ткани с первичной локализацией в костном мозге. Опухолевые клетки легко выходят в периферическую кровь, что приводит к формированию характерной гематологической картины.
Гематосаркомы - опухоли из кроветворной ткани с первичной внекостномозговой локализацией и выраженным местным опухолевым ростом.
Лейкозы и гематосаркомы связаны между собой генетическим родством клеток, из которых они происходят, и могут переходить друг в друга. Все лей-
козы на основании морфологических особенностей опухолевых клеток, составляющих субстрат опухоли, делят на острые и хронические. При острых лейкозах субстратом опухоли служат так называемые бластные клетки, при хронических - созревающие и зрелые клетки.
Разделение острых лейкозов на отдельные формы основано на цитохимических особенностях бластных клеток, в которых с помощью цитохимических исследований обнаруживают определенные вещества и ферменты. Различные формы хронических лейкозов выделяют на основе морфологических свойств клеток, составляющих опухоль.
Выделяют хронические лейкозы миелопролиферативной и лимфопролиферативной этиологии. Подобное разделение основано на том, какая клеткапредшественница - миелоили лимфопоэза - подвергается мутации.
Гематосаркомы в зависимости от типа клеток, составляющих опухоль, делят на лимфогранулематоз (с обязательным присутствием клеток БерезовскогоШтернберга и Ходжкина) и так называемые нелимфогранулематозные (неходжкинские) лимфомы. Среди последних в зависимости от морфологии опухолевых клеток выделяют лимфоцитарные (зрелоклеточные), лимфобластные (малодифференцированные) и гистиоцитарные (ретикулосаркомы) лимфомы.
Этиология
Причины возникновения гемобластозов окончательно не ясны . Мужчины болеют чаще, чем женщины (3:2), а у детей и лиц пожилого и старческого возраста гемобластозы регистрируют чаще, чем у лиц зрелого возраста. Установлен ряд факторов, влияющих на заболеваемость гемобластозами.
• Радиационный фактор: воздействие ионизирующей радиации в атмосфере, облучение по поводу различных заболеваний, применение радиоактивных изотопов.
• Химический фактор: воздействие бензола и других токсичных веществ, в том числе лекарственных препаратов цитостатического действия (метотрексат, хлорамбуцил, азатиоприн, циклофосфан и др.) и других лекарственных средств (фенилбутазон, хлорамфеникол и др.).
• Наследственные хромосомные дефекты. Чаще всего наследуется не сам лейкоз, а нестабильность генома, предрасполагающая родоначальные миелоидные или лимфоидные клетки к лейкозной трансформации.
• Обменные нарушения - изменение обмена триптофана (лейкозогенное действие метаболитов триптофана).
• Вирусный фактор (в частности, вирус Эпстайна-Барр).
Все указанные факторы имеют значение при определенных формах гемобластозов, но их несомненное участие в развитии заболевания в каждом конкретном случае не установлено.
Патогенез
Предполагают, что этиологический фактор повреждает ДНК кроветворной клетки, нарушает генетический код и приводит к безостановочному размножению и нарушению дифференцировки той или иной разновидности клеток. В
соответствии с этой точкой зрения в настоящее время общепризнанной считают клоновую теорию патогенеза гемобластозов, как и опухолей вообще. Лейкозные клетки представлены клоном - потомством одной мутировавшей клетки. Полагают, что мутация происходит почти непрерывно, но мутировавшие клетки уничтожает система фагоцитирующих макрофагов или иммунные силы организма. Можно предполагать, что для развития опухоли, в том числе и гемобластоза, необходимо сочетание мутации клеток и ослабления иммунной защиты. Дальнейшее распространение опухоли происходит путем метастазирования этих клеток по кроветворной системе. Предполагают также, что гемобластозы происходят из клеток первого и второго класса схемы кроветворения. Иначе говоря, родоначальником опухолевого процесса кроветворной ткани чаще всего служит клетка-предшественница миелоили лимфопоэза.
Другая патогенетическая особенность многих гемобластозов - постепенное озлокачествление опухолевого процесса, обозначаемое термином «опухолевая прогрессия». Опухолевая прогрессия выражается следующими процессами:
• угнетением нормального кроветворения;
• развитием бластного криза (смена дифференцированных опухолевых клеток недифференцированными);
• возникновением у лейкозных клеток способности к росту вне органов кроветворения;
• уходом лейкозных клеток из-под контроля цитостатических препаратов;
• неодинаковыми свойствами лейкозных клеток в разных очагах лейкозной пролиферации.
Эти признаки опухолевой прогрессии находят клиническое выражение при гемобластозах. Общий патогенез представлен на рис. 5-1.
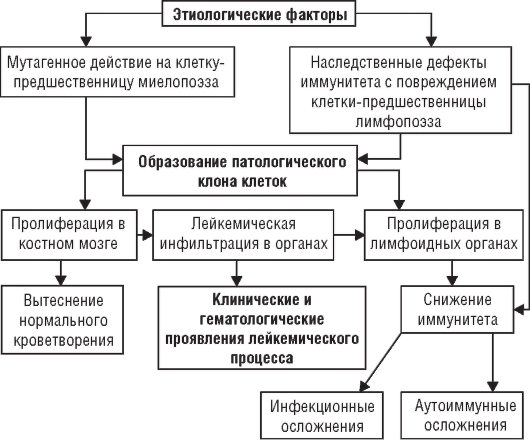
Рис. 5-1. Патогенез лейкоза
ОСТРЫЙ ЛЕЙКОЗ
Острый лейкоз (ОЛ) - опухолевое заболевание кроветворной системы, при котором субстрат опухоли составляют бластные клетки. Опухолевая трансформация осуществляется на самых ранних этапах дифференцировки стволовой кроветворной клетки, и ее дальнейшее созревание не происходит. Термин «острый лейкоз» отражает не временной фактор (длительность течения болезни), а морфологические и цитохимические особенности опухолевых клеток.
ОЛ - одно из наиболее тяжелых заболеваний из группы гемобластозов, занимающее первое место в структуре заболеваемости ими и составляющее приблизительно одну треть их общего числа. ОЛ регистрируют у лиц любого возраста, при этом отмечают два пика заболеваемости - в возрасте 3-4 и 60- 69 лет. Мужчины болеют чаще, чем женщины. В прошлом (до середины 1960-х годов) ОЛ в течение нескольких месяцев приводил к смерти больных, в связи с чем и был предложен термин «острый лейкоз». В настоящее время достигнуты значительные успехи в его лечении: в большинстве случаев (20-40%) удается достигнуть полной ремиссии, которая может продолжаться более пяти лет у 10-20% больных острыми нелимфобластными лейкозами и у 50% пациентов с острыми лимфобластными лейкозами. Фактор времени имеет исключительно большое значение в успехе лечения ОЛ: чем раньше будет диагностировано заболевание и начато лечение, тем больше шансов на его успех.
Этиология
Этиология заболевания неизвестна. Тем не менее признают существование ряда факторов, способствующих развитию ОЛ:
• генетическую предрасположенность (хромосомные аномалии);
• ионизирующую радиацию;
• некоторые токсины и химические соединения (например, бензолы и органические соединения);
• вирусные инфекции, потенцирующие угнетение иммунной системы;
• лекарственные средства - алкилирующие соединения (мелфалан, ломустин) и некоторые другие вещества, применяемые в химиотерапии, могут стимулировать развитие миелодисплазий или миелоидных лейкозов;
• предшествующие заболевания системы кроветворения (рефрактерные формы анемий, пароксизмальная ночная гемоглобинурия, так называемые миелодисплазии).
Эпидемиологические исследования показывают, что в семьях больных ОЛ риск заболеваемости повышается почти в 3-4 раза. На значение генетических факторов в развитии ОЛ указывает и увеличение последней при некоторых генетических нарушениях и аномалиях развития (болезнь Дауна, некоторые виды анемий и др.). Предполагают, что роль генетических факторов ограничена формированием предрасположенности к развитию ОЛ, которая в дальнейшем реализуется под действием различных факторов (лучевых, химических, экзо- и эндотоксических воздействий).
Патогенез
Как было отмечено ранее, в основе болезни лежит пролиферация опухолевых бластных клеток в костном мозге, которые затем метастазируют в различные органы (селезенка, печень, головной мозг и др.). ОЛ сопровождается угнетением нормального ростка кроветворения, что связано с несколькими факторами :
• повреждением кроветворного окружения вокруг опухоли;
• вытеснением нормального ростка гемопоэза;
• выработкой бластными клетками ингибиторов, подавляющих рост нормальных кроветворных клеток.
Многоэтапный процесс озлокачествления клеток крови начинается с нарушений экспрессии генов трех типов.
• Онкогены, продукты экспрессии которых могут вызывать злокачественную трансформацию клеток, - результат изменения обычных генов генома - протоонкогенов. Изменения, инициирующие трансформацию протоонкогенов в онкогены, состоят в амплификации, точечной мутации или транслокации. В результате транслокации может существенно измениться характер экспрессии перемещенного гена.
• Антионкогены (гены-супрессоры опухолевого роста) - гены, кодирующие продукцию белков, ингибирующих пролиферацию клеток. В некоторых случаях делеция в хромосоме блокирует эти гены в одной из аллелей. Если происходит делеция или мутация в другой аллели, то начинается нерегулируемый рост клеток.
• Ингибирование апоптоза: большинство злокачественных клеток устойчивы к апоптозу. В норме за продукцию регуляторных белков, предупреждающих апоптоз, отвечает ген BCL-2. В опухолевых клетках его экспрессия вследствие хромосомной транслокации может быть многократно усилена.
Основные патогенетические звенья представлены на рис. 5-1.
Классификация
Опухолевая трансформация при ОЛ происходит на стадиях дифференцировки родоначальных кроветворных клеток. В связи с этим ОЛ в первую очередь разделяют на лимфобластные, т.е. относящиеся к клеткам-предшественницам лимфопоэза, и нелимфобластные, относящиеся к клеткам-предшественницам миелопоэза и составляющие основную массу ОЛ у взрослых. Каждая из этих двух больших групп неоднородна, и на основании цитогенетических и иммунологических характеристик в них выделяют различные формы.
На основе иммунофенотипической характеристики различают следующие варианты острого миелоидного лейкоза:
• острый малодифференцированный лейкоз;
• острый миелобластный лейкоз без созревания;
• острый миелобластный лейкоз с созреванием;
• острый промиелобластный лейкоз;
• острый миеломонобластный лейкоз;
• острый монобластный лейкоз;
• острый эритромиелоз;
• острый мегакариобластный лейкоз.
На основе иммунологической и цитогенетической характеристики выделяют следующие варианты острого лимфобластного лейкоза:
• пре-пре-острый лимфобластный лейкоз;
• пре-В-острый лимфобластный лейкоз;
• В-острый лимфобластный лейкоз;
• Т-острый лимфобластный лейкоз.
Различают несколько стадий течения острого лейкоза.
• I - начальная стадия, чаще всего оцениваемая ретроспективно.
• II - развернутая стадия с четкими клиническими и гематологическими признаками болезни. В ней различают:
- первую «атаку»;
- ремиссию (полную или неполную);
- рецидив болезни;
- второй рецидив и т.д.
• III - терминальная стадия - отсутствие эффекта от цитостатической терапии, выраженное угнетение нормального кроветворения, язвеннонекротические процессы.
Выделение стадий ОЛ требуется, прежде всего, для практической деятельности, так как в разных стадиях проводят различное лечение (мощную цитостатическую терапию - в развернутой стадии в период первой «атаки» или рецидива, поддерживающую - в период ремиссии).
Клиническая картина
Симптомы ОЛ могут быть весьма многообразными, в связи с чем их можно представить в виде следующих больших синдромов.
• Гиперпластический синдром, также называемый синдромом лейкемической пролиферации и обусловленный опухолевым ростом как в костном мозге, так и вне его (метастазирование):
- увеличение селезенки, печени, лимфатических узлов (периферические, в средостении, брюшной полости) и миндалин;
- поражения кожи (кожные лейкозные инфильтраты - неспецифическая гемодермия или лейкемиды), мозговых оболочек (нейролейкоз или нейролейкемия), почек, миокарда и легких.
• Анемический синдром.
• Геморрагический синдром - возникновение от мелкоточечных и мелкопятнистых единичных или редких высыпаний на коже и слизистых оболочках до обширных кровоизлияний и профузных кровотечений (носовые, маточные, почечные, желудочно-кишечные и пр.).
• Интоксикационный синдром - снижение массы тела, лихорадка, поты, выраженная слабость.
На первом этапе диагностического поиска можно отметить ряд вариантов начала заболевания и его дальнейшего течения. Примерно в половине случаев (преимущественно у лиц молодого возраста) отмечают острое начало заболевания. Обычно это активные, энергичные люди, не привыкшие уделять
внимание своему самочувствию и обращающиеся к врачу лишь в крайних случаях. Острое начало болезни протекает под видом ангины, гриппа или острого респираторного заболевания. У некоторых больных, помимо выраженной интоксикации и лихорадки, развивается тяжелый приступ болей в животе, сопровождающийся диспептическими расстройствами. Таких пациентов нередко направляют в инфекционное отделение с подозрением на заболевания тифопаратифозной группы.
У 10% больных заболевание начинается с профузных кровотечений (носовые, маточные, желудочно-кишечные). Некоторые пациенты впервые обращаются к врачу по поводу гиперпластического гингивита и язвенного стоматита. Примерно в 20% случаев начальная стадия ОЛ проходит мимо внимания врача и самого больного, что обусловлено неспецифическими симптомами, выраженными в весьма незначительной степени. Тем не менее при ретроспективной оценке развития болезни удается установить, что еще до обращения к врачу больной отмечал нарастающую слабость, повышенную утомляемость, боли в костях, мышцах и суставах, незначительное увеличение лимфатических узлов или единичные кровоизлияния в кожу. На эти признаки он не обращал внимания, объясняя их мелкими травмами, и лишь при нарастании числа «синяков» обратился к врачу. Боли в костях и суставах иногда носят упорный характер, и больные могут находиться под наблюдением врача с ОА или РА. Обычно это связано с тем, что не было своевременно выполнено исследование крови.
Наконец, у 52% больных явных изменений общего состояния нет, а заболевание обнаруживают при случайном исследовании крови (например, при диспансерном обследовании, оформлении санаторно-курортной карты и пр.).
Более реальная возможность диагностики ОЛ возникает в развернутом периоде заболевания, но и в это время клиническая картина весьма вариабельна, так как те или иные синдромы выражены в разной степени. Синдром опухолевой интоксикации становится значительно выраженным и манифестирует повышением температуры тела, резкой слабостью, потливостью и прогрессирующим снижением массы тела.
Гиперпластический синдром выражается в увеличении лимфатических узлов и селезенки, обнаруживаемом самим больным. При метастатических поражениях жалобы пациентов весьма разнообразны (сильная головная боль, кашель, одышка, упорный радикулит, боли в животе, рвота, понос, парестезии и кожный зуд) и нередко «уводят» мысль врача от ОЛ, давая возможность предполагать самостоятельные заболевания различных органов.
При нарастании анемического синдрома в клинической картине начинают преобладать признаки циркуляторно-гипоксического синдрома, что дает основание предположить любую форму анемии.
Точно так же весьма существенными могут быть признаки геморрагического синдрома (обширные геморрагии, кровотечения).
Некоторые больные могут сообщить, что им раньше уже диагностировали ОЛ и проводили цитостатическую терапию. В таком случае ухудшение состояния можно связать с ранее обнаруженным заболеванием.
Таким образом, на первом этапе диагностического поиска можно иметь достаточно данных для углубленного обследования больного и обязательного проведения исследования крови.
На втором этапе диагностического поиска в развернутой стадии болезни можно обнаружить симптомы вышеуказанных синдромов. Гиперпластический синдром манифестирует:
• увеличением лимфатических узлов (чаще - шейных) с одной или обеих сторон, безболезненных (характерно для острого лимфобластного лейкоза), имеющих плотноватую консистенцию;
• нерезко выраженным увеличением селезенки (плотноватая, безболезненная или слегка чувствительная, выступает из-под реберного края на 3-6 см);
• увеличением печени (плотноватая, чувствительная, пальпируют на 2-4 см ниже реберного края).
Эти изменения не считают специфическими для ОЛ и обнаруживают при других заболеваниях системы крови. Поражение других органов сопровождается разнообразными симптомами.
При поражении кожи на ней обнаруживают плотноватые инфильтраты розоватого или светло-коричневого цвета, обычно - множественные (лейкемиды).
В случае поражения легких отмечают симптомы бронхиальной обструкции (ослабление дыхания, удлинение выдоха, сухие хрипы) и очаговые инфильтративные изменения (притупление перкуторного звука, ослабленное или жесткое дыхание, сухие и влажные хрипы). Отличить специфический лейкозный пневмонит от бактериальной пневмонии, нередко осложняющей ОЛ, трудно.
При поражении миокарда обнаруживают небольшое расширение границ сердца, тахикардию и глухие тоны, в тяжелых случаях - признаки сердечной недостаточности.
Поражение ЖКТ при физикальном обследовании может манифестировать болезненностью при пальпации в эпигастральной области.
При поражении ЦНС (нейролейкоз) обнаруживают ригидность затылочных мышц, симптом Кернига, нарушение функции черепных нервов, снижение мышечного тонуса и другие симптомы.
Эти явления дают основание подозревать не ОЛ, а скорее самостоятельное заболевание того или иного органа.
Анемический синдром характеризуется бледностью кожного покрова, тахикардией, систолическим шумом во всех точках и снижением АД. Геморрагическому синдрому свойственны кожные геморрагии петехиально-пятнистого характера.
Следует иметь в виду, что указанные многообразные клинические признаки могут отсутствовать, и физикальное обследование в этих случаях не предоставляет никакой информации, поэтому предположение об ОЛ на втором этапе может и не возникнуть. Решающим для диагностики заболевания считают третий этап.
Задачи третьего этапа диагностического поиска - установление диагноза ОЛ и определение его формы, а также обнаружение органных поражений и осложнений.
Для подтверждения опухолевой трансформации костномозгового кроветворения необходимо провести исследование периферической крови и костного мозга. При ОЛ клиническая картина становится вполне определенной, когда
«плацдарм» кроветворения существенно заменен бластными клетками. Считают, что в развернутой стадии болезни общая масса опухоли составляет 1012 клеток на 1 м2 площади тела.
Число лейкоцитов в периферической крови может колебаться от низких цифр до высоких (гиперлейкоцитоз). В настоящее время установлено, что верхняя граница может достигать 20-50х109/л. Весьма часто (в 50% случаев) регистрируют лейкопению. Основной признак ОЛ - присутствие в крови опухолевых бластных клеток (бластемия). Их обнаруживают в мазках крови в количестве от 5-10 до 80-90%. Характерно так называемое лейкемическое зияние - очень малое количество зрелых гранулоцитов (сегментоядерных) и практически полное отсутствие палочкоядерных, юных форм и метамиелоцитов. В алейкемической фазе болезни бласты в периферической крови единичные или вообще отсутствуют. В этих случаях диагноз ставят по результатам исследования пунктата костного мозга, в котором обнаруживают значительное увеличение содержания бластных клеток (увеличение в стернальном пунктате бластных клеток более 30% полностью подтверждает диагноз ОЛ).
Существует правило: необходимо в течение всей жизни больного ОЛ хранить пунктаты его костного мозга и мазки периферической крови, так как под влиянием проводимой цитостатической терапии картина крови и костного мозга становится нетипичной для ОЛ: в препаратах появляются так называемые терапевтические бласты с более грубым ядром, и их практически нельзя отличить от лимфоцитов. При повторных обращениях больного в другие лечебные учреждения диагноз ОЛ можно подтвердить лишь при изучении первичных препаратов.
Другие гематологические показатели не имеют самостоятельного диагностического значения, хотя анемию и тромбоцитопению, сопутствующие ОЛ, нередко можно рассматривать как косвенные критерии диагноза, особенно если эти изменения сочетаются с лихорадкой неясной этиологии и спленомегалией.
Анемия (обычно нормохромного и макроцитарного типа) усиливается по мере прогрессирования заболевания. Ее рассматривают в качестве признака угнетения нормального кроветворения. То же происхождение имеет и тромбоцитопения. В некоторых случаях анемия и тромбоцитопения имеют аутоиммунное происхождение, что сопровождается умеренной желтушностью, ретикулоцитозом, повышенным содержанием непрямого билирубина в крови и положительной пробой Кумбса.
В некоторых случаях возможно развитие так называемой малопроцентной формы ОЛ. Для нее характерны незначительно выраженные клинические симптомы, склонность к цитопении (особенно к анемии) с увеличением числа бластных клеток до 20% как в крови, так и в костном мозге. Эти изменения могут сохраняться несколько месяцев, но в конечном итоге развивается типичная картина ОЛ.
Бластные клетки при всех формах ОЛ характеризуются крупными размерами, большим ядром, занимающим почти всю клетку, и нежно-сетчатым строением хроматина с крупными единичными ядрышками. Цитоплазма клеток представлена узким ободком голубоватого или серо-голубого цвета с единичными мелкими гранулами или без грануляции.
Обычно морфологическое исследование бластных клеток в мазках крови, окрашенных гематологическими красителями, не позволяет с уверенностью дифференцировать формы ОЛ. Исключение составляют бласты при остром промиелоцитарном лейкозе. Для них характерны обильная крупная фиолетовая зернистость цитоплазмы с тельцами Ауэра и различная форма ядер клеток.
Различить бластные клетки при разных формах ОЛ с большой достоверностью можно по цитохимическим свойствам.
Для определения острого лимфобластного лейкоза наиболее характерна PAS-реакция (окраска на гликоген), обнаруживающая в цитоплазме крупные гранулы, чаще - в виде ожерелья. Для острого недифференцированного лейкоза в первую очередь характерна положительная реакция на пероксидазу, а для его различных форм - этот же показатель в сочетании с положительной реакцией на неспецифическую эстеразу (острый монобластный лейкоз и острый миеломонобластный лейкоз), кислые мукополисахариды (острый промиелоцитарный лейкоз) и др.
Для дифференциальной диагностики острого лимфобластного лейкоза большое значение имеют иммунологические методы (обнаружение поверхностных иммуноглобулинов, способность к розеткообразованию и др.). Наконец, все большее значение начинают приобретать методы определения поверхностных клеточных дифференцировочных антигенов, которые возникают и исчезают в характерном для каждого типа клеток сочетании по мере их созревания. Для проведения таких исследований используют специфические моновалентные антисыворотки.
Определение степени органных поражений проводят с помощью лабораторно-инструментальных методов и морфологических исследований (при необходимости).
Последние становятся особенно актуальными в периоде ремиссии, так как позволяют оценить ее полноту. С этой целью выполняют пункцию костного мозга, увеличенных лимфатических узлов, селезенки, печени, инфильтратов кожи и увеличенного яичка.
Обнаружение бластных клеток в пунктатах, а также в спинномозговой и плевральной жидкости указывает на соответствующее органное поражение и исключает полную ремиссию после проведенного лечения.
При подозрении на поражение лимфатических узлов средостения и корней легких проводят томографию и рентгенографию грудной клетки. В случае лейкозного пневмонита на рентгенограмме отмечают усиление легочного рисунка и обнаруживают мелко- и крупноочаговые тени по всей легочной ткани.
При лейкемической инфильтрации миокарда возможны изменения на ЭКГ в виде снижения вольтажа и образования отрицательных зубцов Т. Тем не менее они могут быть связаны не только со специфической инфильтрацией миокарда, но и с миокардиодистрофией, обусловленной опухолевой интоксикацией, а также кардиотоксическим действием цитостатических препаратов, применяемых при лечении ОЛ.
При лейкемической инфильтрации печени УЗИ позволяет обнаружить увеличение и мелкоочаговые изменения эхогенности органа. Изотопная сцинтиграфия в подобных случаях также помогает определить увеличение органа и снижение поглощения радиофармацевтического препарата с его диффузно-
неравномерным накоплением. Эти изменения носят неспецифический характер и свидетельствуют лишь о диффузных изменениях ткани печени, которые могут развиться в результате неспецифического реактивного гепатита, а также токсического гепатита лекарственной (цитостатические средства) этиологии. В последнем случае правильной интерпретации указанных показателей будут способствовать данные анамнеза, физикального обследования (нарастающее увеличение размеров печени, желтуха) и биохимических реакций, обнаруживающих значительную гиперферментемию и гипербилирубинемию.
Специфическое поражение почек при УЗИ может выражаться в увеличении органа (обычно - двустороннем) и присутствии диффузных эхоструктур в его ткани. В анализе мочи - умеренная протеинурия, гематурия и цилиндрурия. Эти изменения могут быть признаком опухолевой интоксикации. При нейролейкемии отмечают характерные изменения спинномозговой жидкости: высокий цитоз, повышенное содержание белка и обнаружение в мазках бластных клеток. Серьезное осложнение цитостатической терапии - развитие агранулоцитоза, о котором судят по обнаружению в крови резкой лейкопении (до 1 х109/л и менее) и изменению лейкоцитарной формулы в виде уменьшения числа нейтрофилов (вплоть до полного исчезновения) и относительного увеличения количества лимфоцитов.
Диагностика
Несмотря на то что клиническая картина ОЛ очерчена довольно ярко, признаков, патогномоничных только для этого заболевания, практически нет. В связи с этим даже такие, казалось бы, характерные для гемобластозов симптомы, как увеличение селезенки, печени, лимфатических узлов и анемия, позволяют врачу лишь заподозрить ОЛ и срочно провести исследование крови. Морфологические методы - исследование костного мозга и периферической крови - позволяют диагностировать ту или иную форму ОЛ. Тем не менее в настоящее время наиболее точным методом диагностики служит цитохимическое исследование. Преимущество иммунофенотипирования бластов состоит в том, что его можно проводить параллельно с цитохимическим исследованием. Оно позволяет определить с помощью моноклональных антител существование или отсутствие кластеров дифференцировки бластных клеток (CD-маркеры).
Существует ряд правил, которые следует помнить любому врачу, чтобы не пропустить ОЛ. Обязательным считают динамическое исследование крови при всех рефрактерных к лечению и рецидивирующих ангинах, респираторных заболеваниях и гриппе, особенно если эти заболевания сопровождаются лимфаденопатией, геморрагическими симптомами, а также артралгиями. Особая гематологическая настороженность должна быть при всех случаях лимфаденитов и гиперпластических гингивитов. Назначение таким больным различных физиотерапевтических и тепловых процедур без предварительного исследования крови может принести вред.
Дифференциальная диагностика
При установлении диагноза ОЛ, особенно при лейкопении и отсутствии гранулоцитов, следует проводить дифференциальную диагностику с рядом заболеваний .
Агранулоцитозы не сопровождаются бластозом костного мозга и редукцией эритроидного роста кроветворения. Геморрагический синдром отмечают редко, селезенка не увеличена.
Для гипопластических анемий не характерно увеличение лимфатических узлов и селезенки. При стернальной пункции в мазке не обнаруживают увеличение количества бластных клеток. Большое значение для диагностики имеет трепанобиопсия: преобладание в трепанате жировой ткани свидетельствует о гипопластической анемии.
При диффузных заболеваниях соединительной ткани и хронических активных гепатитах возможны лимфаденопатия, увеличение селезенки, анемия, тромбоцитопения и нейтропения. В пунктате костного мозга количество бластов может быть повышено до 10-20%. В таких случаях необходимы трепанобиопсия крыла подвздошной кости и повторное исследование крови и костного мозга. Кроме того, правильной оценке симптомов помогает учет всех данных клинической картины.
С симптомами ОЛ сходна клиническая картина инфекционного мононуклеоза: острое начало, лихорадка, ангина, увеличение печени, селезенки и лимфатических узлов, а также лейкоцитоз с присутствием широкопротоплазменных мононуклеаров в гемограмме. В отличие от ОЛ отсутствуют анемия и геморрагический синдром, а увеличенные болезненные лимфатические узлы локализуются по заднему краю грудиноключично-сосцевидной мышцы. В мазке крови вместо бластных клеток обнаруживают 60-70% мононуклеаров, большинство из которых составляют средне- и широкоплазменные лимфоциты. Увеличено количество моноцитов, тромбоцитопения отсутствует. Заболевание не сопровождается осложнениями и в легких случаях проходит без всякого лечения.
Существенные трудности возникают при дифференциальной диагностике ОЛ и хронического миелолейкоза (ХМЛ), дебютирующего бластным кризом. Клинические признаки, исследование крови и костного мозга не позволяют дифференцировать заболевания. В таких случаях существенную пользу приносит исследование хромосом. Обнаружение Ph-хромосомы свидетельствует о
ХМЛ.
Лейкемоидные реакции - значительное повышение количества лейкоцитов со сдвигом лейкоцитарной формулы влево. Причиной их возникновения могут быть тяжелые инфекционные и воспалительные заболевания, прием некоторых медикаментов, интексикации и травмы. В отличие от ОЛ, при лейкемоидных реакциях бласты отсутствуют.
Формулировка развернутого клинического диагноза должны учитывать:
• форму ОЛ;
• стадию заболевания;
• существование внекостномозговых поражений;
• существование осложнений.
Лечение
Цель современного лечения ОЛ у взрослых - достижение длительной выживаемости без возникновения рецидивов болезни. Это оказалось возможным при условии внедрения программного лечения, представленного поэтапным
уничтожением лейкозных клеток с помощью различных комбинаций цитостатических средств, а также комплексом мероприятий, направленных на борьбу и предупреждение развития агранулоцитоза, тромбоцитопении, анемии и ДВСсиндрома. Использование таких программ позволяет добиться ремиссии у 60- 80% взрослых больных и полного выздоровления 20-30% пациентов. Принципы лечения ОЛ:
• лечение начинают сразу же после установления диагноза;
• лечение должно быть дифференцированным и зависеть от морфологического и цитохимического типа ОЛ;
• одновременно назначают несколько препаратов (полихимиотерапия);
• следует стремиться к достижению лейкопении;
• в стадии ремиссии необходимо проводить поддерживающую терапию. Лекарственные препараты не способствуют превращению лейкемических
клеток в нормальные, а уничтожают их. Основная задача лечения - избавление (санация) организма от лейкемических клеток. Ее выполнения достигают посредством применения большого количества химиопрепаратов с различным механизмом действия.
Антилейкемические средства наиболее активны по отношению к делящимся клеткам, причем некоторые из них активны в каком-то определенном периоде митоза (фазово- и циклоспецифические препараты - меркаптопурин, цитарабин, метотрексат), другие - в течение всего митотического цикла (циклонеспецифические - циклофосфан, винкристин, преднизолон).
Существуют определенные этапы проведения химиотерапии:
• достижение ремиссии (индукция) - проводят при первой «атаке» ОЛ и рецидивах болезни;
• закрепление ремиссии (консолидация) - 2-3 курса;
• противорецидивное лечение (поддержание ремиссии) - проводят в течение всего периода ремиссии;
• профилактика нейролейкемии.
Невыполнение одного из этапов такого программированного лечения и необоснованные изменения схем химиотерапии приводят к неизбежному развитию рецидива и лишают больного шансов на полное выздоровление. Следует отчетливо представлять, на основании каких признаков можно говорить о наступлении ремиссии или рецидива.
Ремиссия характеризуется следующими показателями, сохраняющимися на протяжении не менее 1 мес:
• в костном мозге число бластных клеток не превышает 5%, представлены все ростки кроветворения с нормальными признаками созревания, обнаруживают мегакариоциты, клеточность костного мозга составляет более
20%;
• в периферической крови абсолютное число нейтрофилов составляет не менее 1,5х109/л, тромбоцитов - не менее 100х109/л, бластные клетки отсутствуют;
• нет очагов экстрамедуллярного лейкемического роста (включая отсутствие нейролейкемии).
Рецидив диагностируют при обнаружении в пунктате костного мозга 5-20% бластов (повторное обнаружение бластов в костном мозге в количестве более
5% подтверждает рецидив), а также любого экстрамедуллярного лейкемического поражения (даже без вовлечения в патологический процесс костного мозга).
При назначении полихимиотерапии следует иметь в виду, что те или иные морфологические варианты ОЛ чувствительны к комбинациям определенных препаратов.
При остром миелобластном лейкозе индукцию ремиссии проводят с помощью программы «7+3»: цитарабин (капельно по 100 мг/м2 каждые 12 ч в течение 7 дней), даунорубицин (внутривенно струйно или капельно быстро по 45 мг/м2 (60 мг/м2) в сутки в течение 3 дней). Помимо химиотерапии, обязательным компонентом считают так называемую сопроводительную терапию, направленную на профилактику осложнений, связанных с применением цитостатиков (переливание эритроцитарной массы и свежезамороженной плазмы, стерилизация кишечника неадсорбирующимися антибиотиками, гидратационная терапия, противорвотные препараты). Индукционная терапия предполагает проведение двух аналогичных курсов полихимиотерапии, консолидация ремиссии - два курса «7+3».
Поддерживающее лечение осуществляют с помощью курсов «7+3», проводимых с интервалом 6 нед в течение года с заменой даунорубицина на тиогуанин (внутрь в дозе 60 мг/м2 2 раза в день). На фоне поддерживающей терапии следует проводить контрольные стернальные пункции (1 раз в 3 мес). Профилактику нейролейкемии осуществляют посредством эндолюмбального введения цитостатиков (метотрексат, цитарабин) в сочетании с преднизолоном. Кроме этого можно проводить облучение головы.
При локализации очагов лейкемической инфильтрации в средостении, глотке и яичке проводят рентгенотерапию этих областей.
При остром лимфобластном лейкозе лечение носит постоянный, а не курсовой характер, и зависит от группы риска (стандартный или высокий).
К группе стандартного риска относят больных с пре-пре-В-, пре-В- и Т-клеточным лейкозом в возрасте от 15 до 35 лет и от 51 до 65 лет, не лечившихся по поводу данного заболевания ранее, с числом лекоцитов менее 30х109/л и при достижении ремиссии в течение 28 дней лечения.
К группе высокого риска относят больных с пре-пре-В-клеточном острым лимфобластным лейкозом, лимфобластным и Рh+-острым лимфобластным лейкозом в возрасте от 15 до 50 лет, общим пре-пре-В- и Т-клеточным острым лимфобластным лейкозом в возрасте от 35 до 50 лет при экспрессии миелоидных маркеров на лимфобластах, числом лейкоцитов более 30х109/л и при отсутствии ремиссии на 28-й день лечения.
При стандартном риске индукция ремиссии состоит из комбинации винкристина, преднизолона, даунорубицина, аспарагиназы и циклофосфамида, вводимых внутривенно и внутрь, и шести люмбальных пункций с интратекальным введением метотрексата, преднизолона и цитарабина.
Консолидацию (закрепление) ремиссии проводят в течение пяти дней на 13-й, 17-й и после проведения реиндукции на 31-й, 35-й нед лечения. Применяют два препарата - этопозид и цитарабин.
Реиндукцию ремиссии проводят с 21-й по 26-ю нед лечения и далее - через 3 мес после последнего курса консолидации с интервалом 3 мес в те-
чение двух лет. Препараты и их дозы аналогичны таковым при индукции ремиссии.
Поддерживающее лечение проводят метотрексатом и меркаптопурином через 3-4 нед после последнего курса консолидации в течение двух лет.
Профилактику нейролейкемии осуществляют с помощью эндолюмбального введения метотрексата, цитарабина и преднизолона или облучения головы с обоих латеральных полей.
В последние годы для лечения рецидива ОЛ или его профилактики применяют трансплантацию костного мозга (аллогенного или аутологичного, полученного в период ремиссии) после предварительного введения больших доз циклофосфана (по 50 мг на 1 кг массы тела в течение 4 дней) и однократного тотального облучения в дозе 10 Гр.
Инфекционные осложнения ОЛ весьма грозные, в связи с чем следует своевременно проводить активное лечение антибиотиками широкого спектра действия в достаточных дозах. Профилактика инфекционных осложнений (особенно у больных с гранулоцитопенией): тщательный уход за кожей и слизистой оболочкой полости рта, помещение больных в специальные асептические палаты, стерилизация кишечника с помощью неадсорбируемых антибиотиков (канамицин, неомицин).
При развитии геморрагического диатеза проводят переливание тромбоцитарной массы (1-2 раза в неделю) или свежей цельной крови.
Прогноз
В процессе лечения можно достичь:
• полной клинико-гематологической ремиссии (клиническая компенсация без признаков лейкозной инфильтрации селезенки, печени и других органов, нормальный или близкий к норме анализ крови, в пунктатах костного мозга число бластных клеток не превышает 5%, а общее число лимфоидных и бластных клеток - не более 40%);
• частичной клинико-гематологической ремиссии (клиническая компенсация или улучшение, небольшие изменения в анализе крови с увеличением числа зрелых клеток, исчезновением или резким уменьшением числа бластных клеток в крови и пунктате костного мозга);
• выздоровления (состояние полной клинико-гематологической ремиссии с безрецидивным течением на протяжении пяти лет и более).
При остром лимфобластном лейкозе программное лечение позволяет более чем у 50% детей добиться полной клинико-гематологической ремиссии в течение пяти лет.
Ремиссии достигают у 74-79% взрослых больных. Их продолжительность в среднем составляет два года. Прогностически наименее эффективно лечение всех форм ОЛ, сопровождающихся парциальной цитопенией и панцитопенией.
Профилактика
Первичной профилактики ОЛ не существует. Вторичная профилактика сводится к тщательному контролю состояния больного и правильному проведению противорецидивного лечения. Больных ОЛ ставят на диспансерный учет.
ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ
Среди хронических лейкозов выделяют миелопролиферативные и лимфопролиферативные заболевания. К числу хронических миелопролиферативных процессов относят ХМЛ, эритремию (истинную полицитемию), идиопатический миелофиброз (сублейкемический миелоз) и эссенциальную тромбоцитемию (хронический мегакариоцитарный лейкоз, геморрагическая тромбоцитемия).
Для всей группы миелопролиферативных заболеваний (лейкозов) характерен дефект (мутация) на уровне полипотентной стволовой клетки. В дальнейшем он проецируется на следующий класс стволовых клеток (олигопотентных) - клетки-предшественницы смешанной культуры. Они дают начало трем линиям миелоидного кроветворения - эритроцитарной, гранулоцитарной и мегакариоцитарной. Развивается миелоидная пролиферация - основной признак, характеризующий субстрат этих заболеваний, при этом продукция клеток происходит из одного или нескольких ростков миелоидного кроветворения (эритробластического, гранулоцитарного и мегакариоцитарного). Все сказанное объединяет, казалось бы, внешне различные заболевания миелопролиферативной этиологии - эритремию, ХМЛ, идиопатический миелофиброз и мегакариоцитарный лейкоз (тромбоцитемию).
К числу хронических лимфопролиферативных заболеваний относят хронический лимфолейкоз (его различные формы), а также группу парапротеинемических гемобластозов - заболеваний, при которых опухолевые клетки секретируют патологический белок (парапротеин). К ним относят множественную миелому (миеломная болезнь), болезнь Вальденстрема (макроглобулинемия) и болезнь тяжелых цепей.
Далее будут рассмотрены наиболее распространенные хронические лейкозы: ХМЛ, эритремия, хронический лимфолейкоз (ХЛЛ) и множественная миелома (ММ).
Хронический миелолейкоз
ХМЛ - хроническое миелопролиферативное заболевание, при котором отмечают усиление образования гранулоцитов (преимущественно нейтрофилов, а также промиелоцитов, миелоцитов, метамиелоцитов), служащих субстратом опухоли. В большинстве случаев закономерный исход болезни - бластный криз, характеризующийся образованием большого количества бластных клеток, рефрактерностью к лечению и заканчивающийся летально.
Этиология и патогенез
Причина патологического роста клеток - мутация клетки-предшественницы миелопоэза (частично детерминированная полипотентная клетка). Это доказывает обнаружение у больных ХМЛ специфического маркера - патологической Ph-хромосомы (филадельфийской) в клетках миелоидного, эритроидного, моноцитарного и тромбоцитарного ряда. Ph-хромосома - распространенный клеточный маркер, подтверждающий происхождение всего патологического клона клеток при ХМЛ от одной материнской. Несмотря на то что лейкозными
становятся все три ростка костного мозга, в развернутой стадии ХМЛ происходит безграничный рост, как правило, одного ростка - гранулоцитарного. В костном мозге существенно повышается продукция мегакариоцитов, а в периферической крови - тромбоцитов.
По мере течения болезни моноклоновая стадия сменяется поликлоновой, что доказывает появление клеток с различным неправильным набором хромосом. В этом выражается закон опухолевой прогрессии, которому подчиняется этот лейкоз.
ХМЛ чаще всего развивается у взрослых в возрасте 25-45 лет, при этом отмечают небольшое преобладание мужчин. ХМЛ - наиболее распространенный из всех лейкозов, составляющий 20% гемобластозов у взрослых. Средняя заболеваемость ХМЛ составляет от 3 до 11 случаев на 1 млн населения. Заболевание чаще всего диагностируют в развернутой стадии.
Классификация
ХМЛ в своем развитии закономерно проходит две стадии - моноклоновую и поликлоновую, которым соответствуют три клинические стадии течения заболевания .
• Стадия I (начальная): миелоидная пролиферация костного мозга и небольшие изменения в крови (до 1-3% бластов) без признаков интоксикации.
• Стадия II (развернутая): выраженные клинико-гематологические нарушения (интоксикация продуктами распада лейкозных клеток, увеличение печени и селезенки, миелоидная пролиферация костного мозга и изменения в крови). В периферической крови - до 10% бластов.
• Стадия III (терминальная) соответствует развитию поликлоновой опухоли: рефрактерность к проводимому цитостатическому лечению, истощение, значительное увеличение селезенки и печени, дистрофические изменения внутренних органов, выраженные изменения крови (анемия, тромбоцитопения). Для терминальной стадии ХМЛ характерно развитие так называемых бластных кризов - присутствие в периферической крови бластных клеток (до 30-90%), в связи с чем заболевание приобретает черты острого лейкоза. Чаще всего в костном мозге и периферической крови бластный криз характеризуется обнаружением миелобластов, но можно встретить и недифференцируемые бластные клетки. При исследовании кариотипа обнаруживают поликлоновость патологических клеток. Одновременно происходит значительное угнетение тромбоцитопоэза, развивается геморрагический синдром. Существует также лимфобластный вариант бластного криза (множество лимфобластов в костном мозге и периферической крови).
Клиническая картина
Клинические признаки ХМЛ представлены большими синдромами.
• Миелопролиферативный синдром, в основе которого лежит миелоидная пролиферация костного мозга, включает:
- общие симптомы, вызванные интоксикацией, пролиферацией лейкозных клеток в костном мозге, селезенке и печени (потливость, слабость,
снижение массы тела, тяжесть и боль в области селезенки и печени, оссалгии);
- увеличение печени и селезенки;
- лейкемические инфильтраты в коже;
- характерные изменения костного мозга и периферической крови.
• Синдром, обусловленный осложнениями:
- геморрагический диатез (геморрагии и тромбозы вследствие нарушения прокоагулянтного и тромбоцитарного звена гемостаза);
- гнойно-воспалительные поражения (пневмонии, плевриты, бронхиты, гнойные поражения кожи и подкожной жировой клетчатки), обусловленные резким ослаблением иммунитета;
- мочекислый диатез (гиперурикемия вследствие повышенного распада гранулоцитов).
Различная выраженность синдромов на разных стадиях болезни обусловливает полиморфность клинической картины. Некоторые пациенты не предъявляют никаких жалоб и вполне трудоспособны, другие больные с тяжелыми поражениями внутренних органов истощены и полностью теряют трудоспособность.
На первом этапе диагностического поиска в начальной стадии болезни больные могут не предъявлять жалоб, и заболевание диагностируют лишь на последующих этапах. Жалобы общего характера (слабость, потливость, снижение массы тела) могут возникать при самых разных заболеваниях, поэтому рассматривать их на первом этапе в качестве специфических для ХМЛ нельзя. Лишь позже, при обнаружении других симптомов, указывающих на ХМЛ, их можно будет интерпретировать как выражение миелопролиферативного синдрома.
Тяжесть и боли в области левого и правого подреберий обычно объясняют увеличением селезенки и печени. В сочетании с жалобами общего характера и болями в костях они могут указывать на миелопролиферативный синдром.
В терминальной стадии болезни часть жалоб может быть обусловлена возникновением гнойно-воспалительных осложнений, геморрагического и мочекислого диатеза.
На первом этапе можно получить сведения об изменениях в анализе крови и ранее проводимом лечении цитостатическими препаратами. Следовательно, если в поле зрения врача попадает больной, которому уже ставили диагноз ХМЛ, то последующий диагностический поиск значительно упрощается. Важно выяснить у больных сведения о ранее проводимом лечении и эффективности препаратов, до этого момента улучшавших общее состояние и снижавших количество лейкоцитов. Если они перестали действовать, то следует предположить переход болезни в поликлоновую (терминальную) стадию.
Ha втором этапе диагностического поиска возможно получение сведений, позволяющих высказать предположение:
• о характере патологического процесса, т.е. о существе самого заболевания;
• стадии заболевания;
• возможных осложнениях.
В развернутой и терминальной стадии обнаруживают признаки, в существенной мере подтверждающие предположение о ХМЛ: бледность кожного покрова (обусловлена нарастающей анемией), кожные геморрагии и инфильтраты (более характерно для терминальной стадии ХМЛ). Существенный признак заболевания - спленомегалия (без увеличения лимфатических узлов), сочетающаяся с увеличением печени, что при соответствующих жалобах и данных анамнеза можно расценить как симптом миелопролиферативного синдрома.
При развитии осложнений, например инфаркта селезенки, отмечают ее резкую болезненность при пальпации и шум трения брюшины над селезенкой. Постепенно орган становится плотным (масса составляет 6-9 кг, селезенка нижним полюсом спускается в малый таз).
Наиболее важные данные для диагностики ХМЛ получают на третьем этапе диагностического поиска.
В I стадии болезни в периферической крови обнаруживают лейкоцитоз (более 50х109/л) с нейтрофилезом, гранулоциты на всех стадиях созревания (миелоциты, юные, палочковидные) и эозинофильно-базофильную ассоциацию. Количество тромбоцитов не изменено (иногда немного увеличено). Редко обнаруживают небольшое количество бластов (до 1-3%). Костный мозг богат клеточными элементами с преобладанием клеток гранулоцитарного ряда. Возможно увеличение количества эозинофилов, базофилов и гранулоцитов. Обычно эти изменения в крови обнаруживают случайно (пациент может не предъявлять никаких жалоб, а к врачу обратиться по совершенно иному поводу).
Во II стадии болезни количество лейкоцитов составляет 50-500х109/л, все клетки гранулопоэза увеличены, содержание незрелых форм повышено (промиелоциты составляют 20-30%), бласты составляют до 10% всех клеток, а количество тромбоцитов снижено или увеличено. В костном мозге отмечают выраженную многоклеточность, резко выраженный сдвиг гранулоцитов влево, большое содержание промиелоцитов и число бластов около 10%.
В III стадии болезни количество лейкоцитов невелико (до 50х109/л), обнаруживают множество незрелых форм. Бласты составляют более 10% и среди них обнаруживают клетки уродливой формы. Количество тромбоцитов снижено. В костном мозге число бластов увеличено, эритро- и тромбоцитопоэз угнетен.
Функциональные свойства лейкоцитов и содержание в них ферментов изменены: снижена активность щелочной фосфатазы нейтрофилов, нарушена способность к фагоцитозу. При пункции увеличенной селезенки в развернутой стадии болезни обнаруживают преобладание миелоидных клеток, чего в норме никогда не бывает.
Этот этап считают решающим в идентификации бластного криза, и для него характерно увеличение количества бластных клеток в костном мозге и периферической крови. Суммарное число бластов и промиелоцитов составляет 20% и более, тогда как вне бластного криза оно обычно не превышает 10-15%.
Сцинтиграфия костей помогает обнаружить увеличение «плацдарма» кроветворения. Следует учитывать, что это исследование проводят при неясном диагнозе и его выполнение не считают обязательным для всех больных ХМЛ.
Диагностика
Диагностика ХМЛ в развернутой стадии болезни не представляет трудностей и основана на характерной картине крови, результатах исследования костного мозга, а также обнаружении увеличения печени и селезенки.
Диагностические критерии заболевания:
• лейкоцитоз более 20х109/л;
• присутствие в лейкоцитарной формуле пролиферирующих форм (миелобластов и промиелоцитов) и созревающих гранулоцитов (миелоцитов, метамиелоцитов);
• миелоидная пролиферация костного мозга (по данным миелограммы и трепанобиопсии);
• снижение активности щелочной фосфатазы нейтрофилов (менее 25 единиц);
• расширение «плацдарма» кроветворения (по данным сцинтиграфии костей);
• увеличение размеров селезенки и печени.
При хромосомном анализе клеток костного мозга в 95% метафаз обнаруживают Ph-хромосому. Анализ нуклеотидных последовательностей ДНК методом флюоресцентной гибридизации in situ показывает существование гена BCRABL, а проведение полимеразной цепной реакции - присутствие соответствующей ему матричной РНК.
Дифференциальная диагностика
ХМЛ следует дифференцировать от так называемых лейкемоидных реакций, которые могут возникать при ряде заболеваний (туберкулез, рак, различные инфекционные поражения, почечная недостаточность и др.). Лейкемоидная реакция представлена изменениями в крови и органах кроветворения, напоминающими таковые при лейкозах и других опухолях кроветворной системы, но не трансформирующимися в ту опухоль, на которую они похожи. При лейкемоидной реакции отмечают высокий лейкоцитоз. В периферической крови обнаруживают незрелые нейтрофилы, но базофильно-эозинофильная ассоциация отсутствует. Дифференциальная диагностика основана на обнаружении основного заболевания (рак, туберкулез и др.) и повышения активности щелочной фосфатазы нейтрофилов (вместо ее снижения при ХМЛ). Кроме того, для лейкемоидной реакции характерно увеличение содержания миелоцитов в стернальном пунктате, но Ph-хромосому никогда не обнаруживают.
Лечение
Основная задача лечения любого гемобластоза, в том числе и ХМЛ, - ликвидация или подавление роста патологического клона клеток. Применительно к хроническим лейкозам это не означает, что любого больного, у которого обнаруживают заболевание системы крови, нужно сразу же активно лечить цитостатическими препаратами, подавляющими опухолевый рост.
В начальной стадии болезни при хорошем самочувствии, но несомненных изменениях в периферической крови и костном мозге назначают общеукрепляющее лечение, правильное питание, а также соблюдение режима труда и отдыха (очень важно избегать пребывания под солнцем). Больной должен на-
ходиться под наблюдением врача. Периодически (1 раз в 3-6 мес) необходимо исследовать периферическую кровь.
При возникновении симптомов прогрессирования болезни необходимо проводить цитостатическую терапию, при этом объем лечения зависит от стадии заболевания. При отчетливых признаках опухолевого роста (увеличение размеров селезенки и печени, а также повышение числа лейкоцитов по сравнению с предшествующим периодом болезни) проводят первично сдерживающую терапию. Обычно лечение начинают при содержании лейкоцитов 50-70х109/л. При амбулаторном лечении в невысоких дозах при обязательном гематологическом контроле назначают препараты гидроксимочевины (гидроксикарбамид). После достижения клинической и (или) гематологической ремиссии решают вопрос о поддерживающей терапии.
В развернутой стадии болезни объем химиотерапии зависит от группы риска, определяемой существованием неблагоприятных признаков:
• лейкоцитоз более 200х109/л, бластемия более 3%, сумма бластов и промиелоцитов в крови - более 20%, количество базофилов в крови - более
10%;
• снижение концентрации гемоглобина менее 90 г/л;
• тромбоцитоз более 500х109/л или тромбоцитопения менее 100х109/л;
• спленомегалия (селезенку пальпируют на 10 см ниже реберной дуги и более);
• гепатомегалия (печень пальпируют на 5 см ниже реберной дуги и более). Низкий риск - существование одного признака; промежуточный риск -
присутствие 2-3 признаков; высокий риск - четыре признака и более. При низком и промежуточном риске первоначально назначают монохимиотерапию. При высоком риске с самого начала рекомендуют применение полихимиотерапии.
В развернутой стадии проводят курсовую химиотерапию. Под гематологическим контролем используют иматиниб - ингибитор тирозинкиназ того типа, который продуцирует ген BCR-ABL. Его применение нормализует количество лейкоцитов в крови и вызывает исчезновение Ph-хромосом в клетках костного мозга. Применение этого препарата позволяет увеличить продолжительность хронического течения ХМЛ и уменьшить вероятность трансформации заболевания в острый лейкоз. Препарат назначают в дозе 400 мг/м2 в течение 28 дней. При бластном кризе доза составляет 600 мг/м2 в сутки. Применение препарата приводит к полной ремиссии заболевания без эрадикации опухолевого клона.
Гидроксикарбамид в этот период применяют для контроля количества лейкоцитов в крови больного. Его можно осуществлять с помощью интерферона альфа. Этот цитокин способен предупредить развитие начинающейся бласттрансформации, а его применение увеличивает продолжительность жизни пациентов на 1-2 года. Для предупреждения повышения содержания мочевой кислоты в крови применяют аллопуринол.
Полихимиотерапию проводят курсами при высокой степени риска и в терминальной стадии ХМЛ, при бластном кризе - в объеме, соответствующем лечению ОЛ. Используют препараты, оказывающие цитостатическое действие на пролиферирующие элементы (цитарабин, метотрексат, винкристин, проти-
воопухолевый антибиотик даунорубицин). Курсы полихимиотерапии обычно короткие - 5-14 дней с перерывом 7-10 дней.
Если возраст пациента не превышает 50 лет, то эффективным лечением служит пересадка стволовых гемопоэтических клеток (лучше - от родственников, после соответствующего подбора по HLA-антигенам). У 70% больных хронической фазой болезни такая операция оказывает положительный эффект. В остальных 30% случаев пересадка стволовых клеток приводит к обострению течения заболевания.
При значительном увеличении селезенки иногда проводят облучение рентгеновскими лучами, что приводит к уменьшению ее размеров.
При гнойно-воспалительных осложнениях проводят антибиотикотерапию.
Гемотрансфузии при ХМЛ назначают при выраженном анемическом синдроме, не поддающемся цитостатической терапии, а также при лечении железодефицитной анемии препаратами железа.
Больных ХМЛ ставят на диспансерный учет и проводят периодические осмотры с обязательным гематологическим контролем.
Прогноз
Продолжительность жизни больных ХМЛ в среднем составляет 3-5 лет. У отдельных пациентов она достигает 7-8 лет. Продолжительность жизни после бластного криза редко превышает 12 мес. Применение иматиниба и интерферона альфа существенно изменяет прогноз заболевания к лучшему.
Профилактика
Точных мер предупреждения ХМЛ не существует, в связи с чем можно говорить лишь о вторичной профилактике болезни, которая состоит в предупреждении ее обострений (поддерживающее лечение, исключение инсоляции, простудных заболеваний и др.).
Эритремия (истинная полицитемия)
Эритремия - миелопролиферативное заболевание, хронический, доброкачественно текущий лейкоз, при котором отмечают усиленное образование эритроцитов, нейтрофильных лейкоцитов и тромбоцитов. Источник опухолевого роста - клетка-предшественница миелопоэза.
Заболеваемость эритремией составляет около 0,6 случая на 10 тыс. населения. Одинаково часто болеют как мужчины, так и женщины. Эритремия - болезнь лиц пожилого возраста: средний возраст заболевших - 55-60 лет, но развитие заболевания возможно в любом возрасте.
Этиология
Причины развития заболевания неизвестны.
Патогенез
В основе заболевания лежит опухолевая пролиферация всех трех ростков кроветворения - красного, гранулоцитарного и мегакариоцитарного, но до-
минирует рост красного ростка. В связи с этим основным субстратом опухоли служат созревающие в избыточном количестве эритроциты. Очаги миелоидного кроветворения образуются в селезенке и печени, чего никогда не бывает в норме. Увеличенное количество эритроцитов и тромбоцитов в периферической крови снижает скорость кровотока, повышает вязкость и свертываемость крови и обусловливает возникновение ряда клинических симптомов.
Классификация
Учитывают стадию течения процесса, вовлечение в патологический процесс селезенки и последующую трансформацию эритремии в другие заболевания системы крови.
• Стадия I (начальная): содержание гемоглобина соответствует верхней границе нормы. Характерны небольшое увеличение массы циркулирующих эритроцитов и незначительное увеличение селезенки (вследствие переполнения кровью). Возможно отсутствие изменений в ней. АД нормальное или слегка повышено. В трепанате из подвздошной кости обанруживают очаговую гиперплазию костного мозга. Продолжительность I стадии может превышать пять лет.
• Стадия II (развернутая). Фаза А протекает без миелоидной метаплазии селезенки (простой вариант плеторы без спленомегалии). Характерны тотальная трехростковая гиперплазия костного мозга и отсутствие экстрамедуллярного гемопоэза. Фаза Б протекает с миелоидной метаплазией селезенки. Возникает большой миелопролиферативный синдром: панцитоз в периферической крови, панмиелоз в костном мозге с очаговым миелофиброзом или без него, миелоидная метаплазия селезенки с фиброзом или без него.
• Стадия III (терминальная): перерождение доброкачественной опухоли в злокачественную (миелофиброз с анемизацией, ХМЛ, острый лейкоз). Миелофиброз развивается практически у всех лиц, болеющих на протяжении 10-15 лет и более, и отражает естественную эволюцию заболевания. Признак миелофиброза - цитопения (анемия, тромбоцитопения, реже - лейкопения). Развитие ХМЛ манифестирует нарастанием лейкоцитоза, увеличением или появлением в периферической крови клеток гранулоцитарного ряда (миелоцитов, промиелоцитов), а также присутствием в клетках крови и костного мозга Ph-хромосомы.
Острый лейкоз обычно развивается у больных, получавших лечение цитостатиками и радиоактивным фосфором.
Анемия у больных эритремией может быть связана с частыми кровопусканиями, возможным усиленным депонированием эритроцитов, а также их гемолизом.
Клиническая картина
Эритремия манифестирует двумя большими синдромами.
Плеторический синдром обусловлен увеличенным содержанием эритроцитов, лейкоцитов и тромбоцитов («плетора» - полнокровие). Этот синдром складывается:
• из субъективных синдромов;
• сердечно-сосудистых нарушений;
• изменений лабораторных показателей.
К субъективным симптомам плеторического синдрома относят головную боль, головокружение, нарушение зрения, стенокардитические боли, кожный зуд и эритромелалгию (внезапное возникновение гиперемии с синюшным оттенком на коже пальцев рук, сопровождающееся резкими болями и жжением). Возможно ощущение онемения и зябкости конечностей.
Сердечно-сосудистые нарушения выражаются в изменении окраски кожного покрова и видимых слизистых оболочек по типу эритроцианоза, характерной окраске слизистой оболочки в месте перехода мягкого нёба в твердое (симптом Купермана), АГ, развитии тромбоза и реже - кровоточивости. Помимо тромбозов, возможно возникновение отеков голеней и эритромелалгии. Нарушения кровообращения в артериальной системе могут приводить к тяжелым осложнениям (ИМ, инсультам, нарушению зрения, тромбозу почечных артерий).
Изменения лабораторных показателей в основном обнаруживают при клиническом анализе крови: отмечают увеличение содержания гемоглобина и эритроцитов, повышение гематокрита и вязкости крови, умеренный лейкоцитоз со сдвигом лейкоцитарной формулы влево, тромбоцитоз и резкое замедление
СОЭ.
Миелопролиферативный синдром обусловлен гиперплазией всех трех ростков кроветворения (в костном мозге и экстрамедуллярно). Он включает:
• субъективные симптомы;
• спленомегалию и (или) гепатомегалию;
• изменения лабораторных показателей.
К субъективным симптомам относят слабость, потливость, повышение температуры тела, боли в костях, а также тяжесть или боль в левом подреберье вследствие спленомегалии.
Спленомегалию (увеличение селезенки) объясняют не только миелоидной метаплазией органа (образование очагов экстрамедуллярного кроветворения), но и застоем крови. Реже отмечают увеличение печени.
Среди изменений лабораторных показателей наибольшее диагностическое значение имеет панцитоз, чаще - со сдвигом лейкоцитарной формулы влево. При трепанобиопсии обнаруживают трехростковую гиперплазию костного мозга, а в пунктате селезенки - очаги миелоидной метаплазии органа.
Различная выраженность синдромов на разных стадиях болезни обусловливает чрезвычайную вариабельность клинической картины. Можно наблюдать больных с несомненной эритремией, почти не предъявляющих жалоб и полностью трудоспособных, и пациентов с тяжелым поражением внутренних органов, нуждающихся в проведении лечения и утративших трудоспособность.
На первом этапе диагностического поиска в начальной стадии заболевания больные могут не предъявлять никаких жалоб. По мере прогрессирования болезни они связаны с существованием и выраженностью плеторы и миелопролиферативного процесса. Наиболее часты жалобы плеторического характера, обусловленные повышенным кровенаполнением сосудов и функциональными нейрососудистыми расстройствами (головная боль, эритромелалгия, наруше-
ние зрения и др.). Все эти симптомы могут быть связаны и с другими заболеваниями, что необходимо выяснить при дальнейшем обследовании больного.
Жалобы, обусловленные миелопролиферативным синдромом (потливость, тяжесть в левом подреберье, боли в костях, повышение температуры тела), также неспецифичны для эритремии. Достаточно характерен кожный зуд, который возникает после водных процедур. Этот симптом регистрируют у 55% больных в развернутой стадии заболевания и объясняют гиперпродукцией базофилов и гистаминемией. Аналогична этиология крапивницы, отмечаемой у 5-7% больных.
Перечисленные симптомы имеют значение для определения стадии эритремии: их существование обычно указывает на переход во IIБ или терминальную стадию с развитием миелофиброза как наиболее частого исхода эритремии.
В анамнезе у больных могут быть указания на мозговые инсульты и ИМ. Все это свидетельствует об осложнениях заболевания. Иногда болезнь дебютирует именно этими осложнениями, а истинную причину их развития - эритремию - обнаруживают при обследовании больного по поводу инсульта или
ИМ.
Указания на ранее проводимое лечение радиоактивным фосфором, цитостатиками или кровопусканиями могут навести на мысль о существовании какоголибо опухолевого заболевания крови. Ослабление симптомов плеторического синдрома на фоне лечения указанными средствами позволяет предположить эритремию.
На втором этапе диагностического поиска отчетливые симптомы можно обнаружить лишь во II (развернутой) стадии болезни. Как правило, это признаки плеторического синдрома: эритроцианоз, инъецированные сосуды конъюнктивы («кроличьи глаза»), отчетливая цветовая граница в месте перехода твердого нёба в мягкое. Можно отметить симптомы эритромелалгии: отек кончиков пальцев, стоп и нижней трети голени, сопровождающийся локальной гиперемией и резким жжением.
При обследовании сердечно-сосудистой системы обнаруживают АГ и увеличение левого желудочка. Для развернутой стадии болезни характерны «пестрые ноги» - изменение кожного покрова голеней (преимущественно их дистальной части) в виде образования участков пигментации различной интенсивности, обусловленных нарушением венозного кровообращения.
При пальпации живота можно обнаружить увеличение селезенки, что считают одним из характерных признаков болезни. Увеличение селезенки может быть обусловлено:
• усиленным депонированием элементов крови;
• рабочей гипертрофией органа вследствие усиления его секвестрирующей функции;
• экстрамедуллярным кроветворением (миелоидная метаплазия с преобладанием эритропоэза).
Эти причины часто сочетаются. Обнаруживаемое иногда увеличение печени обусловлено аналогичными факторами, а также развитием фиброза и неспецифического реактивного гепатита. Следует иметь в виду, что гепатомегалия может возникать при злокачественной опухоли печени с развитием вторичного эритроцитоза.
Осложнения эритремии в виде тромбоза сосудов головного мозга манифестируют рядом очаговых симптомов, обнаруживаемых при обследовании ЦНС.
На втором этапе диагностического поиска также нельзя окончательно диагностировать эритремию, так как многие ее симптомы могут присутствовать при симптоматических эритроцитозах. Кроме того, некоторые нарушения, такие, как АГ, спленомегалия и гепатомегалия, характерны для самых разнообразных заболеваний.
В связи с этим третий этап диагностического поиска приобретает решающее значение, так как позволяет:
• поставить окончательный диагноз;
• уточнить стадию эритремии;
• обнаружить осложнения;
• осуществить контроль лечения.
При исследовании периферической крови обнаруживают эритроцитоз, увеличение содержания гемоглобина и гематокрита, что возможно и при симптоматических эритроцитозах. Для диагноза имеет значение сочетание повышения концентрации гемоглобина с эритроцитозом, лейкоцитозом и тромбоцитозом. При подсчете лейкоцитарной формулы обнаруживают нейтрофилез и иногда незрелые гранулоциты.
В крови отмечают увеличение активности щелочной фосфатазы нейтрофилов, содержания витамина В12, способности сыворотки крови к связыванию витамина В12, а также концентрации мочевой кислоты.
Если изменения в периферической крови незначительны или данные неубедительны (например, эритроцитоз не сочетается с тромбоцитозом), то следует провести исследование костного мозга (трепанобиопсию). Присутствие в трепанате тотальной трехростковой гиперплазии костного мозга с преобладанием эритропоэза и обнаружение замещения жировой ткани красным костным мозгом дают возможность поставить окончательный диагноз. Расширение «плацдарма» кроветворения также обнаруживают с помощью радионуклидного сканирования костей с 32Р. При гистохимическом исследовании отмечают повышенную активность щелочной фосфатазы нейтрофилов.
Осложнения
Течение эритремии осложняют:
• сосудистый тромбоз (мозговых, коронарных, периферических артерий);
• геморрагический синдром (кровотечения после малых оперативных вмешательств (например, после экстракции зуба), из сосудов ЖКТ и геморроидальных узлов), обусловленный плохой ретракцией кровяного сгустка вследствие изменения функциональных свойств тромбоцитов;
• эндогенная урикемия и урикозурия (вследствие повышенной гибели клеток на ядерных предстадиях их созревания), манифестирующие симптомами мочекаменной болезни и подагрического артрита.
Исходы болезни - ситуации, указанные в III стадии течения заболевания, - миелофиброз, ХМЛ, ОЛ и анемия.
Диагностика
Эритремию можно заподозрить у лиц со стойким эритроцитозом в сочетании с нейтрофильным лейкоцитозом и тромбоцитозом при отсутствии заболеваний (состояний), которые могли бы вызвать эритроцитоз.
Ниже перечислены диагностические критерии эритремии в развернутой стадии.
• Категория «А»:
- увеличение массы циркулирующих эритроцитов;
- нормальное насыщение артериальной крови кислородом (более 92%);
- увеличение селезенки.
• Категория «Б»:
- лейкоцитоз более 12х109/л (при отсутствии явных причин для его возникновения);
- тромбоцитоз более 400х109/л;
- увеличение активности щелочной фосфатазы нейтрофилов (при отсутствии инфекционного заболевания);
- увеличение ненасыщенной витамин-В12-связывающей способности сыворотки крови.
Диагноз эритремии достоверен при обнаружении трех признаков категории «А» или двух признаков категории «А» и одного признака категории «Б».
Затруднения в установлении диагноза обусловлены развитием так называемого симптоматического эритроцитоза при целом ряде заболеваний. Выделяют абсолютные и относительные симптоматические эритроцитозы. При абсолютных эритроцитозах отмечают увеличение массы циркулирующих эритроцитов и повышенный эритропоэз. Для относительных эритроцитозов характерны уменьшение ОЦК и постоянство массы циркулирующих эритроцитов.
Причины развития симптоматических эритроцитозов:
• генерализованная тканевая гипоксия (легочные, сердечные заболевания, гемоглобинопатии, ожирение и т.д.);
• паранеопластические реакции (опухоли почек, коркового и мозгового вещества слоя надпочечников, гипофиза, яичников и других органов, а также сосудистые опухоли);
• ишемия почек (стеноз почечной артерии, гидронефроз, поликистоз и др.);
• неустановленные причины (заболевание ЦНС, портальная гипертензия). Относительные эритроцитозы возникают при так называемых эксикозах
(обезвоживание вследствие поноса, рвоты, повышенной потливости и др.). Дифференциальная диагностика основана на данных клинической картины. В сложных случаях определяют содержание эритропоэтина в крови. При эритремии оно не повышается, и этот факт имеет большое значение в дифференциальной диагностике.
Формулировка клинического диагноза должна включать следующие сведения :
• стадию заболевания;
• существование осложнений;
• фазу процесса (обострение или ремиссия);
• обнаружение выраженных синдромов (портальная гипертензия, АГ и др.).
Лечение
В развернутой стадии болезни при плеторическом синдроме без лейко- и тромбоцитоза в качестве самостоятельного метода лечения используют кровопускания. Извлекают по 400-500 мл крови за 1 раз через день (в условиях стационара) или два дня (в условиях поликлиники). Для профилактики тромбозов, развивающихся в результате кровопускания, назначают прием ацетилсалициловой кислоты в дозе 0,5-1 г/сут накануне и в день кровопускания, а также в течение 1-2 нед после их окончания. Кроме ацетилсалициловой кислоты назначают и другие дезагреганты - дипиридамол, тиклопидин и пентоксифиллин. Перед кровопусканием целесообразно ввести внутривенно декстран (ср. мол. масса 30 000-40 000) в дозе 400 мл, а также гепарин натрия в дозе 5000 ЕД (через иглу Дюфо). При плохой переносимости кровопусканий, отмечаемой у лиц с выраженным атеросклерозом мозговых сосудов, ограничиваются удалением всего лишь 300 мл крови (2 раза в неделю). При кровопусканиях необходимо снизить гемоглобин до 150 г/л, а гематокрит -
до 42-47%.
При недостаточной эффективности кровопусканий, а также при формах болезни, протекающих с панцитозом и спленомегалией, назначают цитостатическую терапию. Возраст больных более 55 лет расширяет показания к применению цитостатиков. Косвенными показаниями к подобному лечению служат другие признаки миелопролиферативного синдрома (зуд), а также тяжесть заболевания, висцеральные сосудистые осложнения (инсульт, ИМ) и истощение.
Противопоказания к цитостатической терапии - молодой возраст больных, рефрактерность к лечению на предыдущих этапах, а также проведение в прошлом чрезмерно активной цитостатической терапии, связанной с опасением по поводу перехода заболевания в фазу анемии.
Эффект цитостатического лечения следует оценивать через 3 мес после его окончания. Это объясняют тем, что продуцированные до лечения эритроциты живут в среднем около 2-3 мес. Снижение количества лейкоцитов и тромбоцитов наступает значительно раньше (в соответствии со сроками их жизни). Критерий эффективности цитостатической терапии - достижение гематологической ремиссии (полной, когда все показатели крови нормализуются, или частичной, при которой несколько повышенным остается количество эритроцитов, лейкоцитов и/или тромбоцитов).
Из цитостатических препаратов на первом этапе обычно назначают гидроксикарбамид по 45 мг/кг в сутки в 2-3 приема (по 2-3 капсулы в день). Во время лечения необходим контроль числа лейкоцитов. Прием препарата сочетают с применением интерферона альфа (подкожно в дозе 3-5 млн МЕ 3-7 раз в неделю длительно, т.е. не менее года).
У лиц пожилого возраста можно применять изотоп фосфора 32Р-р-излучатель. Он накапливается преимущественно в костной ткани и может подавить миелопоэз на срок до двух лет.
На исходы эритремии (миелофиброз, ОЛ, ХМЛ) воздействуют согласно принципам лечения этих заболеваний: при миелофиброзе применяют анаболические стероиды, цитостатики и переливания эритроцитной массы, при остром лейкозе - полихимиотерапию, а при ХМЛ - цитостатические препараты.
Симптоматическое лечение при приступах эритромелалгии проводят с помощью антиагрегантов и НПВС (ацетилсалициловая кислота, индометацин). АГ и приступы стенокардии лечат в соответствии со стандартными принципами.
При осложнениях эритремии тромбозом сосудов применяют антикоагулянтную и антиагрегантную терапию.
Больных эритремией ставят на диспансерный учет с частотой обращения к врачу и проведением исследования периферической крови 1 раз в 3 мес.
Прогноз
При неосложненном течении эритремии продолжительность жизни может достигать 15-20 лет. Примерно у 30% пациентов со временем развивается миелофиброз, а у 5% - миелолейкоз. Не исключено, что вероятность развития лейкоза увеличивается при лечении 32Р и некоторыми противоопухолевыми препаратами.
Если осложнения со стороны сердечно-сосудистой системы развиваются достаточно рано или же болезнь прогрессирует, то продолжительность жизни больных сокращается. Во всяком случае при прочих равных условиях своевременно начатое лечение увеличивает продолжительность жизни, хотя это происходит не во всех случаях.
Профилактика
Радикальных мер предупреждения болезни не существует, в связи с чем можно говорить лишь о вторичной профилактике, заключающейся в динамическом наблюдении за больными и проведении противорецидивного лечения.
Хронический лимфолейкоз
Хронический лимфолейкоз (ХЛЛ) - опухолевое лимфопролиферативное заболевание, первично поражающее костный мозг, при котором отмечают повышенное образование морфологически зрелых лимфоцитов, служащих субстратом опухоли.
Подобные лимфоциты функционально неполноценны, что выражается в нарушении функций иммунной системы, повышенной склонности к аутоиммунным реакциям и инфекционно-септическим заболеваниям.
ХЛЛ - одна из самых частых разновидностей лейкозов (30% всех случаев). В 95% случаев ХЛЛ имеет В-клеточное происхождение и только в 5% случаев - Т-клеточное. ХЛЛ никогда не возникает у детей, большинство больных - пожилые люди. Около 70% из них заболевают в возрасте 50-70 лет, средний возраст заболевших - 55 лет. Менее 10% пациентов заболевают в возрасте младше 40 лет. Мужчины болеют в 2 раза чаще женщин. Существует наследственно-конституциональная предрасположенность к заболеванию.
Этиология
В происхождении ХЛЛ большое значение имеют наследственная предрасположенность и нарушение иммунологической реактивности. Источник опухоли - клетка-предшественница лимфопоэза. В большинстве случаев субстратом
опухоли служат В-лимфоциты, но в ряде случаев - Т-лимфоциты. При ХЛЛ В-лимфоциты, в норме отвечающие на антиген трансформацией и образованием антител, утрачивают эту функцию. Накапливается масса иммунонекомпетентных клеток, страдают иммунитет и гемопоэз в костном мозге.
Патогенез
Выделяют следующие патогенетические особенности ХЛЛ:
• отсутствуют признаки опухолевой прогрессии (бластный криз очень редко возникает в терминальной фазе);
• нет выраженного морфологического атипизма опухолевых клеток или он крайне редок при волосатоклеточном лимфолейкозе, протекающем злокачественно;
• отсутствуют хромосомные нарушения - цитогенетический критерий злокачественности;
• отсутствует связь с мутагенными факторами (в частности, с ионизирующей радиацией);
• болезнь развивается в определенных этнических группах и характеризуется наследственно-конституциональной предрасположенностью;
• возникают расстройства иммунитета (гуморального и клеточного).
Классификация
В основу классификации положен принцип учета массы опухоли и существование или отсутствие угнетения здоровых ростков кроветворения. В соответствии с этим выделяют следующие стадии (с учетом категории риска):
• 0 - абсолютный лимфоцитоз без видимого увеличения лимфатических узлов (низкий риск);
• I - абсолютный лимфоцитоз и увеличение лимфатических желез (промежуточная категория риска);
• II - абсолютный лимфоцитоз и увеличение печени и (или) селезенки с лимфаденопатией или без нее (промежуточная степень риска);
• III - абсолютный лимфоцитоз и анемия (гемоглобин менее 119 г/л) с увеличением лимфатических узлов, печени и (или) селезенки (высокий риск) или без него;
• IV - абсолютный лимфоцитоз и тромбоцитопения с увеличением лимфатических узлов, печени и (или) селезенки (высокий риск) или без него.
Кроме этого выделяют доброкачественную и прогрессирующую форму ХЛЛ. При доброкачественной форме отмечают незначительное увеличение числа лимфоцитов в крови, очаговый (не диффузный) рост лимфоидной ткани в костном мозге и невысокое содержание пролимфоцитов. При прогрессирующей форме количество лимфоцитов в крови резко увеличено, а в костном мозге обнаруживают диффузную лимфоидную пролиферацию.
В зависимости от особенностей клинической картины болезни выделяют следующие варианты ХЛЛ:
• опухолевый - периферические лимфатические узлы резко увеличены, плотные, малоподвижные, резко выступают над поверхностью кожного покрова;
• селезеночный - в клинической картине доминирует значительное увеличение селезенки, не свойственное ХЛЛ;
• костномозговой - все изменения (лимфоидная гиперплазия) локализованы в костном мозге, лимфаденопатия и спленомегалия практически не выражена;
• пролимфоцитарный - в крови преобладают пролимфоциты;
• волосатоклеточный - при микроскопическом исследовании определяют лимфоциты с отростками протоплазмы в виде нитей («волос»).
Подобная классификация позволяет более четко определять рациональную тактику лечения.
Клиническая картина
Во многих случаях ХЛЛ протекает бессимптомно, и его диагностируют случайно, по результатам клинического исследования крови. Первые признаки ХЛЛ:
• лимфоаденопатия (обычно симметричная, безболезненная);
• потеря массы тела;
• усиленное ночное потоотделение.
В клинической картине ХЛЛ выделяют два больших синдрома.
• Лимфопролиферативный, обусловленный лимфаденопатией, спленомегалией и лимфоидной пролиферацией костного мозга:
- общие симптомы, обусловленные интоксикацией и пролиферацией лейкозных клеток в костном мозге и селезенке (кожный зуд, лихорадка, потливость, боли в костях, селезенке и печени);
- увеличение селезенки и печени;
- лейкемические инфильтраты в коже (лейкемиды);
- симптомы, связанные с увеличением регионарных лимфатических узлов (медиастинальных, мезентериальных);
- характерные изменения в костном мозге и периферической крови.
• Синдром осложнений:
- гнойно-воспалительных;
- аутоиммунных (аутоиммунная гемолитическая анемия). Различная выраженность синдромов на тех или иных стадиях болезни и вариант течения ХЛЛ определяют разнообразную клиническую картину. Все это приводит к тому, что на одних и тех же этапах диагностического поиска у разных больных можно получить самую разнообразную информацию.
На первом этапе поиска в начальной стадии развития заболевания можно не получить никаких данных.
Больные обычно достаточно рано сообщают об увеличении поднижнечелюстных и шейных лимфатических узлов, а затем - подмышечных и паховых. Прогрессирование заболевания приводит к их дальнейшему увеличению, что доставляет неудобства пациенту, точно так же, как и тяжесть в левом подреберье, обусловленная увеличением селезенки. Повышение температуры тела, потливость, снижение массы тела, носовые кровотечения и подкожные геморрагии возникают в развернутой клинико-гематологической стадии болезни.
Повышение температуры тела с преходящей желтухой обычно свидетельствует о развитии аутоиммунного гемолитического криза.
Ухудшение общего состояния (повышение температуры тела, возникновение кашля с выделением мокроты и болей в боку) возможно и при развитии легочных осложнений. Наконец, сведения, сообщаемые больным о ранее проводимом лечении (прием хлорамбуцила в различных дозах), указывают не только на суть заболевания, но и косвенно - на его стадию.
На втором этапе диагностического поиска можно получить информацию, во многом проясняющую диагноз. Прежде всего, обнаруживают увеличенные лимфатические узлы и селезенку (реже - увеличение печени). Такие симптомы, как бледность с легким желтушным оттенком кожи, подкожные геморрагии и похудание, прямого диагностического значения не имеют, но их существование знаменует либо обострение ХЛЛ, либо переход болезни в терминальную стадию. Распространенная лимфаденопатия в сочетании со спленомегалией и нередко с другими симптомами позволяет предположить ХЛЛ.
Окончательный диагноз можно поставить только на третьем этапе диагностического поиска. При исследовании периферической крови обнаруживают лейкоцитоз со значительно увеличенным содержанием В-лимфоцитов (до 80-90%), экспрессирующих антигены CD19+, CD22+ и CD5+. Очень характерно присутствие в мазке телец (теней) Боткина-Гумпрехта (раздавленные при приготовлении мазка неполноценные лимфоциты). При высоком лимфоцитозе можно отметить единичные пролимфоциты, реже - единичные лимфобласты. Распространение лимфоидной ткани в костном мозге может длительно не угнетать продукцию эритроцитов и тромбоцитов. Даже при лейкоцитозе 100х109/л анемия и тромбоцитопения могут отсутствовать. Эти изменения возникают лишь в терминальной стадии. Если эти симптомы преходящи, то следует думать об обострении лейкемического процесса в рамках развернутой стадии болезни.
В крови понижено содержание иммуноглобулинов. В некоторых случаях значительно повышена активность протеинкиназы типа ZAP-70 (плохой прогностический признак).
В пунктате костного мозга обнаруживают увеличенное содержание лимфоцитов (более 30%). Этот признак патогномоничен для ХЛЛ. В пунктате селезенки и лимфатических узлов 95-100% клеток составляют лимфоциты, есть единичные пролимфоциты и лимфобласты.
Особенности течения хронического лимфолейкоза
Характерна склонность к аутоиммунным конфликтам, вызванным образованием антител к собственным нормальным клеткам, - эритроцитам и тромбоцитам (аутоиммунная гемолитическая анемия, аутоиммунная тромбоцитопения). Подобные ситуации не связаны с тяжестью течения лейкемического процесса и могут возникать вне обострения лейкоза. Аутоиммунная гемолитическая анемия - частое осложнение, регистрируемое в 15-30% случаев.
Переход ХЛЛ в терминальную стадию чаще характеризуется развитием лимфосаркомы. Бластный криз отмечают очень редко (3-4%). Саркомный рост лимфатических узлов, определяемый по их интенсивному увеличению, каменистой плотности, инфильтрации и сдавлению окружающих тканей (не свой-
ственно ХЛЛ), сопровождается повышением температуры тела и характерной гистологической картиной.
Скорость прогрессирования ХЛЛ зависит от степени дифференцировки исходной клетки, трансформировавшейся и давшей начало патологическому клону: чем она менее дифференцирована, тем быстрее и тяжелее протекает заболевание и тем хуже прогноз.
Доброкачественная форма ХЛЛ протекает с отсутствием симптомов интоксикации, нормальными размерами периферических лимфатических узлов или их незначительным увеличением, а также небольшим увеличением селезенки, при этом количество лейкоцитов в периферической крови не превышает 30х109/л. Если число лейкоцитов и увеличивается, то это связано с развитием какого-либо неспецифического процесса. В костном мозге определяют лишь очаговую лимфоидную метаплазию. Все эти признаки противоположны симптомам прогрессирующей формы ХЛЛ. Следовательно, врач должен четко дифференцировать две формы течения болезни, что находит свое отражение в подходах к лечению.
Диагностика
Ниже представлены диагностические критерии ХЛЛ.
• Абсолютный лимфоцитоз в периферической крови более 5,0х109/лсозре- локлеточной морфологией лимфоцитов.
• Иммунофенотип лимфоцитов крови, отличающийся следующими характеристиками:
- преобладание В-клеток (на поверхностной мембране лимфоцитов обнаруживают дифференцировочные В-клеточные антигены СD19+, CD20+, CD23+ при наличии CD5+ [Т-клеточный антиген] и отсутствии других пан-Т-клеточных маркеров);
- моноклональность по отношению к экспрессии к- или λ-легких цепей иммуноглобулинов;
- низкая плотность экспрессии поверхностных иммуноглобулинов (sIg).
• Если присутствуют два вышеупомянутых критерия, то исследование костного мозга можно не проводить. Оно требуется в том случае, если абсолютный лимфоцитоз относительно низок и не превышает 5,0х109/л. В пунктате костного мозга с нормальной или повышенной клеточностью должно быть не менее 30% лимфоцитов. Его гистологическое исследование при трепанобиопсии обеспечивает врача прогностически ценной информацией. Так, диффузный тип инфильтрации коррелирует с быстро прогрессирующим течением болезни, а узловой или интерстициальный (недиффузный) тип - с лучшим прогнозом.
Лечение
Комплекс лечебных мероприятий складывается из следующих компонентов .
При отсутствии клинических симптомов и общем хорошем самочувствии, несмотря на клинически ясный диагноз, следует придерживаться выжидательной тактики и ограничиться мероприятиями общего характера: налаживанием
режима труда и быта, нормализацией содержания витаминов в пище, запрещением инсоляции и перегревания, избеганием контакта с гриппозными больными и др.
Важным считают время начала лечения. В настоящее время тактику «наблюдай и жди» применяют только в отношении больных с минимальными симптомами заболевания и лишь до тех пор, пока не возникают любые признаки прогрессирования. Иначе говоря, больные не нуждаются в лечении лишь до тех пор, пока стабильно сохраняется стадия 0-I. Показано, что раннее начало лечения не увеличивает продолжительность жизни больных, поэтому его не считают обязательным.
Показания к незамедлительному началу цитостатической терапии (достаточно соответствия одному из пунктов):
• повышение количества лейкоцитов в периферической крови более 3000х109/л в сочетании с нарушениями микроциркуляции;
• развитие анемии (содержание гемоглобина менее 90 г/л);
• тромбоцитопения менее 30х109/л в сочетании с признаками геморрагического диатеза;
• аутоиммунный гемолиз, сопровождающийся повышением температуры тела;
• значительное увеличение лимфатических узлов, приводящее к сдавлению соседних органов.
Ведущее место в лечении принадлежит хлорамбуцилу, который назначают из расчета 0,2 мг/кг массы тела. Назначают прием препарата внутрь в дозе около 12-16 мг до уменьшения числа лейкоцитов на 50%, после чего дозу уменьшают вдвое и переходят на поддерживающую терапию, применяя хлорамбуцил в дозе 4-6 мг 1 раз в 7-10 дней. Иногда оказывается эффективным добавление к хлорамбуцилу небольших доз преднизолона (по 10-15 мг). Лечение проводят все время, пока пациент отвечает на него, но не менее 8-12 мес. Ответ на терапию получают в 40-70% случаев, но полные ремиссии редки. При возникновении признаков прогрессирования болезни вновь переходят на прием препарата в полной дозе.
Реже парентерально применяют циклофосфамид в дозе 200-400 мг (ежедневно или через день) до достижения суммарной дозы 8-12 г. При необходимости, но не ранее чем через 2-4 нед после окончания предыдущего курса назначают повторное применение препарата.
При устойчивости к хлорамбуцилу или циклофосфамиду проводят полихимиотерапию, включающую винкристин (винбластин) + циклофосфамид + преднизолон. При озлокачествлении опухоли комплексную химиотерапию дополняют доксорубицином.
При аутоиммунном конфликте (гемолитическая анемия, тромбоцитопения) необходимо применять преднизолон по 60-80 мг/сут в сочетании с высокими дозами цитостатических препаратов.
В случае развития инфекционных осложнений назначают антибиотики.
При ХЛЛ антибиотикотерапию следует сочетать со средствами, усиливающими иммунитет (интерферон альфа). Гемотрансфузии проводят при выраженных анемических состояниях, не купируемых приемом препаратов железа,
а также в терминальной стадии или при торпидно текущих инфекционных процессах.
На поздних стадиях ХЛЛ может быть эффективным применение моноклональных антител против CD20+ (ритуксимаб) и антигенов Campath 10 и CD52+ (алемтузумаб).
Прогноз
Продолжительность жизни в отдельных случаях достигает 15-20 лет, а после начала химиотерапии (при прогрессировании болезни) обычно не превышает 4-6 лет. Примерно 50% больных умирают от инфекционных осложнений и 30% - от причин, не связанных с ХЛЛ. Последний иногда трансформируется в агрессивную высокозлокачественную лимфому.
Профилактика
Методов предупреждения развития ХЛЛ не существует, но родственникам больных следует избегать контакта с химическими веществами и инсоляции. Больным ХЛЛ проводят вторичную профилактику, заключающуюся в предупреждении обострений болезни.
Множественная миелома
Множественная миелома (ММ), ранее обозначаемая как миеломная болезнь или плазмоцитома, - опухоль, возникающая на уровне ранних предшественников В-лимфоцитов, при этом моноклональный пул потомков первично трансформированной клетки сохраняет способность к дифференцировке до конечного этапа - плазматических клеток, секретирующих иммуноглобулины. Следовательно, субстратом опухоли служат плазматические клетки. С этим связано и более раннее ее название - плазмоцитома. Так как опухоль продуцирует патологический иммуноглобулин - парапротеин, то ее относят к группе парапротеинемических гемобластозов. Учитывая то, что она происходит из ранних предшественников В-лимфоцитов, ее относят к группе лимфопролиферативных заболеваний.
Заболеваемость в среднем составляет 50 случаев в год на 1 млн населения. ММ представлено 15% всех случаев лимфоидных злокачественных новообразований и 2% всех типов злокачественных опухолей. Мужчины болеют немного чаще женщин. Пик заболеваемости приходится на возраст около 67-71 года. В молодом возрасте (до 40 лет) ММ регистрируют крайне редко. Случаи заболевания детей не зарегистрированы.
Этиология
Причины заболевания, как и этиология опухолей вообще, неизвестны.
Патогенез
В основе заболевания лежит пролиферация плазматических клеток. Плазмоцит (плазматическая клетка) происходит из коротко живущих В-лимфоцитов и обладает способностью вырабатывать неограниченное количество антител,
специфических практически для любого антигена. При ММ все клетки, составляющие массу опухоли, происходят из одной клетки клона, потомки которой повторяют функцию клетки-родоначальницы и в большом количестве секретируют иммуноглобулин лишь одной структуры (моноклоновый иммуноглобулин). Количество нормальных плазматических клеток уменьшается, как и содержание нормальных иммуноглобулинов, выполняющих функцию антител. В связи с этим возникает иммунодефицитное состояние, способствующее развитию инфекционных осложнений. Продуцируемый лейкозными В-лимфоцитами и (или) связанными с ними дополнительными клетками ИЛ-6 потенцирует рост и развитие плазматических клеток, а другие высвобождаемые цитокины (ФНО-а и ИЛ-1) ускоряют резорбцию костной ткани. При генетическом анализе обнаруживают мутации в онкогенах и транслокации в хромосомах.
Первоначально опухоль локализуется в костном мозге. В дальнейшем опухолевые клетки (плазмоциты) метастазируют в органы (селезенку, печень). Увеличенное количество плазматических клеток в костном мозге в дальнейшем вытесняет эритробластический и миелоцитарный ростки костного мозга.
Классификация
В основу современной классификации положены объем опухолевой ткани (стадии течения) и активность патологического процесса (степень агрессивности гемобластоза).
• I стадия (малая масса опухоли): концентрация гемоглобина более 100 г/л, нормальное содержание кальция в крови, нет остеолиза или очагового поражения костей, низкая концентрация IgM. Содержание IgG менее 50 г/л, IgA - менее 30 г/л. Выведение белка Бен-Джонса - менее 4 г/сут. Содержание креатинина в крови не увеличено.
• II стадия (средняя масса опухоли): показатели средние между таковыми в I и III стадии болезни.
• III стадия (большая масса опухоли): концентрация гемоглобина менее 85 г/л, содержание кальция в крови выше нормы, выраженный остеодеструктивный процесс, высокая концентрация IgM при содержании IgG более 70 г/л, IgA - более 50 г/л. Экскреция белка Бен-Джонса с мочой - 12 г/сут. Содержание креатинина в крови повышено.
Активность патологического процесса определяют следующим образом:
• «тлеющая» ММ (малоагрессивная) - признаки прогрессирования отсутствуют в течение многих месяцев или лет;
• медленно прогрессирующая;
• быстро прогрессирующая (агрессивная).
Все эти показатели помогают не только оценить особенности патологического процесса, но и позволяют подобрать оптимальное лечение.
Анатомически (на основании данных рентгенологического исследования скелета и цитологического и патоморфологического анализа пунктатов и трепанатов костей) выделяют следующие формы ММ:
• диффузно-очаговую (наиболее распространенную, около 60% больных);
• диффузную (24%);
• множественно-очаговую (15%);
• редкие формы (склерозирующая, преимущественно висцеральная - 1%).
Выделение анатомических форм оправдано с точки зрения возможности получения при первой же стернальной пункции субстрата болезни (увеличенного числа плазматических клеток).
Клиническая картина
Симптомы заболевания определяют несколько больших синдромов - костномозговой, белковых нарушений и висцеральный.
Костномозговой синдром обусловлен пролиферацией в костном мозге миеломных клеток, что приводит к разрушению костного вещества. В первую очередь деструктивные процессы (остеопороз, остеолиз) развиваются в плоских костях и позвоночнике. Иногда первые очаги разрушения обнаруживают в проксимальных отделах трубчатых костей. Гиперплазия костного мозга вследствие разрастания скоплений миеломных клеток также приводит к вытеснению миелоидных элементов. В результате перечисленных процессов развиваются:
• остеопороз, патологические переломы, гиперкальциемия;
• анемия, лейкопения, тромбоцитопения (реже) в периферической крови;
• миеломноклеточная метаплазия в костном мозге.
Синдром белковых нарушений обусловлен гиперпродукцией моноклонового парапротеина плазматическими клетками, уменьшением секреции нормальных иммуноглобулинов и представлен следующими признаками:
• миелоидной нефропатией;
• параамилоидозом;
• геморрагическим диатезом;
• синдромом повышенной вязкости;
• периферической невропатией;
• синдромом недостаточности антител (с развитием инфекционных осложнений).
Миелоидная нефропатия - наиболее частый и серьезный симптом парапротеинемии. Она приводит к почечной недостаточности, которая занимает одно из первых мест среди причин смерти больных. В основе развивающейся почечной недостаточности лежит нефросклероз. Его причиной служит реабсорбция в канальцах белка, в большом количестве фильтрующегося в клубочках. Это связано с тем, что за счет парапротеина в крови значительно увеличено содержание белка. Реабсорбируемый парапротеин инфильтрирует ткань почки, способствуя развитию склероза. Доказано раннее вовлечение в патологический процесс базальной мембраны капилляров нефрона и мезангиума с их последующим склерозированием. Клинические признаки миелоидной нефропатии складываются из упорной (иногда многолетней) протеинурии и постепенно развивающейся ХПН. Особенность поражения почек - отсутствие отеков и симптомов сосудистого поражения (АГ, ретинопатии).
Амилоидоз LA-типа - тканевый парапротеиноз, регистрируемый в 15% случаев. В отличие от классического вторичного амилоидоза отмечают поражение органов, богатых коллагеном: сосудов (адвентиции), сердца, языка, суставов и сухожилий. Печень, селезенка и почки не страдают. Параамилоидоз не всегда
сопровождается клиническими симптомами и часто бывает лишь патологоанатомической находкой. Тем не менее в ряде случаев можно обнаружить макроглоссию, прогрессирующую сердечную недостаточность, а также упорные боли в суставах с их деформацией. Прижизненная диагностика параамилоидоза затруднена; необходима биопсия кожи, слизистых оболочек (рта, прямой кишки), лимфатических узлов и мышц.
Геморрагический синдром - редкое явление. Кровоточивость из сосудов слизистых оболочек и кожи обусловлена тем, что парапротеин, «окутывая» тромбоциты, затрудняет их адгезию и агрегацию.
Синдром повышенной вязкости - нарушение микроциркуляции вследствие высокой гиперпротеинемии - манифестирует геморрагической ретинопатией, расширением вен сетчатки и нарушениями периферического кровотока вплоть до акрогангрены. При охлаждении тела эти явления могут усиливаться вследствие выпадения криоглобулинов.
Периферическую невропатию регистрируют в 5% случаев. Она выражается в нарушениях тактильной и болевой чувствительности, а также в парестезиях. При гистологическом исследовании обнаруживают дегенеративные изменения нервных волокон.
Синдром недостаточности антител обусловлен резким снижением концентрации нормальных иммуноглобулинов вплоть до их полного исчезновения. Вторичная гипогаммаглобулинемия приводит к выраженной склонности к инфекционным осложнениям, особенно со стороны мочевыводящих путей и бронхолегочного аппарата.
Висцеральный синдром заключается в лейкемической инфильтрации внутренних органов (главным образом, печени и селезенки). В 5-12% случаев при жизни больных обнаруживают гепато- и спленомегалию. Опухолевые плазмоклеточные инфильтраты можно обнаружить практически во всех внутренних органах, но они редко манифестируют клинически и обычно служат патологоанатомическими находками.
Различная выраженность перечисленных синдромов и степени нарушений белкового обмена обусловливает чрезвычайную вариабельность течения болезни. Можно наблюдать пациентов с несомненной ММ, предъявляющих небольшое число жалоб или вообще не отмечающих никаких нарушений. В то же время есть больные, нуждающиеся в проведении постоянного лечения и утратившие трудоспособность вследствие тяжелой инвалидности, связанной с патологическими переломами (прежде всего компрессионными переломами позвоночника).
Заболевание можно обнаружить на разных стадиях течения, но у ряда больных (особенно среди тех, у кого ММ диагностировали рано) можно выделить две стадии болезни:
• относительно доброкачественную, характеризующуюся соматической компенсацией, отсутствием или медленным прогрессированием остеодеструктивного процесса, нормальными показателями крови, стабильно невысоким содержанием патологического иммуноглобулина (парапротеина) и сохранностью нормальных иммуноглобулинов;
• быстро прогрессирующую, при которой нарастает разрушение костей, возникают метастазы во внутренние органы, концентрация парапротеина
резко повышается, а содержание нормальных иммуноглобулинов резко снижается вплоть до развития выраженной гипогаммаглобулинемии, при этом развиваются анемия, лейкопения и повышается количество плазмобластов.
Все вышесказанное обусловливает получение самых разных данных на всех этапах диагностического поиска.
На первом этапе диагностического поиска в начальной стадии болезни больные могут не предъявлять жалоб, и болезнь диагностируют после соответствующего обследования на основании случайного обнаружения протеинурии или значительного увеличения СОЭ (диспансеризация, обращение к врачу по иным причинам), что обычно отмечают в 20% случаев.
Больные могут замечать, что у них в течение многих лет обнаруживают увеличение СОЭ (иногда довольно значительное - до 50-60 мм/ч), при этом тщательное обследование, как правило, направленное на определение злокачественной опухоли, не обнаруживало причины болезни. Как правило, стернальную пункцию или трепанобиопсию не проводили. В половине случаев болезнь дебютирует слабостью, повышенной утомляемостью, снижением массы тела и болями в костях. Иногда заболевание сразу же манифестирует сильными болями в костях или переломами ребер, гребней подвздошных костей, а также компрессионными переломами позвонков. Часто больные страдают вялотекущими пневмониями, которые нередко рецидивируют и плохо поддаются лечению антибиотиками. Также регистрируют заболевания мочевыводящих путей (циститы, пиелиты), характеризующиеся дизурическими расстройствами и упорным субфебрилитетом.
В анамнезе у больных могут быть указания на ранее проводимое лечение цитостатическими препаратами, а также сеансы плазмафереза, после чего их состояние улучшалось.
На втором этапе диагностического поиска в начальных стадиях болезни нередко не обнаруживают никаких патологических изменений. В развернутой стадии заболевания иногда обнаруживают нарушения, обусловленные вышеуказанными синдромами (костномозговым, висцеральным, белковых нарушений). При вялотекущих пневмониях можно отметить участки укорочения перкуторного звука и стойких влажных звонких мелкопузырчатых хрипов. Как правило, обнаруживают болезненность при поколачивании плоских костей. При их деструкции (патологические переломы) образуются участки резкой болезненности и нарушается функция пораженной кости. Неспецифические симптомы - снижение массы тела, субфебрилитет и повышенная потливость.
Следует отметить, что необнаружение симптомов, обусловленных вышеперечисленными синдромами, на втором этапе диагностического поиска отнюдь не отвергает предположение о ММ, но свидетельствует об отсутствии грубых изменений со стороны пораженных органов и систем.
Третий этап диагностического поиска считают решающим для установления диагноза.
При исследовании периферической крови не обнаруживают специфических признаков. У всех больных по мере прогрессирования заболевания развивается анемия, патогенез которой, вероятно, связан с вытеснением нормального кро-
ветворения растущей опухолью. Тем не менее прямой зависимости между степенью анемии и выраженностью костных поражений нет.
Число лейкоцитов и лейкоцитарная формула обычно не изменены. Иногда отмечают нейтропению с относительным лимфоцитозом, реже - умеренный нейтрофилез со сдвигом лейкоцитарной формулы влево и появлением молодых форм гранулоцитарного ряда. При прогрессировании заболевания обнаруживают выраженную лейко- и нейтропению (особенно при лечении цитостатическими препаратами). Часто регистрируют абсолютный моноцитоз.
Мегакариоцито- и тромбоцитопоэз долгое время не изменены. На ранних стадиях иногда отмечают гипертромбоцитоз и увеличение числа мегакариоцитов в пунктате костного мозга.
Характерно значительное увеличение СОЭ (до 60-80 мм/ч).
При анализе миелограммы обнаруживают отчетливую миеломноклеточную пролиферацию: количество миеломных опухолевых клеток превышает 10%, часто отмечают патологические клетки - многоядерные или аномальной формы.
Если диффузного поражения костного мозга нет (лишь «гнездное» поражение), то миелограмма может соответствовать норме. В этой ситуации при подозрении на плазмоцитому (остеолитические очаги, моноклональная иммуноглобулинопатия) необходимо проводить повторные проколы грудины в разных участках, пунктировать или трепанировать гребни подвздошной кости, проводить пункции в местах остеолитических дефектов или костных опухолей.
При биохимическом исследовании закономерно регистрируют гиперпротеинемию: содержание общего белка достигает 10-12 г/л. При электрофоретическом исследовании обнаруживают дополнительную фракцию (М-градиент) в области у-глобулиновой фракции, при этом количество нормальных у-глобулинов резко снижено. Эта дополнительная фракция служит отражением высокой концентрации парапротеина в крови. При исследовании содержания иммуноглобулинов отмечают резкое увеличение концентрации какого-либо класса - IgA, G, Е или D, но не IgM, что свойственно макроглобулинемии Вальденстрема - другому парапротеинемическому гемобластозу, обусловленному гиперплазией короткоживущих В-лимфоцитов. Часто в крови повышено содержание в2-макроглобулина. Чем выше концентрация этого белка, тем хуже прогноз у пациента.
При иммуноэлектрофорезе удается провести более детальное типирование парапротеина при миеломной болезни: определяют класс тяжелых цепей парапротеина - A, G, Е или D, а также тип легких цепей - к (каппа) или λ (лямбда). Возможно развитие особого варианта ММ - так называемой миеломы БенДжонса, парапротеин которой состоит лишь из легких цепей (микромолекулярный вариант болезни).
В моче достаточно часто можно определить разной степени протеинурию. При миеломе Бен-Джонса в ней присутствует одноименный белок. Нагревание мочи приводит к его выпадению в осадок, а дальнейшее нагревание - к растворению.
При рентгенологическом исследовании костей можно обнаружить изменения плоских костей (особенно костей черепа) в виде круглых просветлений в
костной ткани, представляющих участки ее резорбции. Можно также отметить переломы костей, особенно компрессионные переломы тел позвонков.
Следует помнить, что не существует специфических изменений скелета, характерных для ММ. Отсутствие остеодеструкции не исключает ММ, а ее обнаружения недостаточно для установления диагноза, так как для этого необходимы другие признаки, о которых будет сказано ниже.
Гиперкальциемию регистрируют в 20-40% случаев, чаще - в терминальной стадии болезни (особенно при ХПН).
При ХПН присутствуют все ее лабораторные признаки: снижение плотности мочи, уменьшение СКФ и увеличение концентрации креатинина в крови.
Диагностика
Для установления диагноза ММ используют две группы критериев.
• Большие критерии ММ:
- плазмоклеточная инфильтрация костного мозга (по данным трепанобиопсии);
- увеличение количества плазматических клеток в миелограмме более 35%;
- концентрация IgG более 35 г/л, IgA - свыше 20 г/л (по данным электрофореза сыворотки крови), содержание легких цепей иммуноглобулинов при отсутствии признаков амилоидоза более 1,0 г в суточном объеме мочи (по данным электрофореза мочи).
• Малые критерии ММ:
- содержание плазматических клеток в костном мозге около 10-30%;
- присутствие моноклонального иммуноглобулина (по данным электрофореза сыворотки крови), но в меньшей концентрации;
- обнаружение очагов остеолиза;
- содержание иммуноглобулинов, не превышающее для IgM 0,5 г/л, для IgA - 1 г/л, для IgG - 6 г/л.
Диагноз ММ устанавливают при обнаружении одного большого и одного малого или одного большого и двух малых критериев (1+1 или 1+2).
Трудности в диагностике ММ возникают на ее ранних стадиях, когда отсутствует костная деструкция, нет отчетливой миеломноклеточной метаплазии костного мозга, невелик М-градиент при электрофорезе белков сыворотки и нет выраженного снижения содержания γ-глобулинов. Эти стадии течения ММ неотличимы от так называемых эссенциальных (при беременности, у лиц пожилого возраста) и симптоматических (при циррозе печени, диффузных заболеваниях соединительной ткани, злокачественных опухолях, сепсисе) моноклональных гаммапатий. Тщательное исследование позволяет исключить реактивную гаммапатию. Этому также способствует динамическое наблюдение за больными. Следует помнить, что на ранних стадиях болезни, когда больной попадает в поле зрения врача, правильный диагноз может быть поставлен через несколько лет после обнаружения парапротеина в крови.
Дифференциальная диагностика
ММ необходимо дифференцировать от ряда заболеваний и состояний.
• Макроглобулинемия Вальденстрема - одна из опухолей лимфатической системы, рассматриваемая в рамках парапротеинемических гемобластозов, так как речь идет о пролиферации в системе лимфоцитов (источник продукции IgM). Этим заболеванием страдают преимущественно мужчины (до 70%) в возрасте около 60 лет. Клиническая картина чрезвычайно сходна с ММ и обусловлена лейкемической пролиферацией лимфоидных элементов костного мозга, печени, селезенки и лимфатических узлов, накоплением в сыворотке крови парапротеина, тяжелую цепь которого относят к М-классу. Костно-деструктивный процесс развивается редко, болевой синдром обычно отсутствует. Характерна гепато- и спленомегалия. Увеличение печени, селезенки и лимфатических узлов связано с разрастанием лимфатических элементов. Картина костного мозга характеризуется увеличением лимфоцитов, но повышено и количество плазматических клеток. Все остальные синдромы при макроглобулинемии Вальденстрема достаточно выражены, но в отличие от ММ поражение почек обнаруживают редко, что, вероятно, связано с отсутствием гиперпротеинемии и протеинурии. Главное отличие макроглобулинемии Вальденстрема от ММ состоит в обнаружении парапротеина класса IgM.
• Доброкачественная моноклоновая гаммапатия (синоним: моноклоновая гаммапатия неясной этиологии) - вялотекущее заболевание, характеризующееся стабильным и относительно невысоким (менее 20 г/л) содержанием парапротеина в сыворотке крови. Это состояние регистрируют чаще ММ. Концентрация нормальных иммуноглобулинов не снижена. Отсутствуют поражение костей и белок Бен-Джонса в моче, а число плазматических клеток в костном мозге обычно не превышает 10%. У 10-30% больных это заболевание медленно трансформируется в ММ или лимфому.
• При первичном амилоидозе доля плазматических клеток в костном мозге обычно не превышает 10%, костных поражений нет. В моче может присутствовать белок Бен-Джонса, а в сыворотке крови - небольшое количество парапротеина. Иногда первичный амилоидоз трансформируется
в ММ.
• Солитарная плазмоцитома может развиться как в кости, так и в мягких тканях. В сыворотке крови может присутствовать небольшое количество парапротеина. Иногда плазмоцитома может прогрессировать до ММ.
• Плазмоклеточный лейкоз - тяжелое, быстро прогрессирующее заболевание, при котором в крови циркулирует большое количество плазматических клеток. Прогноз, как правило, неблагоприятный.
Лечение
Современное лечение ММ включает применение цитостатических средств, глюкокортикоидов и анаболических гормонов, восстановительные методы, а также комплекс мероприятий, устраняющих или предупреждающих развитие метаболических нарушений и вторичного иммунодефицита.
Если заболевание диагностируют рано (I, частично II стадия болезни), то при отсутствии клинических симптомов, нормальных показателях крови (СОЭ
не учитывают) и функций почек противоопухолевое лечение начинать не следует. Рекомендована выжидательная тактика с ежемесячным контролем показателей крови, мочи и секреции моноклонального парапротеина. У части таких больных существует «тлеющая» ММ, которая в течение нескольких лет не прогрессирует и не нуждается в лечении.
Лечение следует начинать при возникновении симптомов нарастания опухолевой массы (снижение концентрации гемоглобина и количества эритроцитов, повышение содержания парапротеина в крови или моче, сильные боли в костях).
При проведении цитостатической химиотерапии следует придерживаться определенных принципов.
• Подбор цитостатического препарата осуществляют с учетом стадии болезни (величины опухолевой массы) и критериев риска.
• Оценку эффективности лечения следует проводить на основании определенных признаков:
- снижение концентрации парапротеина в сыворотке крови более чем на 50%;
- снижение экскреции белка Бен-Джонса более чем на 50%;
- рентгенологические признаки заживления костных деструкций;
- уменьшение площади пораженных опухолью костей.
• Требуется непрерывное лечение с соблюдением доз и интервалов в течение двух лет и более.
Применяют комбинацию цитостатического препарата мелфалана с преднизолоном. Существуют различные подходы к назначению этих лекарственных средств.
У больных с III стадией болезни при отсутствии явных признаков агрессивности (медленно прогрессирующая ММ) проводят пролонгированное лечение с поддерживающей терапией ударными прерывистыми курсами. Мелфалан сочетают с преднизолоном; одновременно назначают анаболические стероиды. Через 4 нед назначают поддерживающее лечение меньшими дозами используемых препаратов.
Еще один вариант пролонгированного лечения - применение винкристина в сочетании с мелфаланом и преднизолоном. Кроме того, возможно использование циклофосфана и преднизолона.
Другая методика - ударная прерывистая терапия - рекомендована больным с медленно прогрессирующей ММ I и II стадии. Применяют более короткие курсы лечения теми же препаратами - мелфаланом (циклофосфаном) в сочетании с преднизолоном.
При быстропрогрессирующей ММ с симптомами, указывающими на плохой прогноз, и резистентностью к ранее проводимому лечению назначают полихимиотерапию. В течение 3-4 нед назначают комбинацию винкристина, циклофосфана, мелфалана и преднизолона.
У молодых больных с резистентностью к лечению и отсутствием серьезных соматических заболеваний применяют так называемую интенсивную терапию.
Она включает использование высоких доз мелфалана в сочетании с трансплантацией костного мозга и тотальным облучением.
Для лечения ММ также применяют интерферон альфа, который не имеет самостоятельного значения, но рекомендовано его использование одновременно с химиотерапией, а также в перерывах между курсами. Интерферон альфа подавляет пролиферацию клона опухолевых клеток.
Лечение считают эффективным только у тех больных, у которых обнаруживают стабильность или улучшение показателей красной крови и содержания сывороточного альбумина, а также отсутствие увеличения размеров остеодеструктивных очагов. Эти критерии чрезвычайно важны, так как ориентация на степень снижения концентрации парапротеина не всегда верна: прямая зависимость между опухолевой массой и уровнем секреции парапротеина может быть весьма различной. Эффективность лечения оценивают через 3 мес после его начала. При отсутствии признаков улучшения больных относят к прогностически весьма неблагоприятным или так называемым нереагирующим.
Назначение локальной лучевой терапии рекомендовано во всех случаях угрозы патологических переломов позвоночника, крестцово-подвздошной области, бедренных и берцовых костей, даже при отсутствии болевого синдрома. Локальное облучение применяют при ограниченных опухолевых узлах в костях и мягких тканях, а также радикулярных болях, связанных со сдавлением корешков спинного мозга опухолью. Сочетать лучевое лечение и химиотерапию не рекомендуют.
На ранних стадиях ММ некоторым больным рекомендуют трансплантацию донорских стволовых клеток (с обычными условиями).
При инфекционных осложнениях следует применять антибиотики, не обладающие нефротоксичностью. При выраженной протеинемии и парапротеинемии проводят плазмаферез. При поражении костной ткани (переломы и др.) необходимо применять комплекс средств, улучшающих костную репарацию (внутримышечно миокальцик*э или прием внутрь препаратов кальция). Бифосфонаты весьма эффективны при лечении поражений костей, сопровождающих ММ, и могут увеличить продолжительность жизни пациентов. При переломах костей проводят иммобилизацию и вытяжение на щите (особенно при компрессионных переломах позвоночника).
Прогноз
Больные с I стадией ММ могут жить многие годы без какого-либо лечения. При развитии III стадии заболевания средняя продолжительность жизни больного составляет 2-3 года. Современное комбинированное лечение увеличивает ее: удается восстановить активность пациентов и поддерживать их удовлетворительное состояние. Больные погибают вследствие развития ХПН или инфекционных осложнений.
АНЕМИИ
Анемия - состояние, характеризующееся уменьшением содержания гемоглобина в единице объема крови, вследствие снижения его общей концентрации в организме. В большинстве случаев анемия сопровождается снижением количества эритроцитов в единице объема крови. От истинной анемии следует отличать гидремию - разжижение крови за счет тканевой жидкости.
Необходимо отметить, что на сегодняшний день единой общепризнанной классификации анемий не существует.
Поскольку в основе развития анемий лежат различные патологические процессы, с патогенетической точки зрения предложено разделять все анемии на следующие группы.
• Анемии, обусловленные нарушением синтеза гемоглобина:
- железодефицитная анемия (нарушение синтеза гема);
- сидероахрестическая анемия (нарушение синтеза порфирина);
- анемия хронических заболеваний.
• Анемии вследствие нарушения образования и созревания эритроцитов (дисэритропоэтические анемии):
- В12-дефицитная анемия;
- фолиеводефицитная анемия.
• Анемии, обусловленные нарушениями пролиферации клеток костного мозга (гипопролиферативные анемии):
- идиопатическая гипопластическая (апластическая) анемия;
- вторичная гипопластическая (апластическая) анемия (вследствие действия лекарственных средств, токсинов, ионизирующей радиации и др.);
- миелофиброз (первичный или вторичный);
- замещение клеток костного мозга опухолевыми клетками (миелофтизис);
- миелодисплазия.
• Анемии вследствие усиленного разрушения эритроцитов (гемолитические анемии):
- аутоиммунные гемолитические анемии;
- наследственная микросфероцитарная гемолитическая анемия (болезнь Минковского-Шоффара);
- талассемия;
- пароксизмальная ночная гемоглобинурия.
• Анемии вследствие дефицита эритропоэтина.
• Анемии со смешанным механизмом развития.
Каждый из указанных патогенетических вариантов анемических состояний имеет различную этиологию (например, железодефицитная анемия может возникать при мено-, метроррагиях, кровотечениях из ЖКТ, при беременности, нарушении всасывания железа и др.). В ряде случаев самый тщательный диагностический поиск не позволяет обнаружить лежащее в основе анемии заболевание. В этом случае следует говорить об идиопатической форме анемии. Именно поэтому при обследовании больного с предполагаемой анемией необходимо:
• определить патогенетический вариант анемии;
• определить заболевание, лежащее в основе анемии. Симптомы анемий чрезвычайно разнообразны и определяются:
• патогенетическим вариантом анемии;
• этиологией;
• изменениями в организме, которые обусловлены его реакцией на гипоксию тканей, вызванной нарушением дыхательной функции крови (доставка кислорода тканям), - циркуляторно-гипоксическим синдромом.
Циркуляторно-гипоксический синдром манифестирует слабостью, повышенной утомляемостью, одышкой при физической нагрузке, сердцебиениями, «анемическим» шумом в крупных сосудах, увеличением ОЦК и ускорением кровотока. В большей или меньшей степени он выражен при всех видах анемических состояний, а степень его выраженности зависит от уровня гипоксии тканей, что, в свою очередь, определяется кислородной емкостью крови.
Согласно классификации ВОЗ, в зависимости от тяжести выделяют:
• анемии легкой степени тяжести (концентрация гемоглобина не ниже 90 г/л);
• анемии средней степени тяжести (концентрация гемоглобина в пределах 90-70 г/л);
• анемии выраженной степени тяжести или тяжелые (концентрация гемоглобина менее 70 г/л).
Помимо этого анемии классифицируют в зависимости от средних размеров эритроцитов и степени их насыщения гемоглобином. Последнюю определяют с помощью цветового показателя и по средней концентрации гемоглобина в эритроцитах (Mean Corpuscular Hemoglobin - МСН).
Цветовой показатель отражает степень насыщения эритроцитов гемоглобином. Его рассчитывают, умножив концентрацию гемоглобина (г/л) на 3 и разделив на первые две цифры числа эритроцитов.
В норме величина цветового показателя колеблется от 0,8 до 1,1.
В зависимости от насыщения эритроцитов гемоглобином все анемии можно разделить:
• на нормохромные (цветовой показатель от 0,8 до 1,1 либо МСН от 30 до
36 г/дл);
• гипохромные (цветовой показатель менее 0,8 либо МСН ниже 30 г/дл);
• гиперхромные (цветовой показатель выше 1,1 либо МСН превышает
36 г/дл).
В зависимости от величины среднего объема эритроцитов, все анемии можно разделить:
• на нормоцитарные (средний объем эритроцитов от 80 до 100 мкм3);
• микроцитарные (средний объем эритроцитов менее 80 мкм3);
• макроцитарные (средний объем эритроцитов более 100 мкм3).
ЖЕЛЕЗОДЕФИЦИТНАЯ АНЕМИЯ
Сущность железодефицитной анемии (ЖДА) состоит в недостатке железа в организме (истощение его запасов в органах-депо), вследствие чего нарушается синтез гемоглобина, и каждый эритроцит содержит меньшее, чем в норме, количество гемоглобина. ЖДА регистрируют чаще остальных форм анемий, что можно объяснить множеством обстоятельств, приводящих к дефициту железа в организме.
Этиология
Существует несколько основных причин дефицита железа.
• Скрытые (оккультные) кровотечения:
- желудочно-кишечные (язвенная болезнь, геморрой, рак, диафрагмальная грыжа, НЯК, полипоз желудка и кишечника);
- маточные (дисфункция яичников, фибромиома матки, рак шейки матки, эндометриоз и др.);
- легочные (рак, бронхоэктазы, изолированный легочный гемосидероз).
• Недостаточное потребление железа с пищей.
• Повышенный расход железа:
- беременность, лактация;
- период роста и полового созревания;
- хронические инфекционные заболевания, опухоли.
• Нарушение всасывания железа:
- резекция желудка;
- энтерит;
- спру.
• Нарушение транспорта железа (дефицит белка плазмы крови трансферрина).
Из перечисленных причин следует, что ЖДА чаще развивается у женщин в результате обильных маточных кровотечений и повторных беременностей, а также у подростков.
Патогенез
Важнейшая функция железа в организме - его участие в синтезе гема, служащего составной частью гемоглобина. При дефиците железа прежде всего возникает нарушение синтеза гемоглобина, что приводит к развитию ЖДА. Недостаточное образование гемоглобина служит причиной гипоксии тканей и развития циркуляторно-гипоксического синдрома. Дефицит железа также способствует нарушению синтеза тканевых ферментов, что приводит к изменению тканевого метаболизма, при этом происходит поражение быстро обновляющихся эпителиальных тканей - слизистой оболочки ЖКТ, кожи и ее дериватов. Патогенез ЖДА представлен на рис. 5-2.
В организме здорового взрослого человека общее количество железа составляет 3-4 г, при этом у женщин оно несколько меньше, чем у мужчин, что связано с ежемесячными потерями крови во время менструаций. При нормальном питании с пищей поступает 10-20 мг железа в сутки, но только 10% всасывается в двенадцатиперстной кишке и в верхних отделах тонкой кишки. Примерно такое же количество железа ежедневно образуется вследствие физиологического гемолиза эритроцитов.
Наибольшее количество железа содержится в чечевице, желтке куриного яйца и мясных продуктах (говядина).
Клиническая картина
Клиническая картина болезни, как это вытекает из схемы патогенеза, складывается из следующих синдромов:
• циркуляторно-гипоксического (при достаточной выраженности анемии и кислородного голодания тканей);
Рис. 5-2. Патогенез железодефицитной анемии
• поражения эпителиальных тканей (гастроэнтерологические расстройства, трофические нарушения кожи и ее дериватов);
• гематологического (анемия гипохромного типа и признаки дефицита железа).
Кроме этих синдромов, клиническую картину также определяет заболевание, на основе которого развилась ЖДА (например, язвенная болезнь желудка или двенадцатиперстной кишки с повторными кровотечениями, мено- и метроррагии, какие-либо хронические инфекционные поражения и др.). Имеет значение и стадия течения анемии:
• скрытый дефицит железа, манифестирующий снижением концентрации сывороточного железа при отсутствии уменьшения содержания гемоглобина;
• тканевый сидеропенический синдром (манифестирует гастроэнтерологическими расстройствами, трофическими изменениями кожи и ее дериватов);
• анемия (снижение концентрации гемоглобина).
На первом этапе диагностического поиска при достаточно выраженной анемии можно обнаружить жалобы на слабость, шум в ушах, сердцебиение, одышку при физической нагрузке и ноющие боли в области сердца (признаки циркуляторно-гипоксического синдрома). Очень своеобразны гастроэнтерологические расстройства, представленные извращением вкуса и обоняния, снижением и извращением аппетита (желание есть мел, сухие макароны, зубной порошок), затруднением при глотании и неопределенными болевыми ощущениями в эпигастральной области. Нередко больные отмечают повышение температуры до субфебрильных цифр.
При умеренно выраженной анемии и дефиците железа все указанные жалобы могут быть выражены незначительно или отсутствовать. В анамнезе у таких
больных есть указания на случайно обнаруженное снижение концентрации гемоглобина (например, во время профилактического осмотра). Пациенты могут предъявлять разнообразные жалобы, а также сообщать те или иные сведения о фоновом заболевании (состоянии), обусловившем возникновение дефицита железа и последующей анемии.
На втором этапе диагностического поиска следует активно искать симптомы поражения эпителиальной ткани и трофических расстройств кожи и ее дериватов (волос, ногтей). Так, можно обнаружить сглаженность сосочков языка, сухость и шелушение кожного покрова, ломкость ногтей, сухость и выпадение волос. Циркуляторно-гипоксический синдром манифестирует тахикардией, систолическим шумом над верхушкой сердца и на крупных сосудах (тоны сердца не изменены). На яремных венах можно выслушать шум «волчка». Кожный покров и слизистые оболочки обычно бледные; размеры селезенки, как правило, соответствуют норме. Ее умеренное увеличение обычно отмечают у тех больных, которым проводили многочисленные гемотрансфузии.
На третьем этапе диагностического поиска проводят исследования, результаты которых подтверждают не только существование и выраженность анемии, но и ее патогенетический вариант (обусловленность дефицитом железа).
При исследовании периферической крови обнаруживают снижение концентрации гемоглобина, микроцитоз (увеличение количества эритроцитов малого диаметра) и гипохромию эритроцитов, снижение цветового показателя и среднего содержания гемоглобина в эритроците (массовое и процентное). Содержание ретикулоцитов в норме или повышено. Изменяются показатели обмена железа: уменьшается содержание свободного железа в сыворотке крови и насыщение трансферрина железом; повышается общая железосвязывающая способность сыворотки и концентрация общего трансферрина. Это связано с тем, что в организме снижено содержание железа. Для определения его резервов железа в организме применяют десфераловую пробу. В норме взрослый человек после введения дефероксамина в дозе 500 мг теряет 0,6-1,3 мг железа с мочой. При ЖДА содержание железа в моче после введения препарата значительно ниже (0,2-0,4 мг), что указывает на уменьшение запасов железа в организме. Дефероксамин - продукт метаболизма актиномицетов, способный связывать железо. Известное представление об уменьшении запасов железа в организме можно получить, изучая всасывание радиоактивного железа. При ЖДА оно повышается.
В костном мозге при ЖДА отмечают уменьшение количества сидеробластов - эритрокариоцитов, содержащих железо (как известно, в норме 20-40% эритрокариоцитов костного мозга содержат единичные гранулы железа). В ряде случаев гранулы обнаружить не удается.
При обследовании ЖКТ достаточно часто обнаруживают снижение желудочной секреции (базальной и стимулированной), а также атрофические изменения слизистой оболочки пищевода и желудка.
При выраженном циркуляторно-гипоксическом синдроме можно обнаружить признаки поражения миокарда (миокардиодистрофия вследствие анемии) в виде умеренного расширения сердца (определяют при рентгенологическом
исследовании) и изменений конечной части ЭКГ (снижение амплитуды или отрицательный зубец Т, преимущественно в грудных отведениях).
Диагностика
Выделяют два этапа диагностики ЖДА:
1) сбор доказательств дефицита железа в организме, послужившего причиной анемии;
2) установление причин развития железодефицитного состояния. Критерии дефицита железа и анемии:
• концентрация гемоглобина ниже 120 г/л у мужчин и ниже 116 г/л у женщин;
• снижение цветового показателя ниже 0,8;
• снижение среднего содержания гемоглобина в эритроцитах (менее 24 пг);
• снижение средней концентрации гемоглобина в эритроцитах (ниже 30%);
• увеличение количества микроцитов (эритроцитов диаметром менее 6 мкм)
более 20%;
• снижение концентрации сывороточного железа менее 11,6 мкмоль/л;
• повышение содержания свободного (более 35,8 мкмоль/л) и общего трансферрина (общей железосвязывающей способности сыворотки) более 71,6 мкмоль/л;
• снижение насыщения трансферрина железом (менее 25%);
• усиление всасывания радиоактивного железа;
• положительная проба с дефероксамином (уменьшение содержания железа в моче после введения дефероксамина).
Для установления причины железодефицитного состояния прежде всего необходимо найти источник кровотечения. Для этого, наряду с тщательным клиническим обследованием, требуется проведение эндоскопических (эзофагогастродуоденоскопия, ректоромано- и колоноскопия, бронхоскопия) и других исследований. Женщин обязательно должен осмотреть гинеколог.
Обнаружить скрытые (оккультные) кровотечения очень трудно. Если не удалось установить их источник, то применяют пробу с введением больному его собственных эритроцитов, предварительно меченных 51Сr, а в дальнейшем определяют радиоактивность кала. Ее повышение свидетельствует об источнике кровотечения в ЖКТ.
При хронических инфекционных заболеваниях большое значение имеет определение концентрации свободного трансферрина в крови (латентная железосвязывающая способность сыворотки), которая, в отличие от постгеморрагических анемий, остается нормальной.
Дифференциальная диагностика
ЖДА следует дифференцировать от сидероахрестической анемии и талассемии (один из видов наследственной гемолитической анемии). При сидероахрестической анемии вследствие генетического или приобретенного нарушения обмена порфиринов железо не поступает в эритроидные клетки. В результате этого развивается анемия с резким снижением цветового показателя при повышенном содержании железа в крови. В костном мозге - раздражение красного
ростка и повышенное содержание эритроидных клеток с включением железа. Лечение его препаратами при сидероахрестической анемии безуспешно.
При талассемии (более подробно см. «Гемолитические анемии») отмечают умеренное снижение содержания гемоглобина при значительном уменьшении цветового показателя. Концентрация сывороточного железа повышена. Характерно обнаружение мишеневидных эритроцитов. Одновременно отмечают все признаки гемолитического синдрома.
Формулировка развернутого клинического диагноза ЖДА должна включать следующие компоненты:
• определение характера анемии (в данном случае - железодефицитная);
• указание этиологии заболевания;
• определение стадии процесса (ремиссия или рецидив, который может характеризоваться скрытым дефицитом железа).
Лечение
Воздействуют на этиологические факторы (удаление источника кровотечения, борьба с инфекционным поражением, противоопухолевое лечение, профилактика врожденного дефицита железа) и проводят патогенетическую терапию (ликвидация дефицита железа, борьба с серьезными расстройствами гемодинамики) .
Рацион больных ЖДА должен включать продукты, богатые железом, но следует учитывать не только содержание железа в них, но и степень всасывания микроэлемента. Наибольшее количество железа содержат мясные продукты (говядина, телятина). Содержащееся в них гемовое железо всасывается на 25-30%. Всасывание железа из рыбы ниже (до 10%), из растительных продуктов - всего 3-5%. Таким образом, ликвидацию дефицита железа осуществляют с помощью приема внутрь или парентерального введения препаратов железа. Необходимо, чтобы суточная доза двухвалентного железа (только оно подвергается всасыванию) составляла 100-300 мг. В связи с этим при выборе препарата железа и определении его суточной дозы необходимо ориентироваться не только на общее содержание в нем железа, но и на концентрацию двухвалентного железа. Естественно, что предпочтительнее назначать средства с высоким содержанием последнего. Это связано с удобством их приема больными (1-2 раза в сутки). Входящие в состав многих лекарственных форм аскорбиновая и янтарная кислота, фруктоза и цистеин усиливают всасывание железа. Для улучшения последнего препараты железа следует принимать до приема пищи.
Основной принцип лечения препаратами железа - их длительное применение в достаточных дозах. Только в этом случае можно получить стойкий результат. Довольно давно применяют препарат железа сульфат в сочетании с аскорбиновой кислотой (по 15-20 драже в сутки). В настоящее время существуют препараты железа, содержащие двухвалентное железо в существенно большей концентрации, что позволяет обойтись приемом препарата 1-2 раза в день. Сорбифер дурулес* принимают по 1-2 таблетки в день, железа сульфат - по две таблетки в день. Представляет интерес новый препарат железа сульфат + фолиевая кислота + цианокобаламин, содержащий кроме сульфата железа аскорбиновую кислоту в дозе 100 мг, цианокобаламин в дозе 10 мкг и фолиевую кислоту в дозе
5 мг. Препарат можно принимать по 1-2 таблетки в сутки. Железа сульфат + серин - назначают по одной капсуле 2-3 раза в день или в виде сиропа (по одной чайной ложке на 12 кг массы тела). Сироп нельзя назначать больным сахарным диабетом, так как он содержит много сахара. В лекарственном средстве поливитамины + минералы железо содержится в микродиализных капсулах, что обеспечивает постоянство скорости его высвобождения (плазменной концентрации препарата) в течение суток. Для парентерального введения используют железа гидроксид полиизомальтозат и фербитол*. Железа гидроксид полиизомальтозат для внутримышечного введения выпускают в ампулах по 2 мл, содержащих 100 мг железа (соединение окиси трехвалентного железа с полиизомальтозой), а для внутривенного введения - в ампулах по 5 мл коллоидного раствора, в котором железо связано с натриево-сахаратным комплексом. Новый препарат для внутримышечного введения - железа гидроксид полимальтозат, для внутривенного введения - железа гидроксид сахарозный комплекс.
При назначении препаратов железа в достаточной дозе на 7-10-й день после начала лечения отмечают увеличение количества ретикулоцитов в периферической крови (ретикулоцитарный криз). Прирост концентрации гемоглобина начинается через 3-4 нед после его начала, но в ряде случаев это может произойти на 6-8-й нед. Лечение следует проводить не менее 3 мес. После достижения ремиссии больным с продолжающимися кровотечениями (например, при меноррагиях) следует рекомендовать поддерживающее лечение тем же препаратом (ежемесячно по 7-10 дней).
Ряду пациентов назначают парентеральное введение препаратов железа. Основные показания:
• тошнота, рвота (непереносимость препаратов железа при приеме внутрь, что не позволяет продолжать дальнейшее лечение);
• нарушение всасывания при патологических изменениях кишечника (энтериты, резекция тонкой кишки, синдром недостаточного всасывания);
• нежелательность приема внутрь препаратов железа больными с заболеваниями ЖКТ (обострение язвенной болезни желудка или двенадцатиперстной кишки, БК, НЯК);
• необходимость более быстрого насыщения организма железом (особенно в ситуациях, когда планируют оперативное вмешательство).
При аллергических реакциях на парентеральное введение препаратов железа и непереносимости их приема внутрь следует проводить трансфузии эритроцитной массы. Переливание крови способствует быстрому увеличению содержания гемоглобина, но его утилизация при этом значительно ограничена. Кроме того, существует опасность заражения больных инфекционным мононуклеозом, сывороточным гепатитом и др. В связи с этим гемотрансфузии проводят лишь по жизненным показаниям (при подготовке к оперативному вмешательству, выраженных гемодинамических нарушениях, связанных с анемией). В последнем случае следует стремиться не к нормализации концентрации гемоглобина путем гемотрансфузий, а к улучшению общего состояния больного.
Прогноз
Ликвидация причины потери крови, а также систематический прием препаратов железа приводят к полному выздоровлению. У женщин с обильными ма-
точными кровотечениями необходим систематический контроль содержания гемоглобина (как правило, их ставят на диспансерный учет).
Профилактика
Лица, подверженные опасности развития дефицита железа (недоношенные дети, дети от многоплодной беременности, девушки в период полового созревания при быстром росте, женщины с обильными менструациями, беременные), должны употреблять продукты с достаточным содержанием железа (прежде всего, говядину). Им рекомендовано периодическое исследование крови для определения скрытого дефицита железа и анемии.
СИДЕРОАХРЕСТИЧЕСКАЯ АНЕМИЯ
Сидероахрестическая анемия (САА) - железонасыщенная или сидеробластная анемия, при которой эритроциты содержат мало железа (гипохромны) вследствие его неиспользования костным мозгом для синтеза гемоглобина.
Этиология и патогенез
В основе развития сидероахрестических анемий лежит нарушение синтеза гема. Железо и белок, необходимые для синтеза гемоглобина, есть, но отсутствует достаточное количество протопорфирина. Вследствие этого не происходит синтез гема - основного компонента молекулы гемоглобина. Гем - соединение порфириновых колец (протопорфирина) с атомом железа. Соединяясь с глобином, он образует молекулу гемоглобина.
При САА уменьшается образование порфиринов и возникает избыток железа. Уменьшение образования первых обусловлено врожденным или приобретенным дефицитом ряда ферментов. Накопление железа в организме приводит к его отложению во внутренних органах.
Выделяют две основные наследственные формы САА: пиридоксинзависимую (дефицит пиридоксальфосфата, в связи с чем эффективно назначение пиридоксина) и пиридоксинрезистентную (регистрируют крайне редко). Существует непосредственный ферментный дефект (дефицит гемсинтетазы, обеспечивающей включение железа в молекулу гема).
Приобретенные формы чаще обнаруживают в пожилом возрасте, заболевание не носит семейного характера. САА чаще возникает при лечении туберкулостатическими препаратами, приводящем к истощению запасов пиридоксальфосфата, свинцовой интоксикации и алкоголизме. Возможно развитие идиопатических форм САА.
Клиническая картина
При наследственных формах заболевание начинается уже в раннем детстве . На первом этапе диагностического поиска обнаруживают жалобы, обусловленные циркуляторно-гипоксическим синдромом. В анамнезе - указания на бледность, слабость, увеличение печени и селезенки. Дети быстро устают, плохо учатся и имеют плохую память. Взрослые отмечают слабость
и снижение толерантности к физической нагрузке, возникающее после длительного лечения основного заболевания (туберкулеза) и воздействия профессиональных вредностей (контакт со свинцом). В анамнезе есть указания на обнаружение низкой концентрации гемоглобина и неэффективное лечение препаратами железа.
На втором этапе диагностического поиска в период обострения можно обнаружить бледность кожного покрова и видимых слизистых оболочек, у части больных - умеренное увеличение печени и селезенки. В связи с этим у таких пациентов обычно предполагают хроническое заболевание печени (чаще всего - хронический гепатит).
Отложение железа во внутренних органах может привести к возникновению ряда своеобразных симптомов. Так, отложение железа в поджелудочной железе вызывает развитие сахарного диабета, в печени - цирроза печени, в сердце - сердечной недостаточности, в половых железах - возникновение евнухоидизма.
Основным в установлении диагноза считают третий этап диагностического поиска. При лабораторном исследовании обнаруживают снижение концентрации гемоглобина в сочетании с низким цветовым показателем и нормальным или повышенным количеством ретикулоцитов. В крови определяют высокое содержание железа, а в пунктате костного мозга - повышенное количество сидеробластов (клетки костного мозга с включениями железа в виде гранул). Дефицит ферментов, участвующих в обмене порфиринов, уточняют посредством определения содержания продуктов распада последних в моче. Повышение концентрации железа в организме подтверждают с помощью пробы с дефероксамином (после введения препарата с мочой выделяется повышенное количество железа). При биопсии печени и селезенки можно обнаружить признаки гемосидероза. Общая железосвязывающая способность сыворотки у таких больных снижена.
Лечение
Назначение препаратов железа неэффективно и еще больше увеличивает содержание железа в крови, способствуя гемосидерозу органов. Проведение гемотрансфузий не рекомендовано. Назначают прием внутрь пиридоксина в дозе 50-200 мг/сут или его внутримышечное введение по 100 мг 2 раза в неделю на протяжении 2 мес. Наиболее эффективно использование кофермента пиридоксаль фосфата, так как иногда блокируется возможность трансформации пиридоксина в пиридоксальфосфат. При наследственных формах лечение пиридоксином необходимо периодически повторять.
Для уменьшения гемосидероза органов и снижения концентрации сывороточного железа, ориентируясь на его содержание и присутствие сидеробластов в костном мозге, назначают дефероксамин (внутривенно по 500-1000 мг с перерывами).
В12-ДЕФИЦИТНАЯ АНЕМИЯ
Сущность В12-дефицитной анемии (В12ДА) состоит в нарушении образования ДНК и РНК в связи с нехваткой в организме витамина В12 (цианокобаламина), что приводит к нарушению кроветворения, появлению в костном мозге мегалобластов, внутрикостномозговому разрушению эритрокариоцитов, снижению количества эритроцитов и гемоглобина, лейкопении, нейтропении и тромбоцитопении, а также к изменениям в ряде органов и систем (ЖКТ, ЦНС).
Этиология
В12ДА регистрируют значительно реже, чем ЖДА. Дефицит витамина В12 в организме может носить либо приобретенный, либо наследственный характер, т.е. быть генетически обусловленным.
Ниже перечислены причины развития В12ДА.
• Нарушения всасывания витамина В12.
• Приобретенные формы дефицита витамина В12:
- нарушение секреции гастромукопротеина (внутреннего фактора) в желудке;
- атрофия париетальных клеток слизистой оболочки желудка;
- антитела к париетальным клеткам слизистой оболочки желудка;
- антитела к гастромукопротеину или комплексу «гастромукопротеин + витамин В12»;
- органические поражения желудка (гастрэктомия, опухоли желудка, распространенный полипоз желудка);
- органические заболевания тонкой кишки (резекция кишечника, илеит, БК, спру).
• Наследственные формы дефицита витамина В12:
- наследственный дефицит внутреннего фактора (гастромукопротеина);
- генетически обусловленные нарушения всасывания комплекса «гастромукопротеин + витамин В12» в энтероцитах (болезнь ИмерслундГресбека);
- наследственный дефицит и функциональные аномалии транскобаламина II.
• Повышенный расход витамина В12:
- беременность;
- изменения кишечной микрофлоры при дивертикулезе кишечника;
- инвазия широкого лентеца.
• Уменьшенное потребление витамина В12:
- неправильное питание;
- отсутствие в рационе продуктов животного происхождения;
- строгое вегетарианство.
Сходную с В12ДА гиперхромную анемию вызывает дефицит фолиевой кислоты, который возникает:
• при ее повышенном расходе (беременность);
• вскармливании детей козьим молоком;
• нарушении всасывания (органические заболевания кишечника, алкоголизм);
• приеме некоторых лекарственных препаратов (противосудорожные, противотуберкулезные средства, фенобарбитал, контрацептивы и др.).
Патогенез
Витамин B12 (цианокобаламин) не синтезируется в организме человека и поступает только с продуктами питания. Больше всего его содержится в мясе, яйцах, молоке, сыре, печени и почках. Запасы витамина B12 у взрослого человека достаточно велики, составляют 2-5 мг и преимущественно находятся в печени. Для развития дефицита цианокобаламина вследствие нарушений всасывания либо поступления витамина B12 с пищей требуется от 6 мес до 3-5 лет.
Чистый витамин B12 не способен всасываться в кишечнике. Для всасывания в подвздошной кишке цианокобаламин должен предварительно соединиться с так называемым внутренним фактором - гликопротеином (гастромукопротеин), синтезируемым париетальными клетками слизистой оболочки желудка.
Перенос всосавшегося витамина B12 кровью в костный мозг для участия в кроветворении осуществляется с помощью специфических транспортных белков - транскобаламинов I, II и III.
Витамин B12 состоит из двух коферментов - метилкобаламина и дезоксиаденозилкобаламина. Дефицит первого кофермента обусловливает нарушение синтеза ДНК, вследствие чего нарушается деление и созревание клеток красного ряда и происходит их избыточный рост без утраты ядра. Большие клетки, содержащие ядра, называют мегалобластами. Они не созревают до мегалоцитов (гигантские эритроциты без ядер) и легко гемолизируются, еще находясь в костном мозге. Дефицит витамина B12 вызывает нарушение роста клеток лейкоцитарного и тромбоцитарного ряда, но это не так заметно сказывается на их морфологии и количестве клеток, как нарушения эритропоэза.
При недостатке второго кофермента нарушается обмен жирных кислот, вследствие чего в организме происходит накопление токсичных продуктов - пропионовой и метилмалоновой кислоты: развивается поражение заднебоковых канатиков спинного мозга - фуникулярный миелоз (рис. 5-3).
Клиническая картина
Клиническая картина В12ДА, как это вытекает из схемы патогенеза, складывается из следующих синдромов:
• циркуляторно-гипоксического (при достаточной выраженности анемии и кислородного голодания тканей);
• гастроэнтерологического;
• неврологического;
• гематологического (анемия гиперхромного типа).
Кроме этих синдромов, клиническую картину также определяет заболевание, на основе которого развилась В12ДА.
На первом этапе диагностического поиска при достаточно выраженной анемии могут возникать симптомы, обусловленные циркуляторно-
Рис. 5-3. Патогенез В12-дефицитной анемии
гипоксическим синдромом (слабость, повышенная утомляемость, одышка при физической нагрузке, боли в области сердца, сердцебиение). В случае нерезкого кислородного голодания тканей эти жалобы могут отсутствовать. Снижение аппетита, отвращение к мясу, боли и жжение в кончике языка, чувство тяжести в эпигастральной области после приема пищи, а также чередование поноса и запора обусловлены поражением ЖКТ и, в частности, выраженной секреторной недостаточностью желудка. При поражении ЦНС больные жалуются на головную боль, неустойчивую походку, зябкость, чувство онемения в конечностях и ощущение «ползания мурашек». Выраженность этих жалоб не всегда соответствует степени анемии. В период ремиссии заболевания они могут отсутствовать. Весьма существенно, если все перечисленные жалобы предъявляет немолодой человек; в таких случаях вероятность обнаружения В12ДА повышается.
В семейном анамнезе у больных с предполагаемой В12ДА могут быть указания на это заболевание среди родственников. Одна из причин развития анемии - злоупотребление алкоголем.
Данные анамнеза могут помочь в определении патогенетического варианта анемии. Ее развитие после пребывания больного возле больших водоемов и употребления в пищу сырой или недостаточно обработанной рыбы заставляет предположить в качестве возможной причины дифиллоботриоз. Если заболевание возникло у пожилого человека, страдающего хроническим гастритом, и развивается медленно, то можно думать о В12ДА. Если нарушения со стороны ЖКТ сочетаются со снижением массы тела и быстро прогрессируют, то в ка-
честве причины заболевания следует предположить злокачественное новообразование.
Наконец, сведения об успешном лечении больного цианокобаламином позволяют с большой уверенностью рассматривать имеющиеся признаки как симптомы В12ДА.
На втором этапе диагностического поиска симптомы могут быть обусловлены поражением ЖКТ и ЦНС. Кроме того, вероятность существования у пациента В12ДА повышает обнаружение ряда неспецифических признаков. Так, при В12ДА отмечают бледность кожного покрова в сочетании с небольшой иктеричностью склер и одутловатостью лица. Масса тела таких больных, как правило, нормальная или повышенная. При ее снижении в качестве возможной причины В12ДА нередко рассматривают злокачественную опухоль. Аналогичное значение имеет обнаружение увеличенного плотного лимфатического узла (возможно, метастаза опухоли). Циркуляторно-гипоксический синдром манифестирует так же, как и при ЖДА (расширение границ сердца влево, тахикардия, систолический шум, шум «волчка» на яремных венах).
Несомненное диагностическое значение имеет обнаружение при обследовании признаков глоссита (сглаженные сосочки языка вплоть до их полной атрофии - «полированный» язык). Печень несколько увеличена, можно пропальпировать селезенку. Тем не менее все эти симптомы не обязательны для В12ДА. Отмечают нарушения глубокой чувствительности, нижний спастический парапарез (картина псевдотабеса). Следует отметить, что нарушения со стороны нервной системы возникают далеко не во всех случаях, поэтому их отсутствие не исключает существования В12ДА.
Таким образом, результаты второго этапа в сочетании с анамнестическими данными и жалобами больного дают основание лишь заподозрить В12ДА. Окончательный диагноз ставят после проведения серии лабораторных исследований.
На третьем этапе диагностического поиска при исследовании периферической крови обнаруживают следующие изменения: снижение количества эритроцитов (менее 3х1012/л), повышение цветового показателя (более 1,1), среднего содержания гемоглобина в эритроците (более 34 пг) и среднего объема эритроцита (более 120 мкм3). Эритроцитометрическая кривая сдвинута вправо - увеличено количество макроцитов, присутствуют мегалоциты (эритроциты диаметром более 12 мкм). Форма эритроцитов изменена (пойкилоцитоз). Отмечают единичные мегалобласты.
Дополнительный признак - присутствие нейтрофилов с гиперсегментированными ядрами.
Если в картине периферической крови не обнаруживают характерных признаков, то выполняют стернальную пункцию. Последняя позволяет обнаружить в костном мозге мегалобластический тип кроветворения.
Важным считают определение содержания сывороточного железа: при В12ДА оно может соответствовать норме или быть повышенным в связи с усиленным гемолизом эритроцитов. В этих случаях увеличено содержание непрямого билирубина. При исследовании желудочного сока часто обнаруживают гистами-
ноустойчивую ахилию (характерный признак анемии Аддисона-Бирмера), а эндоскопически - атрофию слизистой оболочки желудка.
Другие инструментальные методы исследования помогают обнаружить признаки миокардиодистрофии (развивается на фоне выраженной анемии), а также уточнить этиологию заболевания.
Диагностика
В диагностике В12ДА выделяют два этапа:
1) доказательство дефицита витамина В12 в качестве причины анемии;
2) установление причин дефицита витамина В12. Критерии В12ДА:
• снижение содержания эритроцитов (менее 3,0х1012/л);
• повышение цветового показателя (более 1,1);
• повышение содержания гемоглобина в эритроцитах (более 34 пг);
• увеличение среднего объема эритроцита (более 120 мкм3);
• сдвиг эритроцитометрической кривой вправо (увеличение количества макроцитов, присутствие мегалоцитов - эритроцитов диаметром более 12 мкм);
• обнаружение в мазках пунктата костного мозга элементов мегалобластного кроветворения;
• повышение содержания сывороточного железа более 30,4 мкмоль/л;
• снижение радиоактивности мочи после приема витамина B12, меченного радиоактивным кобальтом.
Для установления причины анемии следует проводить рентгенологическое, эндоскопическое (опухоль желудка, дивертикулез тонкой кишки), гельминтологическое исследование (инвазия широким лентецом), функциональное исследование печени с биопсией (хронический гепатит, цирроз) и определение нейтрального жира в кишечнике (спру).
В12ДА следует дифференцировать от фолиеводефицитной анемии. При дефиците фолиевой кислоты обнаруживают макроцитарную гиперхромную анемию, а в костном мозге - мегалобласты. Следует отметить, что дефицит фолиевой кислоты регистрируют значительно реже. В отличие от В12ДА, при фолиеводефицитной анемии содержание фолиевой кислоты в крови и эритроцитах снижено. Кроме того, при воздействии на препарат костного мозга ализарином красным окрашиваются только мегалобласты, связанные с дефицитом витамина В12, и не окрашиваются мегалобласты, связанные с дефицитом фолиевой кислоты.
Течение
Возможно резкое обострение заболевания. В таких случаях развивается коматозное состояние: потеря сознания, снижение температуры тела и АД, одышка, рвота, арефлексия и непроизвольное мочеиспускание. Между развитием коматозного состояния и снижением концентрации гемоглобина нет четкой зависимости (у больных с его резко сниженным содержанием кома отсутствует). Главную роль в патогенезе комы играют быстрый темп и степень снижения гемоглобина, а также резкая ишемия и гипоксия ЦНС.
Формулировка развернутого клинического диагноза должна учитывать:
• этиологию В12ДА (отдельно следует выделять анемию, вызванную болезнью Аддисона-Бирмера);
• стадию процесса (рецидив или ремиссия);
• выраженность отдельных синдромов (как правило, при неврологических расстройствах, обусловленных фуникулярным миелозом).
Лечение
Комплекс лечебных мероприятий при В12ДА следует проводить с учетом этиологии, выраженности анемии и существования неврологических нарушений. При лечении следует ориентироваться на следующие положения.
• Непременное условие лечения В12ДА при глистной инвазии - дегельминтизация (для изгнания широкого лентеца по определенной схеме назначают никлозамид или папоротника мужского корневища).
• При органических заболеваниях кишечника и поносе следует применять ферментные препараты (панкреатин, гемицеллюлаза + желчи компоненты + панкреатин), а также закрепляющие средства (карбонат кальция в сочетании с дерматолом).
• Нормализации кишечной микрофлоры достигают приемом ферментных препаратов (панкреатин, гемицеллюлаза + желчи компоненты + панкреатин), а также подбором диеты, способствующей ликвидации синдромов гнилостной или бродильной диспепсии.
• Сбалансированное питание с достаточным содержанием витаминов белка и безусловным запрещением употребления алкоголя - непременное условие лечения В12ДА и фолиеводефицитной анемии.
• Патогенетическое лечение осуществляют с помощью парентерального введения цианокобаламина (ликвидация его дефицита), нормализации показателей центральной гемодинамики и нейтрализации антител к гастромукопротеину (внутреннему фактору) или комплексу гастромукопротеин + витамин В12 (применение глюкокортикоидов). Цианокобаламин вводят внутримышечно в дозе 200-500 мкг 1 раз в день ежедневно в течение 4-6 нед до наступления гематологической ремиссии. Критерии последней: резкое увеличение количества ретикулоцитов в периферической крови (ретикулоцитарный криз) и трансформация мегалобластического кроветворения в нормобластическое. Развитие ретикулоцитарного криза на 5-6-й день лечения - ранний критерий его эффективности. В процессе лечения цианокобаламином количество эритроцитов увеличивается быстрее, чем содержание гемоглобина, поэтому цветовой показатель обычно снижается. После нормализации костномозгового кроветворения и состава крови (обычно - через 1,5-2 мес) препарат вводят 1 раз в неделю в течение 2-3 мес, а затем - 2 раза в месяц в тех же дозах, что и в начале курса, в течение полугода). В дальнейшем больных ставят на диспансерный учет и с профилактической целью вводят им цианокобаламин (1-2 раза в год короткими курсами по 5-6 инъекций или ежемесячно по 200-500 мкг пожизненно).
При симптомах фуникулярного миелоза в значительных дозах вводят цианокобаламин (по 500-1000 мкг ежедневно в течение 10 дней, а затем 1-3 раза в неделю до исчезновения неврологических нарушений).
Гемотрансфузии проводят лишь при значительном снижении гемоглобина и возникновении симптомов коматозного состояния. Рекомендовано введение эритроцитарной массы в дозе 250-300 мл (5-6 трансфузий).
Преднизолон (по 20-30 мг/сут) назначают при заболевании аутоиммунной этиологии.
Прогноз
В настоящее время применение цианокобаламина сделало прогноз В12ДА благоприятным. При полноценном лечении больные живут длительное время.
Профилактика
Мер первичной профилактики не существует. У лиц, имеющих ранее перечисленные этиологические факторы, следует периодически исследовать кровь для своевременного обнаружения анемии.
Гемолитические анемии
Гемолитические анемии (ГА) - обширная группа заболеваний, значительно различающихся по этиологии, патогенезу, клинической картине и лечению. Основной патологический процесс, объединяющий эти заболевания в одну группу, - повышенный гемолиз. Он может происходить внутриклеточно (в макрофагах селезенки, как обычный физиологический процесс) и непосредственно в сосудах (внутрисосудистый или внеклеточный гемолиз). В норме продолжительность жизни эритроцита составляет 100-120 дней, но при гемолитических анемиях она сокращается до 12-14 дней.
Усиленный гемолиз, происходящий в клетках мононуклеарно-фагоцитарной системы (главным образом в селезенке), манифестирует следующими симптомами:
• в крови увеличивается содержание свободного (непрямого) билирубина, с чем связано желтушное окрашивание кожи и слизистых оболочек разной степени выраженности;
• гепатоциты перерабатывают избыточное количество непрямого билирубина в прямой билирубин, вследствие чего возникает интенсивное окрашивание желчи (плейохромия) и развивается склонность к образованию камней в желчном пузыре и протоках;
• в кишечнике, куда поступает желчь, в большом количестве образуется стеркобилиноген и уробилиноген, в связи с чем возникает интенсивное окрашивание каловых масс;
• в моче увеличивается содержание уробилина;
• общее количество эритроцитов уменьшается, увеличивается число ретикулоцитов в периферической крови и содержание эритробластов и нормоцитов в костном мозге.
Признаки усиленного внутрисосудистого гемолиза:
• увеличение концентрации свободного гемоглобина в крови;
• выделение свободного гемоглобина с мочой в неизмененном виде или в виде гемосидерина (моча красного, бурого или почти черного цвета);
• отложение гемосидерина во внутренних органах (гемосидероз).
Классификация
Все гемолитические анемии разделяют на две большие группы - наследственные и приобретенные. Наследственные ГА обусловлены генетическими дефектами эритроцитов, которые становятся функционально неполноценными и легко разрушаются. Приобретенные ГА - следствие воздействия на нормальные эритроциты различных факторов (образование антител, гемолитических ядов, механические воздействия и др.), приводящих к их разрушению.
• Наследственные ГА:
- связанные с нарушением мембраны эритроцитов (гемолитическая микросфероцитарная анемия или болезнь Минковского-Шоффара, овалоцитоз, стоматоцитоз);
- связанные с нарушением активности ферментов (глюкозо-6- фосфатдегидрогеназы (Г-6-ФД), пируваткиназы, глутатионредуктазы и др.) в эритроцитах;
- связанные с нарушением структуры или синтеза цепей глобина (талассемия, серповидноклеточная анемия и др.).
• Приобретенные ГА:
- связанные с воздействием антител (изоиммунные, аутоиммунные);
- связанные с изменением структуры мембраны эритроцитов вследствие соматической мутации (пароксизмальная ночная гемоглобинурия или болезнь Маркиафавы-Микели);
- связанные с механическим повреждением мембраны эритроцита (протезы клапанов сердца, маршевая гемоглобинурия);
- обусловленные химическими повреждениями эритроцитов (гемолитические яды, свинец, тяжелые металлы, органические кислоты);
- обусловленные недостатком витамина Е;
- связанные с воздействием паразитов (малярия).
Частота возникновения тех или иных ГА весьма различна. Так, наследственный микросфероцитоз в Европе регистрируют с частотой 0,03%, а в Японии и Африке значительно реже. Частота развития анемии, обусловленной дефицитом Г-6-ФД, высока в странах средиземноморского бассейна, на Ближнем Востоке и на Кавказе. Ночная пароксизмальная гемоглобинурия - редкое заболевание. В дальнейшем будет рассмотрена диагностика трех видов наиболее распространенных ГА: наследственного микросфероцитоза, талассемии и аутоиммунной ГА.
Наследственный микросфероцитоз (болезнь Минковского-Шоффара)
В основе микросфероцитарной ГА (наследственного микросфероцитоза) лежит дефект оболочки эритроцита, наследуемый по аутосомно-доминантному типу. Сферические ригидные эритроциты не могут менять форму при продвижении по узким капиллярам, особенно в синусах селезенки, в связи с чем теряется часть оболочки эритроцита, и происходит гемолиз. Мембрана эритроцита пропускает внутрь повышенное количество ионов натрия, накопление которых способствует усилению расхода АТФ и глюкозы на последующее выведение их из клетки. Это также приводит к укорочению продолжительности жизни эритроцита.
Клиническая картина
Клиническая картина заболевания определяется гемолитическим синдромом и сопутствующими врожденными аномалиями скелета и внутренних органов . Болезнь течет волнообразно: «спокойные» периоды прерывают гемолитические кризы, провоцируемые развитием неспецифических инфекционных поражений, во время которых гемолиз резко интенсифицируется и все симптомы болезни усиливаются.
На первом этапе диагностического поиска можно получить информацию о жалобах больного на периодически возникающую легкую желтушность кожного покрова и преходящую слабость. При тяжелом течении заболевания регистрируют гемолитические кризы, обычно возникающие спонтанно либо под влиянием инфекционных возбудителей, переутомления, травмы и переохлаждения; отмечают озноб, повышение температуры тела, боли в мышцах, в области печени и селезенки. Резко усиливается желтуха, моча и кал темнеют. Если симптомы криза выражены не столь резко, но желтуха выражена вполне отчетливо, то таких больных нередко госпитализируют в инфекционные больницы с подозрением на вирусный гепатит, где обычно диагноз не подтверждают. Постоянная желтушность вне кризов у таких больных может послужить основанием для предположения о хроническом гепатите.
На втором этапе диагностического поиска обнаруживают лимонножелтое окрашивание кожи, усиливающееся до более интенсивной желтухи в период гемолитического криза. У части больных можно отметить врожденные аномалии (башенный череп, заячья губа, пороки сердца). При выраженной анемии регистрируют циркуляторно-гипоксический синдром (анемический систолический шум, тахикардия, снижение АД, шум «волчка» на яремных венах и др.). Гемолиз происходит в селезенке, поэтому со временем орган увеличивается. Данные второго этапа диагностического поиска скорее исключают ряд заболеваний печени, способных стать причиной желтухи, а не подтверждают наследственный микросфероцитоз.
Решающим считают третий этап диагностического поиска, во время которого обнаруживают синдром гемолиза, протекающий у больных наследственным микросфероцитозом с некоторыми особенностями.
Общий анализ крови позволяет определить снижение содержания гемоглобина и эритроцитов. Основной морфологический признак заболевания - присутствие в крови большого количества мелких круглых эритроцитов (микросфероцитов). Их диаметр уменьшен, а осмотическая резистентность значительно снижена. Гемолиз начинается при концентрации хлорида натрия 0,6-0,8%, а при его содержании около 0,4% происходит полный гемолиз. В норме же он начинается при концентрации 0,42-0,46%, а становится полным при содержании хлорида натрия около 0,30-0,32%.
Усилен аутогемолиз: во время инкубации эритроцитов в течение 48 ч при температуре 37 °С гемолизируется не менее 30%, тогда как в норме - лишь 3-4% клеток. Положительны пробы с АТФ и дектрозой: их добавление к эритроцитам уменьшает аутогемолиз. Продолжительность жизни эритроцита, определяемая с помощью эритроцитов, меченных 51Сr, при наследственном микросфероцитозе укорочена.
В крови определяют и другие признаки гемолиза: ретикулоцитоз и увеличение концентрации непрямого билирубина. В кале повышено содержание стеркобилина, а в моче - уробилина. При длительном течении болезни холецистография и УЗИ позволяют обнаружить в желчном пузыре и протоках конкременты.
Диагностика
Критерии заболевания:
• спленомегалия;
• камнеобразование в желчном пузыре;
• гипергенераторная анемия и желтуха в период криза;
• микросфероциты в мазке крови;
• снижение осмотической резистентности эритроцитов после инкубации цельной крови в стерильных условиях в течение суток при 37 °С в случаях, когда количество сфероцитов превышает 1-2% общего числа эритроцитов;
• усиление спонтанного гемолиза после 49-часовой инкубации крови в стерильных условиях до 10-50% (в норме лизируется менее 4%), при этом аутогемолиз удается предотвратить добавлением к эритроцитам до начала инкубирования декстрозы.
Таким образом, как и при других видах анемии, диагностика заболевания основана преимущественно на данных третьего этапа, но и результаты второго этапа поиска имеют значение.
Формулировку развернутого клинического диагноза осуществляют в следующей последовательности:
• наименование ГА;
• фаза (обострение (гемолитический криз) или ремиссия);
• состояние внутренних органов (спленомегалия, желчнокаменная болезнь, возможные аномалии скелета и других органов).
Лечение
Единственный эффективный метод лечения - спленэктомия, после чего патологический гемолиз прекращается, хотя эритроциты и имеют дефектную
оболочку. Выполнение этого вмешательства рекомендовано при тяжелом течении болезни и частых гемолитических кризах. При резкой анемии допустимо переливание эритроцитарной массы.
Препараты железа, цианокобаламин и глюкокортикоиды применять не следует вследствие их неэффективности (механизмы развития анемии не связаны с дефицитом железа и витамина В12, а гемолиз не связан с противоэритроцитарными антителами).
Талассемия
В основе талассемии лежит нарушение синтеза одного вида цепей глобина, что связано с наследственным дефектом транспортной РНК или гена-регулятора. Может быть нарушен синтез различных цепей глобина, в связи с чем различают α-, β- и γ-талассемию. Возможно сочетание нарушения синтеза различных цепей. Чаще всего обнаруживают изменения β-цепей, в связи с этим содержание нормального гемоглобина А, в состав которого входят две α-цепи и две β-цепи, уменьшается, а концентрация гемоглобина F и А2 увеличивается. Эритроциты, содержащие аномальный гемоглобин, легко разрушаются, секвестрируются и гемолизируются в узких капиллярах селезенки. Этому способствует повышение проницаемости мембраны эритроцита.
Клиническая картина
Различают два варианта течения β-талассемии:
• большая талассемия (анемия Кули), регистрируемая в детском возрасте (гомозиготная форма);
• малая талассемия, регистрируемая у взрослых (гетерозиготная форма). Ниже рассмотрены этапы диагностического поиска при малой талассемии. На первом этапе диагностического поиска можно обнаружить жалобы
на повышенную утомляемость, слабость и головные боли. Они неспецифичны для этого вида ГА, как и преходящая желтушность, которая тем не менее должна обратить на себя внимание врача. В анамнезе - указания на периоды повышения концентрации билирубина, обнаружение увеличенной селезенки, а также безуспешность лечения препаратами железа. Такие же симптомы могут быть и у родственников больного, что указывает на наследственный характер заболевания.
На втором этапе диагностического поиска в периоды обострения возможно обнаружение умеренной желтушности кожного покрова, а у половины больных - увеличение селезенки. В связи с этим у таких пациентов предполагают существование хронического заболевания печени (чаще всего - хронического гепатита).
Основным для установления диагноза считают третий этап диагностического поиска. При лабораторном исследовании обнаруживают признаки гемолиза (увеличение содержания непрямого билирубина и ретикулоцитов, уробилинурию), а также характерные симптомы талассемии:
• гипохромную анемию в сочетании с высокой концентрацией сывороточного железа;
• характерные мишеневидные эритроциты;
• увеличение числа малых фракций гемоглобина (гемоглобин А2 и F).
Диагностика
Диагностические критерии талассемии:
• выраженная гемолитическая анемия с гипохромными микроцитарными эритроцитами;
• анизоцитоз, множество мишеневидных, а также капле- и сигарообразных эритроцитов в мазке периферической крови;
• увеличение фракции гемоглобина F, отсутствие фракции гемоглобина А;
• возможно увеличение фракции гемоглобина А2 почти в 2 раза.
Лечение
• Гемотрансфузии + прием дефероксамина (во избежание развития гемосидероза) .
• Прием фолиевой кислоты по 0,005 г 1-2 раза в день при снижении концентрации гемоглобина в связи с инфекционными заболеваниями или во время беременности.
• Прием витаминов группы В (В6, B12) и аскорбиновой кислоты для улучшения эритропоэза (подобное назначение небесспорно).
• Спленэктомия при значительной спленомегалии и распаде эритроцитов в селезенке.
Аутоиммунная гемолитическая анемия
Аутоиммунная гемолитическая анемия (АИГА) - распространенная форма приобретенных ГА. Выделяют два варианта болезни:
• симптоматическую форму, при которой анемия развивается на фоне определенного заболевания (гемобластоза, системного заболевания соединительной ткани, хронического активного гепатита, опухоли, НЯК и др.);
• идиопатическую форму, когда обнаружить определенное заболевание не удается (острое инфекционное заболевание, беременность, роды и травма в анамнезе не служат причиной АИГА, а лишь провоцируют ее обострение).
При АИГА вырабатываются антитела к собственному антигену эритроцитов. Первый этап патогенеза АИГА - изменение антигена эритроцитов под влиянием лекарственных препаратов, вирусов или бактерий. Возможна также соматическая мутация единичного иммуноцита. Дальнейшая реакция антител и антигенов эритроцитов обусловливает развитие гемолиза и анемии.
АИГА может развиваться при участии различного вида аутоантител, вызывающих гемолиз при различной температуре. Различают два типа антител - тепловые (реагируют с эритроцитами при температуре тела не ниже 37 °С) и холодовые (реагируют с эритроцитами при температуре ниже 37 °С). На этом основании выделяют четыре вида АИГА:
• АИГА с неполными тепловыми агглютининами;
• АИГА с тепловыми гемолизинами;
• АИГА с холодовыми агглютининами;
• АИГА с двухфазными гемолизинами.
Образующийся комплекс «эритроцит + антитело» поглощают макрофаги селезенки (внутриклеточный гемолиз). Также могут вырабатываться аутоиммунные антитела к тромбоцитам, что приводит к развитию тромбоцитопении.
Чаще всего регистрируют АИГА, обусловленные тепловыми аутоантителами. Последние принадлежат к IgG и служат неполными тепловыми агглютининами, максимально демонстрирующими свое действие при температуре 37 °С. Гемолиз происходит внутриклеточно и существенно реже - внутри сосудов.
Холодовые аутоантитела принадлежат к IgM и представлены агглютининами. Гемолиз возникает в результате их соединения с эритроцитами и комплементом. Действие антител начинается при низкой температуре (ниже 32 °С): в мелких сосудах дистальных отделов тела (пальцы рук, ног, кончики ушей и носа) образуются крупные конгломераты из агглютинированных эритроцитов, а сами сосуды спазмируются. Гемолиз происходит преимущественно внутриклеточно, но обнаружение гемоглобинурии указывает и на внутрисосудистый гемолиз. При переходе пациента в теплое помещение гемолиз прекращается.
Значительно реже возникают АИГА, обусловленные действием двух других типов аутоантител - тепловых и двухфазных холодовых гемолизинов. При обоих вариантах агглютинации эритроцитов не происходит. Гемолиз возникает при осаждении аутоантител (гемолизинов) на эритроцитах, в связи с чем он происходит внутри сосудов и сопровождается выделением черной мочи (гемоглобинурия).
Под действием тепловых гемолизинов гемолиз происходит в обычных условиях (пребывание на холоде - не обязательное условие). Во время пребывания больного на холоде двухфазные гемолизины осаждаются на эритроцитах, но собственно гемолиз начинается лишь после перехода больного в теплое помещение.
Клиническая картина
Клиническая картина АИГА полиморфна и обусловлена:
• быстротой развития гемолиза (кризовое или более спокойное течение);
• преобладающим патогенетическим механизмом гемолиза (те или иные аутоантитела приводят к гемолизу при различных внешних условиях);
• изменениями в органах (в частности, в печени и селезенке);
• местом, где происходит гемолиз (селезенка, сосудистое русло);
• фоновыми заболеваниями (при вторичных АИГА).
В связи с этим при конечном сходном результате - гемолизе эритроцитов и развитии всех признаков ГА - на всех трех этапах диагностического поиска можно получить совершенно различные данные.
Ha первом этапе диагностического поиска больные с гемолитическими кризами, обычно развивающимися после травм и инфекционных заболеваний, предъявляют жалобы на повышение температуры тела, боли в пояснице, озноб и возникновение желтушности. При АИГА, спровоцированных воздействием холода, отмечают непереносимость низких температур: у больных синеют дис-
тальные участки конечностей, нос и уши. Как правило, они плохо себя чувствуют в холодное время года.
На втором этапе диагностического поиска (без учета симптомов основного заболевания при вторичных формах АИГА) обычно возникают две ситуации:
• в период ремиссии, кроме легкой желтушности и незначительного увеличения селезенки (иногда - печени), можно не обнаружить никаких изменений;
• в период криза симптомы более яркие и представлены повышением температуры тела, более интенсивной желтухой и сосудистыми изменениями по типу синдрома Рейно (особенно при АИГА, провоцируемой действием низких температур).
Информация, полученная на первом и втором этапе, не дает оснований для установления диагноза АИГА, а тем более для идентификации ее серологического варианта. Может возникнуть лишь предположение об этом заболевании (особенно при развитии несомненных гемолитических кризов, синдроме Рейно или выделении черной мочи в период криза). Для уточнения диагноза необходимо доказать аутоиммунность ГА и отвергнуть ряд заболеваний печени и желчных путей, способных сопровождаться сходными симптомами.
На третьем этапе диагностического поиска обнаруживают в большей или меньшей степени выраженный синдром гемолиза (в зависимости от существования или отсутствия гемолитического криза). Чрезвычайно важно обнаружение аутоантител. Основной метод определения неполных тепловых агглютининов - проба Кумбса, основанная на агглютинации антиглобулиновой сывороткой эритроцитов больного с фиксированными на них антителами (прямая проба Кумбса) или агглютинации с помощью антиглобулиновой сыворотки эритроцитов здорового человека, «нагруженных» антителами из сыворотки крови больного (непрямая проба Кумбса). Аутоантитела также обнаруживают с помощью агрегатгемагглютинационной пробы, которая во много раз чувствительнее пробы Кумбса.
Полные холодовые агглютинины обнаруживают путем инкубации при различных температурах эритроцитов донора и сыворотки больного. Агглютинация происходит при определенных разведениях сыворотки и температуре, при этом чем выше температура, при которой возможна агглютинация, тем тяжелее протекает болезнь.
Двухфазные гемолизины определяют с помощью эритроцитов донора, фиксирующих на себе антитела больного при низкой температуре. В дальнейшем при инкубации такой смеси происходит гемолиз эритроцитов. Иногда для обнаружения гемолизинов используют пробу Кумбса: чем более высокая температура требуется для гемолиза, тем тяжелее протекает заболевание.
При формах АИГА, протекающих с выработкой гемолизирующих аутоантител (гемолизинов), в моче присутствуют гемоглобин и гемосидерин, так как гемолиз протекает внутри сосудов. Моча приобретает темную окраску (вплоть до черной).
На третьем этапе диагностического поиска при симптоматических формах АИГА можно обнаружить изменения, обусловленные основным заболеванием:
опухолью, гемобластозом, диффузным заболеванием соединительной ткани, поражением печени и др.
Диагностика
Диагностика АИГА основана на обнаружении сочетания признаков гемолиза и определении аутоантител. Естественно, что в процессе диагностики следует исключить ГА, обусловленные воздействием различных химических средств, малярийного плазмодия, механическим повреждением оболочки эритроцита, а также наследственной этиологии.
Лечение
При назначении лечения учитывают фазу аутоиммунной ГА (ремиссия или гемолитический криз).
В период криза средством выбора служат глюкокортикоиды, которые всегда прекращают или уменьшают гемолиз. В острой фазе назначают большие дозы преднизолона (по 60-90 мг/сут) или эквивалентные дозы других глюкокортикоидов. При наступлении ремиссии их постепенно уменьшают, переводя больного на поддерживающие дозы (по 5-10 мг/сут). Продолжительность гормонального лечения при проведении гематологического и серологического контроля (до исчезновения или существенного уменьшения количества аутоантител) составляет 2-3 мес.
В межприступном периоде можно назначать другие иммунодепрессанты, например аминохинолиновые препараты (хлорохин), которые следует принимать длительно (до одного года).
При плохой переносимости глюкокортикоидов, противопоказаниях к их применению или недостаточной эффективности рекомендовано применение цитостатических иммунодепрессантов (циклофосфамид, метотрексат). Эти средства особенно эффективны при АИГА, связанной с холодовыми агглютининами.
В случаях, когда применение глюкокортикоидов и цитостатических средств не позволяет достичь четкого улучшения, хороший эффект может оказать спленэктомия. При выраженной анемии рекомендовано переливание эритроцитарной массы, но кровь необходимо подбирать индивидуально, с помощью непрямой пробы Кумбса, когда переливаемые эритроциты «нагружают» антителами сыворотки крови больного. Если проба Кумбса с эритроцитами донора отрицательная, то такую кровь можно переливать.
Прогноз
При незначительном гемолизе и отсутствии гемолитических кризов прогноз удовлетворительный. Усиление гемолиза с резким снижением концентрации гемоглобина значительно ухудшает прогноз.
Профилактика
Меры первичной профилактики ГА в настоящее время разработаны недостаточно. При установлении диагноза ГА больных ставят на диспансерный учет
и периодически проводят исследования крови. Кроме того, им запрещают контакт с веществами, способствующими усилению гемолиза.
Пароксизмальная ночная гемоглобинурия
Пароксизмальная ночная гемоглобинурия (ПНГ), также называемая болезнью Маркиафавы-Микели, - приобретенная ГА, сопровождающаяся постоянным внутрисосудистым гемолизом и выделением с мочой гемосидерина.
В основе заболевания лежит выработка патологического клона эритроцитов с повышенной чувствительностью к различным гемолитическим агентам (тромбин, комплемент, снижение рН крови и др.). Предполагают, что заболевание возникает в результате соматической мутации на уровне клетки-предшественницы миелопоэза, что приводит к выработке патологического клона эритроцитов, оболочка которых дефектна. При сканирующей микроскопии такие клетки выглядят своеобразно - их мембрана имеет множество отверстий. Кроме этой патологической популяции эритроцитов, существуют и нормальные клетки, не разрушающиеся при подкислении среды. Патологические клоны эритроцитов, гранулоцитов и тромбоцитов высокочувствительны к действию комплемента вследствие дефицита в их оболочке регуляторного белка, который частично обеспечивает быстрое превращение активного С3-компонента комплемента в неактивную форму.
При ПНГ гемолиз происходит внутри сосудов, поэтому в крови присутствует свободный гемоглобин. Его обнаружение в моче зависит от содержания гемоглобина в крови и концентрации гаптоглобина - белка крови, связывающего свободный гемоглобин. При достаточном содержании последнего и небольшом гемолизе гемоглобинурии не будет, но при прохождении свободного гемоглобина через канальцы почек он разрушается и откладывается в эпителии канальцев в виде гемосидерина, а также выделяется с мочой (гемосидеринурия), что служит важным признаком болезни. Заболевание протекает волнообразно: периоды умеренно выраженного гемолиза чередуются с его резким усилением - гемолитическими кризами, во время которых концентрация свободного гемоглобина в крови резко увеличивается; его также обнаруживают в моче (гемоглобинурия).
Клиническая картина
Клиническую картину заболевания определяет выраженность внутрисосудистого гемолиза, а также возникновение множественных тромбозов мелких сосудов (мезентериальных, верхних и нижних конечностей, почечных, мозговых и сосудов селезенки). Предполагают, что склонность к последним, отмечаемая в периоды гемолитических кризов, обусловлена освобождением при гемолизе эритроцитов (особенно ретикулоцитов) и большого количества веществ, обладающих тромбофилической активностью.
На первом этапе диагностического поиска можно обнаружить жалобы больного на периодически возникающую слабость и легкое желтушное окрашивание склер. Типичный признак болезни - моча черного цвета, обусловленная присутствием в ней гемоглобина и гемосидерина. Нередко гемоглобинурия
возникает в ночное время, что объясняется наступающим во время сна физиологическим ацидозом, а также активацией других факторов, усиливающих гемолиз. Во время гемолитических кризов, кроме черной мочи, больные также отмечают приступы болей в животе (капиллярные тромбозы мезентериальных сосудов). В период криза у пациентов может повышаться температура тела. В анамнезе могут быть указания на госпитализацию в инфекционное отделение с подозрением на вирусный гепатит после возникновения желтушности кожного покрова. В отношении части больных такие предположения сохраняются, если присутствует постоянная желтушность, а четко выраженных кризов с отхождением черной мочи нет.
На втором этапе диагностического поиска в период гемолитического криза отмечают бледность кожи с небольшим желтушным оттенком. При развитии достаточно выраженной анемии обнаруживают циркуляторногипоксический синдром (тахикардия, анемический систолический шум, снижение АД, шум «волчка» на яремных венах) и небольшое увеличение селезенки. Возможно увеличение печени, но это не постоянный признак. Данные второго этапа позволяют исключить ряд заболеваний печени, способных вызвать желтуху. В связи с тромбозами сосудов у части больных во время гемолитического криза определяют признаки острого живота (резкие боли, симптомы раздражения брюшины). Нередко это приводит к выполнению хирургических вмешательств, что связано с подозрением на острый аппендицит, прободную язву или острый холецистит.
Решающим в диагностике считают третий этап диагностического поиска, во время которого обнаруживают синдром внутрисосудистого гемолиза.
При общеклиническом исследовании крови обнаруживают снижение концентрации гемоглобина и количества эритроцитов. В период обострения болезни содержание гемоглобина существенно уменьшается (до 30-50 г/л), возвращаясь к норме в период ремиссии. Число эритроцитов уменьшается соответственно снижению концентрации гемоглобина, поэтому цветовой показатель долго остается близким к 1,0. Он снижается, если больной теряет с мочой много железа в виде гемоглобина и гемосидерина. Содержание ретикулоцитов умеренно повышено (до 2-4%). Количество лейкоцитов чаще всего умеренно снижено, а тромбоцитов - соответствует норме или умеренно снижено.
Исследование костного мозга позволяет обнаружить признаки гемолитической анемии - раздражение красного ростка при нормальном количестве миелокариоцитов.
Концентрация железа в крови снижается вследствие гемоглобинурии, при этом гемосидерин постоянно обнаруживают в моче. Тем не менее снижение содержания железа не считают признаком ПНГ.
Концентрация билирубина в большинстве случаев повышается незначительно или не изменяется.
Нарушение структуры оболочки эритроцита обнаруживают с помощью пробы Хэма (кислотная проба) и сахарозной пробы. При пробе Хэма эритроциты больного гемолизируются в свежей подкисленной сыворотке (нормальные эритроциты не подвержены гемолизу). При сахарной пробе их гемолиз происходит при добавлении к сыворотке сахарозы.
Диагностика
Диагностика заболевания основана преимущественно на данных третьего этапа диагностического поиска (положительный результат кислотного и сахарозного теста), а также на особенностях клинической картины болезни (приступы болей в животе, выделение темной мочи, волнообразное течение болезни). ПНГ необходимо дифференцировать от приобретенной АИГА, при которой в сыворотке крови присутствуют гемолизины, обусловливающие внутрисосудистый гемолиз. Последний также манифестирует гемоглобинемией, гемоглобинурией и гемосидеринурией. Дифференциальная диагностика также основана на обнаружении гемолизинов, для чего выполняют пробу Кумбса.
Лечение
Патогенетических методов лечения ПНГ не существует. Тяжесть анемии обусловлена интенсивностью гемолиза и ответной реакцией клеток эритроидного ряда. При анемии целесообразно переливание эритроцитов, предварительно трижды отмытых изотоническим раствором хлорида натрия. Такие эритроциты переливают 1 раз в 4-5 дней в дозе 200-400 мл. Многие больные нуждаются в таких переливаниях на протяжении нескольких месяцев.
В последнее время применяют средства, обладающие антиоксидантным действием и способствующие стабилизации мембран эритроцитов. Витамин Е назначают в дозе 3-4 мг/сут.
Препараты железа назначают при его значительной потере или выраженном дефиците.
Для борьбы с тромбозами, как правило, в небольших дозах применяют гепарин натрия (по 5000 ЕД 2-3 раза в день под кожу живота), а также антикоагулянты непрямого действия.
Прогноз
Продолжительность жизни пациентов составляет от одного года до семи лет с момента возникновения первых признаков заболевания. Кроме того, описаны случаи значительной продолжительности жизни больных. Редко регистрируют полную ремиссию и даже полное выздоровление.
ГИПОПЛАСТИЧЕСКИЕ (АПЛАСТИЧЕСКИЕ) АНЕМИИ
Сущность гипопластической (апластической) анемии состоит в резком угнетении костномозгового кроветворения, что сопровождается снижением количества эритроцитов, лейкоцитов и тромбоцитов. Известна также парциальная форма гипопластической анемии с угнетением образования только эритроцитов.
Апластическая анемия (АА) - относительно редкое заболевание. Его регистрируют с частотой около 0,5 случая на 100 тыс. населения. По мере увеличения возраста пациентов от одного года до 20 лет число случаев болезни увеличивается. Различий в частоте заболевания среди лиц в возрасте от 20 до 60 лет не обнаружено, но после достижения возраста 60 лет число больных
увеличивается. В некоторых семьях существует генетическая предрасположенность к болезни.
Этиология
К резкому угнетению костномозгового кроветворения приводят различные причины :
• внешние факторы, оказывающие миелотоксическое действие (инфекционные заболевания, ионизирующая радиация, цитостатические препараты и другие лекарственные средства, различные химические вещества);
• внутренние (эндогенные) причины (влияние токсических веществ при уремии, гипотиреозе и др.);
• аутоагрессия и образование антител к кроветворным клеткам;
• идиопатические формы, когда не удается обнаружить никаких причин развития анемии (у 50% больных).
Патогенез
Механизм развития гипоплазии (аплазии) костного мозга окончательно не выяснен. Предполагают несколько механизмов развития АА:
• поражение полипотентной стволовой клетки костного мозга;
• подавление кроветворения, связанное с воздействием иммунных механизмов (клеточных, гуморальных);
• нарушения функционирования элементов микроокружения;
• дефицит факторов, стимулирующих кроветворение.
Содержание веществ, непосредственно участвующих в процессе кроветворения (железо, витамин B12, протопорфирин), не снижено, но они не могут быть использованы кроветворной тканью.
Клиническая картина
Клиническая картина болезни чрезвычайно разнообразна, в основном связана с цитопенией и зависит от степени ее выраженности. Существуют переходные формы заболевания - от частичного угнетения кроветворения до выраженной аплазии костного мозга.
Клиническая картина представлена тремя основными синдромами: цитопеническим, септико-некротическим и геморрагическим. Их различная выраженность обусловливает разнообразие данных, получаемых на различных этапах диагностического поиска.
На первом этапе диагностического поиска обнаруживают такие неспецифические признаки, как повышенная утомляемость и слабость. Часто больные адаптируются к анемии и обращаются к врачу лишь при развернутой картине болезни. Геморрагический синдром манифестирует различными кровотечениями (носовыми, маточными) и образованием кровоподтеков.
На втором этапе в начальных стадиях болезни, а также при хроническом течении обнаруживают лишь умеренную бледность кожного покрова и видимых слизистых оболочек, иногда - кровоподтеки. При остром течении кроме выраженной бледности отмечают значительный геморрагический синдром, не-
кроз слизистых оболочек и высокую лихорадку. Различные воспалительные заболевания (пневмония) сопровождаются характерными симптомами. Печень и селезенка обычно не увеличены, но при обнаружении антител к эритроцитам (аутоиммунная форма болезни) возможны умеренная спленомегалия, а также легкая желтушность кожи и склер, что связано с существованием гемолитического компонента.
Основным считают третий этап диагностики. В периферической крови определяют выраженную анемию (обычно - нормохромную). Содержание гемоглобина снижается до 20-30 г/л. Количество ретикулоцитов уменьшается, что свидетельствует о гипорегенераторном состоянии костного мозга. Характерны выраженная лейкопения и гранулоцитопения. Число лимфоцитов не изменено. Количество тромбоцитов иногда снижается до нуля. В большинстве случаев значительно увеличивается СОЭ (до 30-50 мм/ч).
В костном мозге уменьшено содержание миелокариоцитов (клеток красного костного мозга, содержащих ядро), увеличено число лимфоцитов, тучных и плазматических клеток. Мегакариоциты могут полностью отсутствовать. При гистологическом исследовании костного мозга обращает на себя внимание почти полное исчезновение костномозговых элементов и их замещение жировой тканью.
Содержание железа в крови у большинства больных увеличено, а насыщение трансферрина железом достигает практически 100%.
Диагностика
Диагностика заболевания основана на данных цитологического анализа периферической крови и цитоморфологического исследования костного мозга. При подозрении на АА обязательно выполнение трепанобиопсии. Необходимо провести дифференциальную диагностику с рядом заболеваний, сопровождающихся цитопенией: дебютом ОЛ, метастазами рака в костный мозг и костномозговой формой ХЛЛ. Панцитопения у пожилых людей также может быть признаком В12ДА, но при этом в костном мозге обнаруживают мегалобласты. При патоморфологическом изучении трепаната отмечают гиперплазию костного мозга в противоположность аплазии при АА. Следует подчеркнуть, что диагноз АА - диагноз исключения, устанавливаемый лишь в том случае, если перечисленные причины развития аплазии полностью исключены.
Течение
Согласно международным критериям, принято выделять две основные формы АА. Критерии тяжелой формы: количество нейтрофилов в периферической крови менее 0,5х109/л, тромбоцитов - менее 20х109/л, ретикулоцитов - менее 10%. При исследовании костного мозга отмечают выраженное снижение клеточности или умеренную гипоплазию с количеством гемопоэтических клеток менее 30%. Если показатели периферической крови соответствуют хотя бы двум критериям, а результаты исследования костного мозга - одному, то можно диагностировать тяжелую форму АА. При нетяжелой форме заболевания прогноз намного лучше (все перечисленные показатели изменены существенно меньше).
Формулировка развернутого клинического диагноза АА (гипопластической анемии) должна включать следующие компоненты:
• название анемии (в данном случае АА);
• характер течения (острый, подострый, хронический);
• наиболее выраженные синдромы (геморрагический, септико-некротический);
• осложнения.
Лечение
Лечение АА - непростая задача, включающая:
• ликвидацию (ограничение) контакта больного с лицами, страдающими инфекционными заболеваниями (в том числе и неспецифическими);
• устранение воздействия этиологического фактора (например, лекарственных или химических средств, если доказана связь между их приемом и развитием болезни);
• трансфузионную терапию;
• спленэктомию;
• введение иммуноглобулина антитимоцитарного;
• применение глюкокортикоидов и циклоспорина;
• трансплантацию костного мозга.
Объем перечисленных мероприятий при разном течении болезни неодинаков, но проведение части из них обязательно для всех больных.
Больного АА, особенно при содержании гранулоцитов менее 0,5х109/л, изолируют в специально выделенную палату с боксом, где медицинский персонал меняет обувь и одежду. Кожу пациента обрабатывают антисептическим мылом; обязательна санация ротовой полости. Для ликвидации кишечной микрофлоры назначают неадсорбируемые антибактериальные препараты. Для уменьшения менструальных кровопотерь рекомендовано длительное применение средств, уменьшающих кровопотерю или полностью прекращающих менструации (гормональные комбинированные препараты).
При глубокой анемии переливают эритроцитарную массу или отмытые эритроциты. При количестве тромбоцитов 20х109/л или геморрагическом синдроме проводят трансфузии тромбоцитарной массы.
При нетяжелой форме эффективна спленэктомия, так как селезенка - орган, в котором образуются антитела, участвующие в цитотоксических реакциях. Эффект спленэктомии возникает не сразу.
При тяжелой форме АА эффективен иммуноглобулин антитимоцитарный. Препарат вводят внутривенно в течение 5-8 дней. Пятилетняя выживаемость после лечения им составляет 50-70%. Применение иммуноглобулина антитимоцитарного сочетают с назначением циклоспорина - препарата, влияющего на функцию лимфоцитов посредством подавления образования и секреции лимфокинов. Возможно, иммуносупрессивный эффект циклоспорина обусловливает подавление продукции антител против клеток костного мозга и периферической крови. Сначала назначают внутривенное введение препарата, а затем - прием внутрь.
При тяжелой форме АА рекомендована трансплантация костного мозга от HLA-совместимого донора. Показания к ней ограничены возрастом (не старше 40 лет).
Прогноз
При нетяжелой форме АА прогноз более благоприятный. Выполнение спленэктомии и применение иммуноглобулина антитимоцитарного обусловливает пятилетнюю выживаемость около 80% больных. При тяжелой форме заболевания прогноз значительно хуже: в течение 6 мес умирают 50% больных; в течение года живут лишь 20% пациентов.
Профилактика
Исключают контакт с различными внешними факторами (ионизирующая радиация, бензол, цитостатические средства). Меры первичной профилактики идиопатической формы аплазии костного мозга неизвестны.
ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ
Геморрагические диатезы (ГД) - группа врожденных или приобретенных болезней и синдромов, основным клиническим признаком которых служит повышенная кровоточивость - склонность к повторным кровотечениям или кровоизлияниям, возникающим спонтанно или после незначительных травм.
Повышенная кровоточивость в клинической картине внутренних болезней может быть:
• основным признаком заболевания (например, тромбоцитопенической пурпуры, гемофилии, синдрома Ослера-Рандю и др.);
• синдромом, служащим частью какого-либо определенного заболевания (например, цирроза печени, СКВ, ИЭ и др.);
• осложнением проводимого лечения (прямые и непрямые антикоагулянты, фибринолитические препараты).
Выраженность кровоточивости может варьировать в больших пределах - от незначительных кровоизлияний в кожу или слизистые оболочки до массивных кровоизлияний в полость сустава и межмышечные пространства, а также фатальных кровотечений из внутренних органов (кишечник, желудок, матка и др.).
В развитии кровоточивости принимают участие следующие факторы:
• тромбоцитарное звено гемостаза;
• плазменное звено гемостаза;
• состояние сосудистой стенки;
• фибринолитическая активность крови.
При различных ГД ведущая роль какого-либо из перечисленных факторов весьма различна. В ряде случаев главным является какой-либо один механизм, в то время как в других ведущей служит комбинация тех или иных факторов.
Классификация
В соответствии с ведущим механизмом кровоточивости среди наиболее распространенных ГД выделяют несколько больших групп.
• Тромбоцитопении (снижение количества тромбоцитов) и тромбоцитопатии (нарушение функциональных свойств тромбоцитов).
• Коагулопатии (гемофилии), развивающиеся:
- при недостаточном содержании прокоагулянтов, участвующих в плазменном звене гемостаза;
- недостаточной функциональной активности прокоагулянтов;
- присутствии в крови ингибиторов отдельных прокоагулянтов.
• Ангиопатии (вазопатии) - повреждение сосудистой стенки врожденной этиологии или развивающееся в результате иммуноаллергического или инфекционно-токсического воздействия.
• Избыточный фибринолиз, возникающий:
- при лечении тромболитическими препаратами;
- дефекте ингибитора плазмина или избытке тканевого активатора плазминогена наследственной этиологии.
• ДВС-синдром, представленный сочетанием нарушений различных компонентов гемостаза (тромбоцитопения, коагулопатия и др.).
Клиническая картина
Клиническая картина ГД чрезвычайно разнообразна, что зависит от формы и выраженности геморрагического синдрома, вовлечения в патологический процесс различных органов и систем, а также осложнений. В связи с этим клиническую картину ГД целесообразно представить в качестве сочетания различных видов кровоточивости, синдрома поражения органов и систем, неспецифического синдрома и синдрома нарушений гемостаза, обнаруживаемых с помощью лабораторных методов исследования.
В настоящее время выделяют пять типов кровоточивости, характерных для различных ГД:
• гематомный;
• пятнисто-петехиальный (микроциркуляторный);
• смешанный микроциркуляторно-гематомный;
• васкулитно-пурпурный;
• ангиоматозный.
При гематомном типе преобладают массивные, глубокие, напряженные и весьма болезненные кровоизлияния в крупные суставы, мышцы, подкожную жировую и забрюшинную клетчатку. Они вызывают расслоение и деструкцию тканей, развитие артрозов, контрактур, патологических переломов, костных псевдоопухолей и атрофию мышц. Возникают профузные спонтанные, посттравматические и послеоперационные кровотечения. Этот тип характерен исключительно для наследственных коагулопатий (в частности, для гемофилии А и В).
Пятнисто-петехиальный (микроциркуляторный) тип характеризуется безболезненными не напряженными, не сдавливающими окружающие ткани поверхностными кровоизлияниями в кожу и слизистые оболочки, петехиями, синяками, десневыми, носовыми и маточными кровотечениями. Кровоизлияния возникают при незначительном повреждении сосудов микроциркуляторного русла во время измерения АД, в местах пальпации, при растирании кожи
рукой и т.д. Для этого типа кровоточивости не характерно образование гематом. Мышцы, суставы и другие отделы опорно-двигательного аппарата интактны. Кровотечения при полостных оперативных вмешательствах редки и не склонны к рецидивированию. Этот тип кровоточивости отмечают при тромбоцитопениях и тромбоцитопатиях.
Смешанный микроциркуляторно-гематомный тип кровоточивости характеризуется не просто сочетанием признаков двух вышеперечисленных вариантов геморрагического синдрома, но и рядом присущих только ему качественных особенностей. В клинической картине преобладает петехиальнопятнистая кровоточивость. Гематомы немногочисленны, но достигают очень больших размеров и располагаются преимущественно в подкожной жировой или забрюшинной клетчатке. Кровоизлияния в суставы редки и не ведут к развитию артрозов и атрофии мышц. В зависимости от локализации гематомы могут имитировать картину острого живота, непроходимости кишечника и острого аппендицита. Этот тип кровоточивости отмечают при наиболее тяжелых формах коагулопатий (гемофилии А), ДВС-синдроме и передозировке антикоагулянтов.
Васкулитно-пурпурный тип кровоточивости объединяет все геморрагии, обусловленные воспалительным процессом в микрососудах. Последние возникают на фоне локальных экссудативно-воспалительных процессов и общих иммуноаллергических или инфекционно-токсических нарушений. Геморрагические высыпания на коже, как правило, симметричны (как на конечностях, так и на туловище) и несколько приподняты вследствие воспалительной инфильтрации и отека. Часто возникновению геморрагии предшествуют зудящие высыпания, имеющие вид небольших уплотнений. Эти элементы затем приобретают пурпурный вид вследствие пропитывания кровью. Характерная черта - длительно сохраняющаяся после исчезновения геморрагии сыпь бурого цвета. При других типах кровоточивости такой остаточной пигментации не бывает. Отмечают при геморрагическом васкулите.
Ангиоматозный тип кровоточивости характеризуется отсутствием спонтанных и посттравматических кровоизлияний в кожу, подкожную жировую клетчатку и другие ткани и органы, но возникают весьма упорные кровотечения одной-двух локализаций (носовые, реже - гематурия, легочные и желудочнокишечные кровотечения). Этот тип кровоточивости отмечают при различных формах телеангиэктазии.
Кроме геморрагического синдрома в ряде случаев обнаруживают неспецифический синдром, представленный общими симптомами (лихорадка, потеря массы тела, слабость).
Суставной синдром манифестирует отечностью болезненного сустава и возникает при геморрагическом васкулите и гемофилии. В последнем случае он обусловлен кровоизлиянием в суставы - гемартрозом.
Абдоминальный синдром в виде схваткообразных болей в животе, тошноты (иногда - рвоты) и картины острого живота нередко регистрируют при геморрагическом васкулите и гемофилии.
Почечный синдром манифестирует гематурией, протеинурией, дизурическими явлениями и приступами почечной колики. Его обнаруживают при гемофилии (упорная гематурия) и геморрагическом васкулите.
Анемический синдром: слабость, головокружения, тахикардия, систолический шум в точках выслушивания сердца, артериальная гипотензия - все эти симптомы могут сопутствовать любой форме ГД и зависят от выраженности геморрагического синдрома.
Для определения патогенетического механизма кровоточивости используют лабораторные методы исследования. В настоящее время существует множество методов изучения гемостаза, причем многие из них по сути дела дублируют друг друга, в связи с чем необходимо определение рационального минимума исследований.
Ниже представлен примерный алгоритм использования методов исследования гемостаза, который необходимо применять при ГД любого происхождения.
• Исследование начинают с определения времени кровотечения (в норме - от 2 до 5 мин), которое позволяет оценить участие тромбоцитарного звена гемостаза в патогенезе кровоточивости: при его удлинении роль нарушений тромбоцитарного звена несомненна.
• Определение количества тромбоцитов в периферической крови позволяет сделать более детальное заключение: при его снижении речь идет о тромбоцитопенической пурпуре. Если количество тромбоцитов не снижено, то следует думать о тромбоцитопатии, для чего проводят определение функциональных свойств тромбоцитов (адгезии и агрегации).
• Если же при существовании геморрагических признаков время кровотечения не увеличено, то, вероятно, речь может идти о ГД, обусловленном нарушением плазменного звена гемостаза. С этой целью определяют протромбиновое время (ПВ) и АЧТВ.
• При удлинении ПВ и неизмененном АЧТВ дефект находится во внешнем каскаде свертывания крови (дефицит фактора VII - гипоконвертинемия).
• При удлинении АЧТВ и неизмененном ПВ дефект гемостаза локализуется во внутреннем каскаде (дефицит фактора VIII, IX).
• При удлинении ПВ и АЧТВ речь идет о дефиците фактора X, V, II (протромбин) или I (фибриноген). Для дальнейшего уточнения требуется определение концентрации фибриногена, а также использование так называемых дефицитных сывороток. Это исследование проводят в специализированных лабораториях.
• Если количество и функциональные свойства тромбоцитов не изменены, а показатели коагуляционного гемостаза также соответствуют норме, то следует думать, что ГД обусловлен патологическими изменениями сосудистой стенки или другими причинами, в частности повышенным фибринолизом. Об этом будет свидетельствовать укорочение времени лизиса эуглобулиновых сгустков (в норме - 2-4 ч).
Перечисленные методы исследования системы гемостаза при ГД позволяют обнаружить наиболее распространенные нарушения гемостаза. Его более детальное исследование обычно проводят в специализированных лабораториях.
Далее будет рассмотрена диагностика ГД, обусловленных снижением количества тромбоцитов (тромбоцитопении), патологическими изменениями плазменного звена гемостаза (гемофилии) и сосудистой стенки (ангиопатии), а также ДВС-синдрома.
ТРОМБОЦИТОПЕНИЧЕСКИЕ ПУРПУРЫ
Тромбоцитопенические пурпуры объединяют целую группу заболеваний и синдромов, обусловленных снижением количества тромбоцитов в периферической крови вследствие их повышенного разрушения, недостаточного образования и повышенного потребления. Патологические изменения тромбоцитов служат причиной кровоточивости в 80% случаев, при этом тромбоцитопения - ее наиболее частый признак.
Причины тромбоцитопений можно представить в следующем виде.
• Тромбоцитопении вследствие повышенного разрушения тромбоцитов:
- наследственные формы (обусловленные дефектами ферментов гликолиза или цикла Кребса);
- приобретенные формы;
иммунные (аллоиммунные, трансиммунные, гетероиммунные, аутоиммунные):
V симптоматические;
V идиопатические;
неиммунные (механическое разрушение тромбоцитов при гемангиомах, спленомегалиях различной этиологии, протезах клапанов
сердца).
• Тромбоцитопении вследствие недостаточного образования тромбоцитов (при гемобластозах, гипо- и апластической анемии, В12ДА, ПНГ).
• Тромбоцитопении вследствие повышенного потребления тромбоцитов (ДВС-синдром).
Наиболее разнообразна группа тромбоцитопений, обусловленных повышенным разрушением тромбоцитов. Наследственные формы регистрируют редко (не более 25%), и они связаны с дефектом ферментов гликолиза или цикла Кребса, что обусловливает повреждение мембраны тромбоцита и укорочение его жизни. Приобретенные формы составляют большинство тромбоцитопений, при этом наибольшее значение имеют иммунные формы (особенно аутоиммунные). Аутоиммунные формы связаны с образованием аутоантител (иммуноглобулинов) против собственных тромбоцитов. Продолжительность циркуляции таких клеток в сосудистом русле сокращается с нормальных 7-10 дней до нескольких часов, поскольку их фагоцитируют макрофаги ретикулоэндотелиальной системы.
Аутоиммунные формы тромбоцитопений могут быть симптоматическими и служить частью какого-либо заболевания (СКВ, хронического активного гепатита и др.). Если не удается установить причину их возникновения, то говорят об идиопатической аутоиммунной тромбоцитопении, которую следует расценивать как самостоятельное заболевание.
Аллоиммунные и трансиммунные формы регистрируют исключительно у новорожденных, аутоиммунные и гетероиммунные (образование антител против фиксированного на тромбоцитах чужеродного антигена - вируса, лекарственного средства) формы - у взрослых.
Тромбоцитопения вследствие недостаточного образования тромбоцитов (как правило, при различных заболеваниях системы кроветворения) - часть основного заболевания. Ее выраженность может колебаться в широких пределах.
Тромбоцитопения вследствие повышенного потребления - обязательная часть ДВС-синдрома, и ее выраженность, наряду с коагулопатией потребления, определяет тяжесть течения ДВС-синдрома.
Далее будет описан диагностический поиск при идиопатической тромбоцитопенической пурпуре иммунной этиологии.
Идиопатическая тромбоцитопеническая пурпура
В основе идиопатической тромбоцитопенической пурпуры (ИТП) лежит повышенное разрушение тромбоцитов макрофагами селезенки, что связано с фиксацией на поверхности первых антител (IgG), возможно, продуцируемых лимфоидной тканью селезенки больных и направленных против антигенов собственных тромбоцитов. В результате этого резко укорачивается продолжительность жизни тромбоцитов (до нескольких часов вместо 7-10 дней в норме). В ответ на усиленное разрушение тромбоцитов происходит компенсаторное усиление их продукции в костном мозге в несколько раз. Об этом свидетельствует увеличение числа мегакариоцитов в костном мозге и отсутствие вокруг них тромбоцитов, что указывает на их повышенный выход в кровяное русло, но не на их недостаточную «отшнуровку».
ИТП лежит в основе практически 95% тромбоцитопений. Ежегодно обнаруживают от 10 до 125 свежих случаев ИТП на каждый миллион населения. ИТП считают острой, если она продолжается менее 6 мес. Более длительные случаи ИТП следует расценивать как хроническую ИТП. Острую форму ИТП чаще регистрируют у детей. У 75% заболевших она заканчивается полным выздоровлением. Взрослые чаще страдают хроническим вариантом ИТП, при этом до 5% из них погибают от кровотечений (в основном от кровоизлияния в мозг). ИТП чаще страдают женщины (в 4 раза чаще мужчин) молодого и среднего возраста.
Клиническая картина
Клиническую картину болезни определяет геморрагический синдром. Как правило, болезнь течет волнообразно: «спокойные» периоды прерывают обострения, во время которых геморрагический синдром рецидивирует (усиливается), что сопровождается снижением количества тромбоцитов в периферической крови.
На первом этапе диагностического поиска можно получить информацию о жалобах больного на возникновение кожных геморрагий и кровотечений из слизистых оболочек. Кожные геморрагии образуются после небольших травм или спонтанно и чаще всего локализуются на передней поверхности туловища и на конечностях (чаще - на внутренней поверхности). В местах инъекций лекарственных препаратов могут образоваться более крупные кровоизлияния. Больные также отмечают кровоточивость десен и носовые кровотечения. После экстракции зуба кровотечение возникает сразу же. Реже пациенты отмечают кровохарканье, кровотечения из ЖКТ и гематурию. Женщины предъявляют жалобы на маточные кровотечения или более обильные и длительные менструации. Часть больных могут сообщить, что у них ранее при исследовании крови
обнаруживали тромбоцитопению, причем периоды усиления кровоточивости сопровождались более выраженным снижением количества тромбоцитов. Наконец, пациенты могут сообщить об успешном лечении глюкокортикоидами (преднизолон), приводившем к уменьшению кровоточивости и увеличению числа тромбоцитов. Некоторым больным ранее предлагали спленэктомию. Данные анамнеза могут помочь в первую очередь при выяснении наследования заболевания. Существование аномалий развития у ближайших родственников обследуемого позволяет заподозрить и установить наследственную этиологию тромбоцитопении.
На втором этапе диагностического поиска в период обострения заболевания можно обнаружить пятнисто-петехиальный тип кровоточивости - расположение геморрагий на передней поверхности туловища и конечностях, причем их давность различна: наряду с темно-синими отмечают багровые, зеленоватые и желтые пятна. Пробы на ломкость капилляров (симптомы щипка и жгута) положительны. Изменения со стороны внутренних органов обычно не обнаруживают, но у части больных отмечают увеличение селезенки, особенно если тромбоцитопения сочетается с гемолитической анемией. Увеличение печени не свойственно тромбоцитопении. У некоторых пациентов в период обострения заболевания могут незначительно увеличиваться лимфатические узлы (особенно шейные) и повышаться температура тела (до субфебрильных цифр).
Решающим считают третий этап диагностического поиска, во время которого выясняют этиологию кровоточивости. Прежде всего отмечают увеличение времени кровотечения, а также тромбоцитопению (вплоть до полного исчезновения тромбоцитов из периферической крови). Ретракция кровяного сгустка на высоте тромбоцитопении отсутствует. При числе тромбоцитов, превышающем 50х109/л, геморрагический синдром обнаруживают редко. Отмечают изменения формы тромбоцитов (большие, атипичной формы, со скудной специфической зернистостью).
Показатели коагуляционного гемостаза не изменены (ПТИ, АЧТВ).
Содержание эритроцитов и гемоглобина может быть нормальным. Лишь при частых кровотечениях развивается постгеморрагическая анемия. Увеличение числа ретикулоцитов в крови зависит от интенсивности кровопотери. Количество лейкоцитов у большинства больных нормальное.
Цитологическая картина костного мозга обычно не изменена, но у большинства больных увеличено число мегакариоцитов (преобладают молодые формы).
Диагностика
Диагностика заболевания основана на обнаружении петехиально-пятнистого типа кровоточивости в сочетании с тромбоцитопенией, не вызванной определенными причинами (инфекционным заболеванием, приемом лекарственных средств), нормальным или повышенным количеством мегакариоцитов в костном мозге и отсутствием отшнуровки тромбоцитов. Большое значение в диагностике придают положительному эффекту от приема преднизолона.
При дифференциальной диагностике следует исключить гемобластозы, В12ДА, ГА (болезнь Маркиафавы-Микели) и АА. Следует помнить, что аутоиммунная тромбоцитопения может быть дебютом СКВ.
ИТП (аутоиммунную) следует отличать от иммунной (гетероиммунной) тромбоцитопении, возникающей после перенесенной вирусной инфекции или приема некоторых лекарственных препаратов. Гетероиммунные тромбоцитопении обычно возникают остро у детей и лиц пожилого возраста. После отмены препарата количество тромбоцитов постепенно восстанавливается. Явления кровоточивости обычно выражены незначительно. В отличие от аутоиммунной тромбоцитопенической пурпуры, часто имеющей хроническое течение, в дальнейшем тромбоцитопения не возникает.
Лечение
Базисное лечение аутоиммунной тромбоцитопенической пурпуры, направленное на увеличение количества тромбоцитов, складывается из применения глюкокортикоидов, спленэктомии и назначения иммунодепрессантов (цитостатических) .
Лечение начинают с назначения преднизолона в дозе 1 мг/кг массы тела с ее последующим снижением и постепенной отменой препарата после нормализации количества тромбоцитов и ликвидации клинических и лабораторных признаков заболевания. В ряде случаев один такой курс может привести к длительной ремиссии и даже окончательному излечению. Тем не менее, как правило, после полной отмены преднизолона или даже при попытке снижения дозы наступает рецидив, требующий возврата к исходным дозам.
При неполном и нестабильном эффекте лечения преднизолоном (обычно - через 3-4 мес с момента его начала) возникают показания к спленэктомии или назначению иммунодепрессантов. Спленэктомия оказывает положительный эффект в 80% случаев. Ее результаты лучше тогда, когда нормализация числа тромбоцитов наступала после приема небольшой дозы преднизолона. В зависимости от эффекта спленэктомии, в дальнейшем проводят курсы лечения преднизолоном в существенно меньших дозах, чем до операции. У части больных спленэктомия не дает отчетливый эффект, и тогда назначают цитостатические препараты: азатиоприн (по 2-3 мг/кг массы тела в сутки) или циклофосфан (по 200-400 мг/сут) в течение 3-5 мес.
Альтернатива вышеуказанному лечению - внутривенное введение высоких доз иммуноглобулина. Препарат вводят из расчета 200-400 мг/кг в сутки посредством внутривенной капельной инфузии в течение 1-5 дней. Такое лечение проводят при неэффективности или невозможности применения глюкокортикоидов с целью повышения количества тромбоцитов для обеспечения полноценного гемостаза в период спленэктомии. Длительность эффекта восстановления тромбоцитов при монотерапии высокими дозами иммуноглобулина без последующей спленэктомии колеблется от 14 до 28 дней.
При хронической форме болезни, для которой характерно низкое число тромбоцитов в крови (менее 20х109/л) и которая сопровождается различными признаками кровоточивости, используют препараты, стимулирующие другие звенья гемостаза: этамзилат (антифибринолитический эффект) по 0,25 мг
3-4 раза в сутки внутрь или дицинон* (активирующее действие на синтез тромбопластина - гликопротеина клеточных мембран) по 2-4 мг каждые 4-6 ч. Также применяют замороженную нативную плазму в дозе до 600 мл/сут.
Кровотечения останавливают с помощью самых разных гемостатических средств (гемостатическая губка, тампонада с аминокапроновой кислотой). Гемотрансфузии у больных ИТП проводят только по жизненным показаниям; следует переливать отмытые и индивидуально подобранные эритроциты.
ГЕМОФИЛИИ
Гемофилии - одни из классических форм ГД, известные с древнейших времен. Это генетическое заболевание, наследуемое по рецессивному типу, сцепленному с полом. Ген, ответственный за синтез фактора VIII и IX, расположен в хромосоме Х, вследствие чего гемофилией болеют исключительно мужчины. Женщина заболевает лишь в случае брака между больным гемофилией и женщиной-кондуктором.
Причина кровоточивости заключается в дефиците или недостаточной активности фактора VIII, IX или XI. Заболевание, обусловленное дефицитом фактора VIII, обозначают гемофилией А, дефицитом фактора IX - гемофилией В, дефицитом фактора XI - гемофилией С. Чаще всего регистрируют гемофилию А (85-90%), а гемофилию В - существенно реже (10-15%). Точная частота возникновения гемофилии С неизвестна.
Фактор VIII синтезируется в гепатоцитах и эндотелиальных клетках. Его молекулу кодирует ген, расположенный на длинном плече хромосомы Х, - один из самых больших генов. Фактору VIII принадлежит ключевая роль в процессе образования фибрина, поэтому его количественная или качественная неполноценность способствует кровоточивости.
Клиническая картина
Клиническую картину заболевания определяет гематомный тип кровоточивости: характерны кровоизлияния в крупные суставы, внутримышечные гематомы, обильные и длительные кровотечения при травмах, гематурия. Реже регистрируют легочные и желудочно-кишечные кровотечения, внутричерепные кровоизлияния и забрюшинные гематомы.
На основании определения выраженности снижения концентрации фактора VIII в крови больного выделяют три степени тяжести заболевания. Тяжелую степень болезни диагностируют у тех больных, у которых содержание фактора VIII не превышает 1% нормы. Умеренную степень (заболевание средней тяжести) диагностируют при концентрации фактора VIII в пределах от 1 до 4% нормальной. Заболевание легкой степени определяют при содержании фактора VIII в пределах от 5 до 25% нормы. Болезнь протекает в тяжелой форме лишь в 30-40% случаев.
На первом этапе диагностического поиска удается выяснить, что кровоточивость возникла с раннего детского возраста. Так, при рождении могут образоваться цефалогематомы. Когда ребенок начинает ходить, то при падениях возникают носовые кровотечения, гематомы в области головы и мягких тка-
ней. В старшем возрасте развиваются кровоизлияния в суставы. Одновременно возможно поражение от 1-2 до 6-8 суставов. В анамнезе отмечают указания на возникновение кровотечений после травм и операций, развивающиеся не сразу после вмешательства, как при тромбоцитопенической пурпуре, а спустя 1-5 ч. Длительные кровотечения обычно возникают после экстракции зуба или тонзиллэктомии.
На втором этапе диагностического поиска можно обнаружить поражение опорно-двигательного аппарата, связанное с кровоизлияниями в полость сустава. Выделяют несколько типов суставного поражения:
• острые гемартрозы (первичные и рецидивирующие);
• хронические геморрагически-деструктивные остеоартрозы;
• вторичный иммунный ревматоидный синдром (как осложнение основного процесса).
Острый гемартроз - внезапное возникновение (часто после небольшой травмы) или резкое усиление боли в суставе. Последний увеличивается в объеме, становится горячим на ощупь, кожа над ним гиперемирована. При больших кровоизлияниях определяют флюктуацию. Боль проходит после эвакуации крови из полости сустава и одновременной трансфузии антигемофильной плазмы. При хроническом геморрагическом ОА в «холодном» периоде функция сустава может не нарушаться, но при рентгенологическом исследовании определяют все характерные признаки ОА (сужение суставной щели, остеофиты, деформации). Со временем подвижность сустава нарушается, что сочетается с атрофией мышц, приводящих сустав в движение.
Вторичный ревматоидный синдром выражается в хроническом воспалительном поражении мелких суставов кисти и стопы, в которых ранее не возникало кровоизлияние, с последующей типичной деформацией, болью и выраженной утренней скованностью. С возрастом распространенность и тяжесть всех поражений суставов неуклонно прогрессируют, что приводит к инвалидности. Прогрессирование поражений суставов зависит от частоты развития острых гемартрозов. Очень тяжелы и опасны подкожные, межмышечные и забрюшинные гематомы. Профузные желудочно-кишечные кровотечения могут быть спонтанными или спровоцированы приемом ацетилсалициловой кислоты, бутадиона и других ульцерогенных препаратов.
Кровоизлияния в брыжейку и сальник имитируют острые хирургические заболевания - острый аппендицит, непроходимость кишечника и др.
Единственный критерий диагностики в этой ситуации - быстрый положительный эффект интенсивной заместительной терапии (переливание антигемофильной плазмы).
Характерная черта гемофилии - длительные кровотечения при травмах и операциях, возникающие не сразу после них, а через 1-5 ч. Тонзиллэктомия при гемофилии значительно более опасна, чем полостные хирургические операции, точно так же, как и экстракция зубов. Все эти вмешательства следует проводить при заместительной терапии концентратами антигемофильных препаратов.
На третьем этапе диагностического поиска подтверждают этиологию кровотечений. У больных гемофилией удлинено АЧТВ при нормальном ПТИ.
Количество тромбоцитов соответствует норме, что обусловливает неизмененную длительность кровотечения. Тромбиновое время, указывающее на активность комплекса «гепарин-антитромбин III», также не изменено. Пробы жгута и щипка отрицательные.
Форму гемофилии устанавливают с помощью добавления к плазме больного так называемой бариевой плазмы (плазма здорового человека, смешанная с сульфатом бария, связывающим факторы протромбинового комплекса II, VII, IX и X, но не связывающим фактор VIII). Если удлиненное АЧТВ нормализуется после добавления бариевой плазмы, то речь идет о гемофилии А, если нет - то о гемофилии В. Более точную дифференциальную диагностику проводят при смешивании плазмы больного с образцами плазмы больных с заведомо известной формой гемофилии и отсутствии при этом нормализации свертывания плазмы пациента.
Диагностика
Гемофилию следует предполагать у всех больных с гематомным типом кровоточивости и поражением опорно-двигательного аппарата, а также в случаях упорных поздних кровотечений при хирургических вмешательствах. Имеет значение обнаружение семейных случаев заболевания по мужской линии, а также начало болезни в раннем детском возрасте. Решающими в установлении диагноза гемофилии и определении ее формы считают результаты лабораторных исследований.
Дифференциальная диагностика
Гемофилию необходимо дифференцировать от ангиогемофилии (болезнь Виллебранда) - наследственным заболеванием, обусловленным нарушением синтеза основного компонента фактора VIII, участвующего в тромбоцитарнососудистом гемостазе и обозначаемого как фактор Виллебранда (VIII-ФВ). Полагают, что VIII-ФВ - регулятор синтеза одного из компонентов фактора VIII, в связи с этим при болезни Виллебранда снижено содержание как VIII-ФВ, так и фактора VIII, тогда как при гемофилии снижена лишь концентрация фактора VIII, а содержание VIII-ФВ соответствует норме. При болезни Виллебранда редко обнаруживают гемартрозы. Нарушена адгезивность тромбоцитов, время кровотечения удлинено.
Лечение
Гемостатическое лечение назначают в периоды кровотечения. Основной метод лечения - заместительная терапия, связанная с применением гемопреципитатов, содержащих фактор VIII (антигемофильная плазма, криопреципитат, концентраты фактора VIII). Наиболее эффективным считают криопреципитат, выделяемый из плазмы с помощью криоосаждения, - белковый концентрат, содержащий большое количество фактора VIII. Концентрат фактора VIII вводят внутривенно. При гемофилии В применяют концентраты фактора IX. В экстренных случаях проводят массивные прямые переливания крови не реже 3 раз в сутки. Кровоточащие участки обрабатывают тромбином или аминокапроновой кислотой.
При острых гемартрозах сустав временно иммобилизуют, удаляют кровь из его полости и вводят гидрокортизон. При ревматоидном синдроме назначают прием внутрь преднизолона в дозе 20-40 мг/сут с последующим медленным снижением дозы. В «холодном» периоде поражения суставов назначают физиотерапевтические процедуры и ЛФК.
Профилактика
Предупреждение кровотечений при гемофилии включает систематическое внутривенное введение концентратов фактора VIII, трансфузионную терапию перед оперативным вмешательством и профилактику травм.
АНГИОПАТИИ(ВАЗОПАТИИ)
При ангиопатиях ГД развивается вследствие врожденного нарушения сосудистой стенки (например, наследственная геморрагическая телеангиэктазия - синдром Ослера-Рандю) или иммуноаллергического либо инфекционно-токсического поражения стенки сосуда (например, геморрагический васкулит - болезнь Шенлейна-Геноха).
При наследственной геморрагической телеангиэктазии - синдроме Ослера-Рандю - отмечают очаговое истончение сосудистой стенки вследствие недоразвития субэндотелиального слоя и малого содержания в нем коллагена. В этих случаях сосуд расширен, а его стенка состоит лишь из эндотелия. Мышечный слой подвергается дегенеративным изменениям, эластические волокна полностью или частично отсутствуют. Кровоточивость обусловлена чрезвычайно легкой ранимостью сосудистой стенки в месте ангиэктазии. Отмечают поражение капилляров и посткапиллярных венул. В других участках обнаруживают формирование кавернозных артериовенозных аневризм. Нарушенное строение не позволяет сосудам сокращаться при повреждении, что и приводит к кровоточивости.
Синдром Ослера-Рандю - наследственное заболевание (аутосомно-доминантный тип наследования), поэтому его можно зарегистрировать у лиц разного возраста.
Клиническая картина
Клиническая картина болезни зависит от выраженности изменения сосудов и распространенности поражения. Телеангиэктазии обычно обнаруживают у детей в возрасте 6-10 лет. С возрастом их число и распространенность увеличиваются.
На первом этапе диагностического поиска удается выяснить, что с детских лет у пациента спонтанно или после небольших механических травм возникали носовые кровотечения. Острые респираторные заболевания провоцируют и усиливают их. Носовые кровотечения могут быть весьма упорными и требовать специализированной оториноларингологической помощи (передняя или задняя тампонада полости носа и др.). Иногда отмечают кровохарканье и желудочно-кишечные кровотечения.
На втором этапе диагностического поиска обнаруживают телеангиэктазии на коже и слизистых оболочках, а также их кровоточивость. Телеангиэктазии чаще всего локализуются на губах, крыльях носа, языке, деснах, внутренней поверхности щек и слизистой оболочке полости носа. Иногда их обнаруживают на кончиках пальцев и коже волосистой части головы. Врожденная неполноценность сосудов внутренних органов может манифестировать артериовенозными аневризмами, локализующимися в легких, печени, почках и селезенке. При их расположении в легких возникают одышка и цианотично-красный цвет лица. Такое поражение внутренних органов диагностируют с большим трудом.
Телеангиэктазии могут сочетаться с другими признаками мезенхимальных дисплазий в виде диафрагмальных и паховых грыж, аномалий скелета, гиперэластичности кожи и гипермобильности суставов с их подвывихами.
На третьем этапе диагностического поиска, если кровотечения достаточно обильные и часто повторяются, можно обнаружить гипохромную ЖДА. Количество тромбоцитов и их функциональные свойства не изменены. Длительность кровотечения не изменена. Показатели коагуляционного гемостаза (прежде всего ПТИ и АЧТВ) соответствуют норме.
При риноскопии обнаруживают телеангиэктазии в полости носа. Если по поводу кровотечения из ЖКТ или легких проводят эндоскопическое исследование, то во время него также визуализируют телеангиэктазии. При их расположении в почечных лоханках возникает выраженная в большей или меньшей степени гематурия.
Диагностика
Диагностика синдрома Рандю-Ослера основана на определенных критериях (Curacao, 1999), которые включают:
• носовые кровотечения (спонтанные, повторные);
• телеангиэктазии (множественные);
• висцеральные поражения (желудочно-кишечные с кровотечениями и без них), легочные артериовенозные аневризмы и сосудистые аномалии печени;
• семейный характер;
• аутосомно-доминантный тип наследования.
При этом диагноз врожденного заболевания считают достоверным, если обнаруживают три критерия, вероятным - если есть два критерия, и сомнительным - если определен один критерий.
Лечение
Основные лечебные мероприятия направлены на предупреждение кровотечений и их остановку.
При носовых кровотечениях используют местные гемостатические средства. Рекомендовано применение вазопротекторов (этамзилат), кальция добезилата (парентеральное введение в дозе 10-20 мг/кг 3-4 раза в день или прием внутрь по 250-500 мг 3-4 раза в день в течение 3-4 нед) и ингибиторов фибринолиза (5% раствор аминокапроновой кислоты в дозе 100-200 мл внутривенно капельно). Применяют местно орошение 5% раствором аминокапроновой кис-
лоты, тампонаду носа масляными тампонами, прижигания и локальную лазеротерапию. Существуют хирургические методы лечения (иссечение слизистой оболочки носа с кровоточащими сосудами и телеангиэктазиями), перевязка, эмболизация сосудов, баротерапия и применение эстрогенов. Кровоточащие кожные телеангиэктации с определенным успехом лечат посредством лазерной и криотерапии. Предупреждение носовых кровотечений предусматривает соблюдение диеты, исключающей употребление острых блюд, перца, пряностей и алкоголя. Пища должна быть обогащена витаминами (С, В, А).
Профилактика
Следует избегать повреждения слизистых оболочек и кожи в местах телеангиэктазий. При развитии вследствие повторяющихся кровотечений ЖДА назначают препараты железа. Учитывая вероятность возникновения функциональных нарушений у части больных, рекомендовано ограничить прием лекарственных средств, обладающих антиагрегантным эффектом (например,
НПВС). Прогноз
В большинстве случаев прогноз благоприятный, но при выраженных телеангиэктазиях возможны не только носовые смертельные кровотечения, но и легочно-бронхиальные, желудочно-кишечные и из мочевыводящих путей.
Геморрагический васкулит (болезнь Шенлейна-Геноха) - приобретенная форма поражения сосудов, при которой геморрагический синдром выражен в различной степени, - рассмотрен в главе «Системные васкулиты».
ДИССЕМИНИРОВАННОЕ ВНУТРИСОСУДИСТОЕ СВЕРТЫВАНИЕ КРОВИ
ДВС-синдром - нарушение гемостаза, в основе которого лежит распространенное свертывание крови с образованием большого количества микросгустков и агрегатов клеток крови, что приводит к нарушению микроциркуляции в органах и тканях вплоть до ее полной блокады и развитию выраженных дистрофических изменений.
Этиология
ДВС-синдром развивается в различных ситуациях: при хирургических вмешательствах, акушерских патологических состояниях, сепсисе, злокачественных опухолях, а также при некоторых терапевтических заболеваниях (гемобластозах, ОПН, ХПН, системных васкулитах и остром гемолизе).
Патогенез
Изменение состояния свертывающей и противосвертывающей системы при развитии ДВС-синдрома проходит несколько стадий.
В начальной стадии (стадия гиперкоагуляции) под влиянием различных экзогенных (продукты жизнедеятельности бактерий, змеиный яд, трансфузионные средства и др.) и эндогенных (продукты протеолиза и цитолиза, тканевый
тромбопластин и др.) факторов активируются процессы свертывания крови и агрегации тромбоцитов.
Происходит выпадение тромбов, чему способствует проникновение в кровоток под влиянием одновременной активизации других систем (фибринолитической, калликреин-кининовой) большого количества продуктов белкового распада. Множественное тромбообразование приводит к нарушению микроциркуляции и изменениям функционирования различных органов и систем.
Активация свертывания крови вызывает истощение противосвертывающих механизмов - физиологических антикоагулянтов (система «гепаринантитромбин III») и фибринолитической системы «плазминоген-плазмин». Множественное тромбообразование влечет за собой так называемую коагулопатию потребления (снижение содержания плазменных факторов свертывания) и тромбоцитопению, что обусловливает развитие геморрагического синдрома.
В последние годы экспериментальные исследования и клинические наблюдения многих коагулологов внесли серьезные коррективы в учение о ДВСсиндроме, его патогенез и лечение.
Как известно, ранее этот процесс, сопровождающий все тяжелые катастрофические состояния, долгое время объясняли афибриногенемией, а кровоточивость трактовали как фибринолитическую. В связи с этим внутривенно вводили фибриноген*, назначали аминокапроновую кислоту и другие ингибиторы фибринолиза, что приводило к негативным результатам - полиорганной недостаточности (почечной, печеночной, легочной и т.д.). Одновременно назначали средства, повышающие свертываемость крови, так как считали, что ДВС-синдром связан с реакцией антисвертывающей системы, которая, якобы, первична. Тем не менее пусковой механиз ДВС-синдрома - внутрисосудистое свертывание крови, которое на первых этапах блокировали гепарином. Он хорошо действовал до введения антитромбина III и тромбина. Если же гепарин натрия вводили после этого, то эффект был менее выраженным. Впоследствии выяснили, что только гепарин натрия при таком тяжелом состоянии не действует, независимо от этапов этого процесса - фазы гиперкоагуляции, которая может длиться всего 3-5 мин, или гипокоагуляции.
Кроме того, было установлено, что при ДВС-синдроме содержание фибриногена может быть нормальным, а концентрация факторов свертывания крови снижена. Отмечают уменьшение содержания физиологических антикоагулянтов - протеина С и S, а также антитромбина III, инактивирующих активированные факторы свертывания. Их расход оказывается более выраженным, чем затраты факторов свертывания крови.
Кроме того, установлено, что в большинстве случаев острый ДВС-синдром (даже акушерский) носит септический характер, поскольку с помощью специальных исследований была обнаружена бактериемия, усугубляющая тромбоцитопению и ферментопатию.
К числу органов-мишеней относят кишечник (при ожоговой болезни, синдроме раздавливания тканей и др.). Инфекционный процесс сопровождается повреждением эндотелия, увеличивается содержание тромбомодулина, связывающего и инактивирующего тромбин. Вместе с инактивированным тромбином он активирует противосвертывающие вещества (протеин С), таким образом меняя свои свойства и становясь стимулятором противосвертывающей активности.
При развитии бактериемии отмечают неглубокую тромбоцитопению (до 80х109/л), но тромбоциты функционально неактивны, «заблокированы».
Клиническая картина
Клиническая картина ДВС-синдрома включает симптомы основного заболевания, гемокоагуляционного шока, геморрагический синдром различной степени выраженности, признаки нарушения микроциркуляции в органах и системах с разной степенью их недостаточности.
Принято различать острый ДВС-синдром (развитие продолжается в пределах суток), подострый (развивается в течение нескольких суток или недели) и хронический (протекает многие недели и месяцы). В течении ДВС-синдрома условно выделяют четыре стадии:
• гиперкоагуляция и агрегация;
• переходная с нарастающей коагуляцией, тромбоцитопенией и разнонаправленными сдвигами в различных коагуляционных тестах;
• гипокоагуляция;
• восстановительная.
Гемокоагуляционный шок развивается при быстром поступлении в кровоток большого количества тканевого тромбопластина (гликопротеид клеточных мембран) или других веществ с аналогичным механизмом действия. Возникает острое нарушение гемодинамики с падением АД и центрального венозного давления.
Геморрагический синдром манифестирует локальными кровотечениями, петехиально-пятнистым типом геморрагии, гематомами в местах инъекций и кровотечениями из органов. Иногда вытекающая из раны или полости органа кровь не образует полноценных сгустков или вообще не свертывается.
Нарушение микроциркуляции характеризуется острой почечной недостаточностью, часто сопровождающейся гемолизом, а также острой печеночной или легочной недостаточностью. Сочетание поражения почек и печени называют гепаторенальным синдромом. Нарушение микроциркуляции в головном мозге выражается в головокружении, обмороке и нарушении сознания вплоть до коматозного состояния.
Диагноз уточняют на третьем этапе диагностического поиска. В начальной фазе количество тромбоцитов остается нормальным или незначительно сниженным, усиливаются их адгезивные и агрегационные свойства. Повышается содержание фибриногена, уменьшается АЧТВ, снижается фибринолитическая активность. В период выпадения тромбов и развития коагулопатии потребления отмечают снижение количества тромбоцитов и содержания фибриногена. При возникновении геморрагий число тромбоцитов резко снижается, а фибринолитическая активность увеличивается.
Диагностика
Диагностика ДВС-синдрома основана на данных клинической картины (геморрагический синдром, нарушение микроциркуляции, недостаточность функций органов и систем) и лабораторных исследований. Речь, естественно, идет об остром и подостром ДВС-синдроме (хронический ДВС-синдром диагности-
руют по результатам лабораторных исследований и рассматривают в качестве одного из возможных патогенетических механизмов тех или иных заболеваний, например ХГН и др.).
Лечение
Лечение ДВС-синдрома - сложная задача, что связано с быстротой развития симптомов, их тяжестью и опасностью для жизни. Прогрессирующий геморрагический синдром, шок с резким падением АД, ухудшение, а иногда и полная утрата функций различных органов требуют быстрого проведения лечебных мероприятий.
Лечение ДВС-синдрома должно включать меры, направленные на устранение причины его развития (борьба с инфекционным поражением, лечение основного заболевания), борьбу с шоком и коррекцию гемостаза.
Трансфузии свежезамороженной плазмы - один из основных методов лечения острого ДВС-синдрома (гепарин натрия добавляют для того, чтобы плазма не свернулась). Свежезамороженная плазма содержит антитромбин III, плазминоген, факторы свертывания и естественные антиагреганты. Ее получают методом плазмафереза крови донора и замораживают в течение 30-40 мин, при этом активность антитромбина III и плазминогена возрастает на 200%. Хранение плазмы в холодильнике или при комнатной температуре снижает ее активность на 20-40% в сутки. Оттаивание свежезамороженной плазмы осуществляют при температуре не выше 25 °С, после чего ее вводят внутривенно струйно.
В настоящее время используют супернатантную плазму, имеющую меньшую тенденцию к свертыванию, когда ее вводят больному с тромбинемией. Это связано с тем, что в ней содержится меньше фибриногена, фактора VIII, фибронектина и фактора Виллебранда. Таким образом, существует возможность замещения естественных физиологических антикоагулянтов.
Плазмаферез и плазмозамену, позволяющих устранять активность свертывания и удалять продукты паракоагуляции, широко используют при иммунокомплексном синдроме, тканевом распаде, некрозе, выраженном геморрагическом синдроме, септическом шоке, ожогах, синдроме раздавливания тканей. Эти методы улучшают гемодинамику и предупреждают развитие острой почечной недостаточности.
При обнаружении бактериемии (в большинстве случаев острого ДВСсиндрома) необходимо назначать антибиотики стерилизации кишечника.
Целесообразно вводить концентраты тромбоцитов. Показаний к переливанию цельной крови нет, за исключением случаев, когда гемотрансфузия необходима по жизненным показаниям, а в учреждении отсутствует эритроцитарная масса. Трансфузии последней осуществляют в редких случаях - лишь при острой кровопотере (более 1 л крови), так как в крови донора содержится много активаторов свертывания крови.
Поскольку острый ДВС-синдром - своеобразный протеолитический взрыв, то на поздних этапах для блокады протеолиза рекомендуют в больших дозах вводить антипротеазы (например, по десять ампул апротинина).
Для защиты эндотелия от бактериальных эндотоксинов используют эндотелиопротекторы (интерлейкины, цитокины, факторы некроза опухолей, медиаторы воспаления).
В настоящее время проводятся работы по получению антиэндотоксинового гамма-глобулина, на который возлагают большие надежды.