Биология: учебник / Пехов А.П., -, 2010. - 664 с.
|
|
|
|
ГЛАВА VIII НОРМАЛЬНАЯ И ПАТОЛОГИЧЕСКАЯ НАСЛЕДСТВЕННОСТЬ ЧЕЛОВЕКА
Наследственность человека, как и наследственность других живых существ, является менделевской наследственностью, ибо признаки человека генетически детерминированы и их передача от поколения к поколению происходит на основе законов наследственности, открытых и обоснованных Г. Менделем.
МЕТОДЫ ИЗУЧЕНИЯ НАСЛЕДСТВЕННОСТИ ЧЕЛОВЕКА
Применительно к человеку классический генетический анализ исключен из-за невозможности экспериментальных скрещиваний, длительности времени между поколениями и малым количеством потомства на пару (семью). Поэтому для изучения нормальной и патологической наследственности используют другие методы.
1. Генеалогический метод (метод родословных). В медицинской генетике этот метод называют клинико-генеалогическим. Генеалогия - это учение о родословных. Поэтому смысл данного метода заключается в изучении наследственности человека путем учета и анализа распределения наследственных признаков в семьях, т. е. в изучении наследственности человека по родословным. Метод сводится к изучению родственных связей и передачи признаков среди близких и дальних родственников, прямых и непрямых.
Исследование того или иного признака в семье начинают с того члена семьи, который представляет интерес или является больным (исходный пациент, или пробанд). Потомки одних и тех же родителей, происходящие из разных зигот (братья и сестры), получили название сибсов. Родословные составляют путем учета возможно большего количества родственников, используя для обозначения поколений, мужчин, женщин, браков, типов зиготности и т. д., различные символы, перечень которых приводится на рис. 65.
С помощью этого метода возможно установление наследственного характера признака, типа и частоты наследования того или иного
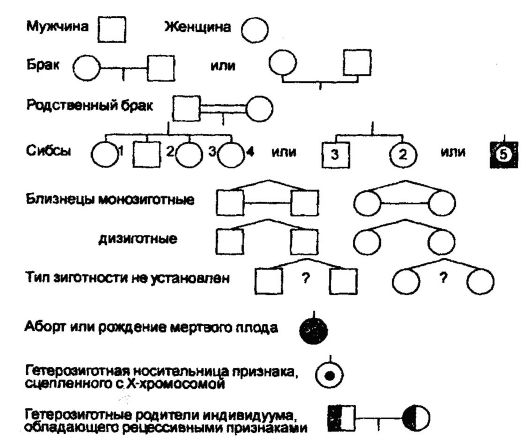
Рис. 65. Символы, используемые при составлении родословных человека
признака, сцепленности признака с полом, а также определение зависимости или независимости распределения признаков. Анализируя родословные, можно обнаружить различия между близким сцеплением и аллелизмом. На рис. 66 приводится в качестве примера родословная с доминантным наследованием, а на рис. 67 - родословная, демонстрирующая независимое распределение неаллельных генов. Метод характеризуется относительно большой разрешающей способностью. Однако он имеет и недостаток, связанный с трудностями сбора сведений о проявлении того или иного признака у родственни- ков пробанда, поскольку люди плохо знают свою родословную.
2. Цитогенетический метод. Этот метод заключается в цитологическом анализе кариотипа человека в норме и патологии. С его помощью исследуют нарушения, изменяющие количество и структуру хромосом.
Цитогенетический метод основывается на данных о хромосомах. В соответствии с денверовской классификацией хромосомы обозна- чают номерами, увеличивающимися по мере уменьшения размеров хромосом. В соответствии с рекомендациями IV Международного
конгресса по генетике человека в Париже (1971) при описании добавочных хромосом их число помещают после общего числа хромосом и половых хромосом со знаком «+» или «-» перед номером вовлеченной аутосомы. Например, запись (формула) 47,ХХ+21 означает кариотип женщины с трисомией по 21-й паре.
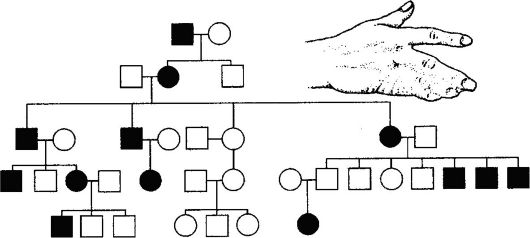
Рис. 66. Родословная, позволяющая проследить доминантное наследование
синдактилии
Напротив, кариотип мужчины с экстрахромосомой X обозначают как 47,XXY. Знак плюс или минус помещают, сопровождая хромо- сомный символ, чтобы указать удлинение или укорочение хромосомного плеча. Буква q символизирует длинное плечо, а р - короткое. Например, запись 46,XY, 1 q+ указывает на увеличение длины длинного плеча хромосомы ? 1. Кариотип 47,XY, +14p+ символизирует мужчину с 47 хромосомами, включая дополнительную хромосому (? 14) с повышением в длине ее короткого плеча. Сокращения def (дефишенс), dup (дупликация), r (кольцо, возникающее после воссоединения двух разрывов в хромосоме), inv (инверсия) и t (транслокация) обозначают аберрации хромосом. Номера хромосомы или хромосом помещают после сокращения в скобках. Например, запись 46,ХХ, r (18) означает кариотип женщины с 46 хромосомами, включая r-хромосому ? 18. Формула 46,Х, inv (X q) есть кариотип женщины с 46 хромосомами, включая одну нормальную Х-хромосому и изохромосому X (с двумя генетически идентичными плечами) для длинного плеча хромосомы X. Банды помечают числами в порядке от центромеры вдоль короткого плеча ( р) и длинного плеча (q) хромосомы.
Главная ценность цитогенетического метода заключается в том, что он позволяет установить связь между нарушениями кариотипа
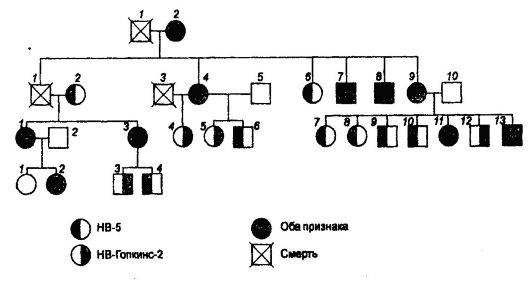
Рис. 67. Родословная, демонстрирующая независимое распределение
хромосом
и изменениями фенотипа, т. е. связь между нарушениями в определенной хромосомной паре и определенным наследственным дефектом. Это в свою очередь помогает найти принадлежность гена к опре- деленной группе сцепления. Основное преимущество этого метода заключается в его простоте. Однако данный подход имеет существенные ограничения. Прежде всего с его помощью могут быть исследованы только крупные нарушения в структуре хромосом, видимые с помощью светового микроскопа. Следовательно, это ограничивает количество анализируемых генетических детерминантов. Далее, этот подход может обеспечить изучение генотипов лишь на уровне групп сцепления.
3. Популяционный метод. Этот метод основан на законе Харди- Вайнберга и заключается в изучении распространения генов в популяциях человека. В условиях свободного скрещивания частота, с которой возможна встреча двух аллелей в диплоидном организме, равна произведению частот каждого аллеля. Если относительную частоту доминантного аллеля А в двухаллельной системе обозначить р, относительную частоту рецессивного аллеля а обозначить q и если р + q=1, то при свободном скрещивании частота трех генотипов составляет значения: АА = р2, Аа = 2pq и аа = q2. Следовательно, зная о равновесии по Харди-Вайнбергу, можно определить влияние названных выше факторов на относительные частоты этих трех генотипов в поколениях. Как видно, данный метод позволяет изучать
не только географическое распространение и частоту тех или иных генов, но и влияние на эти показатели разных факторов.
4. Близнецовый метод. Этот метод заключается в изучении генетических закономерностей, присущих однояйцовым (монозиготным) и разнояйцовым (дизиготным) близнецам. Обычно сопоставляют монозиготных партнеров с дизиготными, а результаты анализа близнецовой выборки сравнивают с результатами анализа общей популяции. Метод позволяет выяснить наследственную предрасположенность в проявлении ряда признаков и заболеваний, установить коэффициент наследуемости и степень влияния факторов внешней среды на проявление признаков. Успех в использовании этого метода чаще связан с изучением тех признаков, которые не подвержены резкому влиянию со стороны внешних факторов, например группа крови, пигментация глаз и др. Недостаток метода связан с неполнотой сведений о пренатальном и постнатальном развитии близнецов.
5. Перенос генов. Под этим названием различают группу методов, позволяющих перенос генов от одних клеток к другим.
Гибридизация соматических клеток - это метод, основанный на том, что соматические клетки животных способны к гибридизации, при которой образуются гибриды клеток, в ядрах которых содержится набор хромосом обеих сходных клеточных линий, т. е. гибриды являются полиплоидами. В процессе роста гибриды могут терять отдельные хромосомы. Для гибридов, полученных из скрещиваний соматических клеток человека с соматическими клетками млекопитающих, характерно то, что преимущественно теряются человеческие хромосомы. Следовательно, наблюдение одновременной потери той или иной хромосомы и признака указывают на локализацию гена, контролирующего признак в данной хромосоме. В исходных скрещиваниях можно использовать также клетки человека с частично удаленными из них хромосомами. Метод имеет ограничения, определяемые невозможностью экспрессии чужеродных генов в гибридах.
Перенос хромосом - это метод, позволяющий выделение хромосом и трансформацию ими клеток.
Перенос ДНК - это метод трансформации клеток очищенной ДНК, позволяющий переносить одновременно около 50 генов.
6. Молекулярно-генетические методы. Эти методы связаны с выделением ДНК, рестрикционным картированием, клонированием сегментов длиной до 50 000 пар оснований и секвенированием отдель-
ных генов. Надежно вошли в практику методы выделения кДНК и полимеразная цепная реакция (ПЦР).
Кроме того, ряд молекулярно-генетических методов направлен на разделение, идентификацию и измерение генных продуктов (белков). Подсчитано, что в клетках человека синтезируется около 30 000 разных белков. Поэтому ставится задача создать каталог белков и построить карту белков человека.
7. Моделирование наследственных болезней. Этот метод основан на законе Н.И. Вавилова о сходных рядах наследственности и заклю- чается в моделировании наследственных болезней на животных, у которых встречаются отдельные из этих болезней, например гемофилии на собаках. Кроме того, используют «сконструированные» линии лабораторных животных, обладающих теми или иными мутантными генами. Например, для изучения болезни Леша-Найяна используют белых мышей, полученных введением в их эмбрионы культивируемых клеток с дефектом по гипоксантин-фосфорибозилтрансферазе.
На основе результатов изучения наследственности человека создают генетические, молекулярные и белковые карты. В ходе реализации проекта «Геном человека» (1990-2003) был расшифрован весь геном человека.
НОРМАЛЬНАЯ НАСЛЕДСТВЕННОСТЬ
У человека ДНК локализована в хромосомах и митохондриях. Количество ДНК в соматических клетках составляет несколько мил- лиардов нуклеотидных пар.
Кариотип человека показан на рис. 68 и 69. Нормальное диплоидное число хромосом у человека равно 23, из которых 22 пары хромосом являются аутосомами, а 23-я пара у мужчин представлена половыми хромосомами XY (см. рис. 68), у женщин - XX (см. рис. 69). Обычно хромосомы человека распределяют по группам, что зависит от положения центромеры, определяющей длину отрезков (плеч) по обеим сторонам центромеры. Как уже отмечено, различают телоцентрические, акроцентрические, субметацентрические и метацентрические типы хромосом.
Расшифрован генетический код человека. Количество генных локусов в геноме человека составляет примерно 32 000, из которых идентифицировано около 2200 и картировано около 1000. Установлено 24 группы сцепления, из которых 22 соответствуют ауто-
сомам, а две - хромосомам X и Y. Около 40% наших генов не имеют видимых эффектов. Некоторые гены кодируют больше чем один протеин. Основное количество идентифицированных генов локализовано на аутосомах, меньшее - на Х- и Y-хромосомах. В частности, наиболее «заселенными» генами являются хромосомные пары 1, 6, 7, 11, 16 и Х-хромосома, длина которой составляет около 100 000 000 пар оснований. У человека имеется около 100 импринтинговых генов, располагающихся на хромосомах 7, 18, 15. Предполагается существование генов долголетия.
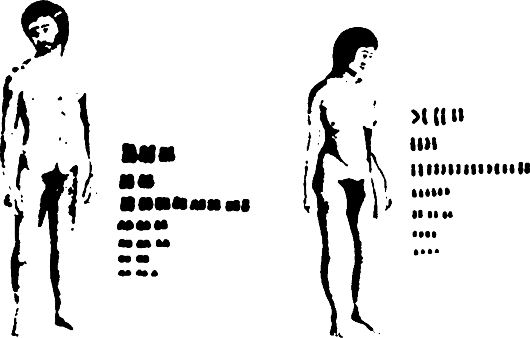
Рис. 68. Кариотип нормального Рис. 69. Кариотип нормальной
мужчины женщины
Гены определяют физическое и психическое здоровье человека. Поэтому человек наследует все признаки, которые характер- ны для него как для живого существа. Любой индивид наследует от своих родителей телосложение, рост, массу тела, форму головы, овал лица, особенности скелета, строение и цвет кожи и волос, восприимчивость или устойчивость к тем или иным болезням, походку, манеру держаться, способность к тем или иным наукам, музыке, группу крови, способность клеток к биохимической активности и т. д. Следовательно, каждый признак, каждая структура, каждая функция человеческого организма детерминирована генами. Так же
как и в случае других живых существ, у человека одни гены начинают действовать еще на ранних этапах онтогенеза, другие - на поздних (рис. 70, табл. 17).
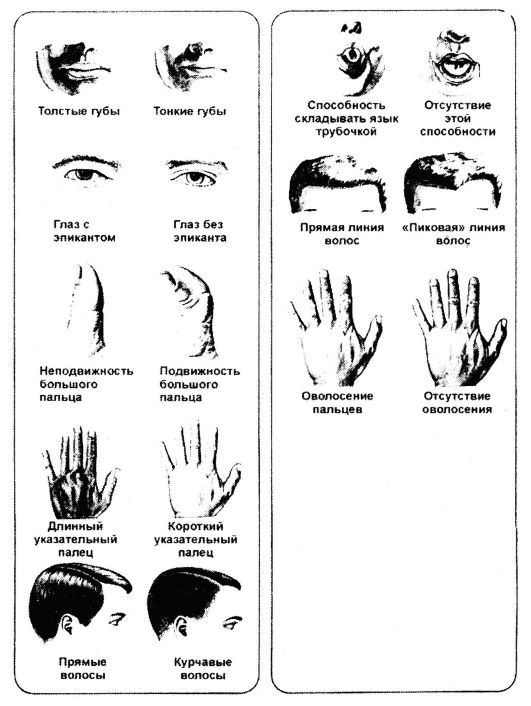
Рис. 70. Нормальные признаки человека
Таблица 17. Наследование некоторых признаков человека
Признаки | Типы наследования | |
доминантный | рецессивный | |
1 | 2 | 3 |
Форма черепа | Короткий (брахицефалия) | Длинный (долихоцефалия) |
Размер глаз | Большие | Маленькие |
Цвет глаз | Карие | Голубые |
Разрез глаз | Прямой | Косой |
Тип глаз | Монголоидный | Европеоидный |
Острота зрения | Близорукость | Нормальная |
Размер носа | Крупный | Средний или маленький |
Форма носа | Острый и выступающий вперед | Нормальный |
Форма носа | Узкий («сухой») | Широкий |
Форма носа | Выпуклый с горбинкой («орлиный») | Прямой |
Форма носа | Прямой | Вогнутый |
Размеры хрящевых крыльев носа | Покрывает перегородку полностью или почти полностью | Оставляет перегородку открытой |
Ширина ноздрей | Широкие | Узкие |
Высота переносицы | Высокая и узкая | Низкая и широкая |
Ширина ушей | Широкие | Узкие |
Длина ушей | Длинные | Короткие |
Характер мочки уха | Висящая свободно | Срощенная |
Наличие острой верхушки уха (дарвинов- ский бугорок) | Присутствует | Отсутствует |
Длина подбородка | Длинный | Короткий |
Ширина подбородка | Широкий | Узкий и острый |
Форма подбородка | Прямой | Отступающий назад |
Щель между зубамирезцами | Имеется | Отсутствует |
Строение волос | С мелкими завитками | Вьющиеся, волнистые или прямые |
Строение волос | Вьющиеся | Волнистые или прямые |
Окончание табл. 17
Признаки | Типы наследования | |
доминантный | рецессивный | |
Строение волос | Волнистые | Прямые |
Строение волос | Жесткие прямые («ежик») | Мягкие прямые |
Цвет волос | Рыжие | Светло-русые |
Цвет волос | Каштановые | Светло-русые |
Поседение волос | До 25 лет | После 40 лет |
Облысение | У мужчин | У женщин |
Белая прядь волос надо лбом | Имеется | Отсутствует |
Овал лица | Круглое | Продолговатое |
Характер нижней губы | Толстая отвисающая | Нормальная |
Толщина кожи | Толстая | Тонкая |
Цвет кожи | Смуглая | Белая |
Наличие веснушек | Имеются | Отсутствуют |
Рост | Нормальный | Пропорциональная карликовость |
Рост | Диспропорциональная карликовость | Нормальный |
Преобладание руки | Праворукость | Леворукость |
Кисть руки | С 6, 7 пальцами (полидактилия) | С 5 пальцами |
Строение ногтей | Тонкие и плоские | Нормальные |
Цвет ногтей | Голубовато-белые | Обычные |
Узоры на коже пальцев | Эллиптические | Циркулярные |
Тип голоса у мужчин | Бас | Тенор |
Тип голоса у женщин | Сопрано | Альт |
Абсолютный музыкальный слух | Имеется | Отсутствует |
Для человека характерно исключительное генетическое разнообразие. Простое менделевское наследование определяется тем, что тот или иной признак контролируется лишь парой генов. В случае человека известны все типы наследования.
Типичным и наиболее изученным примером менделевской наследственности доминантного типа у человека является наследование
способности ощущать вкус фенилтиокарбамида. В США, например, 70% лиц из исследованных групп ощущают вкус этого вещества, тогда как остальные не способны к этому. Среди чешского населения количество лиц, ощущающих вкус фенилтиокарбамида, составляет 67% от общего числа обследованных. Отдельные индивиды способны ощущать вкус этого вещества даже при наличии его в ничтожных количествах (19 ? 10-8 молекул/мл).
Исследование родителей и детей во многих семьях показало, что способность ощущать вкус фенилтиокарбамида детерминируется доминантным аллелем Т, тогда как неспособность - рецессивным аллелем t. Следовательно, гетерозиготы (Tt) и доминантные гомозиготы (ТТ) по этой паре генов ощущают вкус вещества, причем в одинаковой степени. Если оба родителя не ощущают вкус вещества, то у них никогда не рождаются дети, «чувствующие» это вещество. «Чувствующие» дети появляются лишь в браках, в которых оба родителя или один из родителей ощущает вкус вещества. В семьях, в которых оба или один из родителей является доминантным гомозиготным организмом (ТТхТТ, TTxTt, TTxtt), все дети будут ощущать вкус фенилтиокарбамида. Что касается браков между гетерозиготами (TtxTt), а также между гетерозиготами и рецессивными гомозиготами (Ttxtt), то среди детей в этих браках будут встречаться как ощущающие, так и не ощущающие вкус фенилтиокарбамида, причем в первом случае с классическим менделевским отношением 3:1, во втором - 1:1.
Наряду с доминантным и рецессивным типами наследования нормальных признаков у человека встречается кодоминантное наследование, которое заключается в том, что у гетерозиготных индивидуумов формирование признака детерминируют оба аллеля, т. е. проявляются эффекты обоих аллелей. Типичным примером такого наследования является наследование группы крови. У человека четыре группы крови, определяемых на основании антигенных свойств эритроцитов по так называемой системе АВ0. Ими являются группы крови А, В, 0 и АВ, или II, III, I и IV группы соответственно. Данные о распространении групп крови среди разного населения приведены в табл. 18.
Дифференциация групп крови основывается на наличии в эритроцитах людей антигенов А и В (табл. 19). Эритроциты людей, относящиеся к первой (нулевой) группе крови, не имеют антигенов А и В, но в сыворотке крови таких людей есть антитела, агглюти-
Таблица 18. Распространение групп крови системы АВ0 среди людей
Национальность | Количество людей с группой крови, % | |||
0 (1) | А (II) | В (Ш) | АВ (IV) | |
Австралийцы | 57,0 | 38,5 | 3,0 | 1,5 |
Австрийцы | 42,0 | 40,0 | 10,0 | 8,0 |
Англичане | 46,4 | 43,4 | 7,2 | 3,0 |
Греки | 38,5 | 41,6 | 16,2 | 4,0 |
Индейцы (Северная Америка) | 23.5 | 76,5 | 0 | 0 |
Итальянцы | 47,2 | 38,0 | 11,0 | 3,8 |
Казахи | 33,7 | 26,8 | 30,9 | 8,6 |
Киргизы | 37,6 | 26,5 | 28,5 | 7,4 |
Немцы | 40,0 | 43,0 | 12,0 | 5,0 |
Французы | 43,2 | 42,6 | 11,2 | 3,0 |
Японцы | 27,0 | 41,0 | 18,0 | 14,0 |
Таблица 19. Реакция между сывороткой и эритроцитами от лиц, относящихся к разным группам крови
Группа крови | Сыворотка агглютинирует эритроциты групп | Эритроциты агглютинируются сывороткой групп |
0 (первая) | А, В, АВ | Никакой |
А (вторая) | В, АВ | 0, В |
В (третья) | А, АВ | О, А |
АВ (четвертая) | Никакой | 0, А, В |
нирующие (склеивающие) эритроциты людей, обладающих антигенами А, В или АВ. Эритроциты людей, относящихся ко второй группе крови (группе А), имеют антиген А, а в сыворотке крови таких людей есть антитела, агглютинирующие эритроциты с антигеном В. Эритроциты людей, относящихся к третьей группе крови (группе В), имеют антиген В, а в сыворотке крови таких людей есть антитела, агглютинирующие эритроциты с антигеном А. Наконец, эритроциты людей, относящихся к четвертой группе крови (группе АВ), имеют как антиген А, так и антиген В, но в сыворотке крови таких людей нет антител ни против антигена А, ни против антигена В.
Изучение наследования групп показало, что антигенные свойства эритроцитов контролируются геном, имеющим три аллеля - !А, Ρ
и I0, из которых аллель Р1 контролирует продукцию антигена А, а аллель Ρ - антигена В (табл. 20). У гетерозиготных организмов РР каждый аллель независим и под контролем каждого из них осуществляется синтез антигенов (кодоминантность), но оба аллеля доминируют над аллелем I0, вследствие чего эритроциты людей с первой группой крови (гомозигот по I0) не имеют антигенов А и В. Группа крови А (II) бывает лишь у гомозигот IAIA или гетерозигот IAI0, тогда как группа крови В (III) - у гомозигот РР или гетерозигот PP. При кодоминантности (!АР) отмечается группа крови АВ (IV) (см. табл. 20).
Таблица 20. Наследование групп крови системы АВ0 у человека
Группа крови родителей | Возможные группы крови у детей | |
мать | отец | |
0 | А | 0, А |
0 | В | 0, В |
0 | АВ | А, В |
А | А | 0, В |
А | В | 0, А, В, АВ |
А | АВ | А, В, АВ |
В | В | 0, В |
В | АВ | А, В, АВ |
АВ | АВ | А, В, АВ |
Помимо групповых антигенов системы АВ0, эритроциты человека обладают также антигенами, на основе которых возможна дифференциация других групп крови. Среди них хорошо изучены, например, антигены М и N, детерминирующие парой аллельных генов Мм и MN (соответственно), которые кодоминантны. Вследствие этого индивидуумы, гетерозиготные по обоим аллелям (MMMN), обладают эритроцитами, имеющими как антиген М, так и антиген N, и принадлежат к группе MN. Эритроциты с М-антигеном будут у людей, гомозиготных по аллелю Мм, т. е. ММММ (группа крови М), а с N-антигеном - у людей, гомозиготных по аллелю MN, т. е.
MNMN (группа крови N).
Многие нормальные признаки наследуются сцепленно с полом. В большинстве случаев формирование того или иного признака зависит от действия многих пар генов (полигенных систем) и от взаимодействия их со средой.
НАСЛЕДСТВЕННОСТЬ И ПОВЕДЕНИЕ
Давно замечено, что в популяциях человека индивиды различаются между собой не только физически, но и по поведению. Попытки понять основы таких различий у человека, особенно в применении к его интеллектуальным возможностям, часто приводили к противоречиям, в результате чего одни свойства рассматривали в качестве наследственных, другие - приобретенных. Поэтому в прошлом причины этих различий были объяснены в положении «природа и воспитание» (nature a. rupture).
Между тем изучение признаков человека показало, что в формировании любого из них имеют значение и наследственность и среда. Но могут быть вариации, заключающиеся в том, что одни признаки зависят больше от наследственности, другие - от среды. Например, группа крови зависит полностью от наследственности, но размеры тела и углеводный метаболизм прямо и существенно определяются как генетическими факторами, так и средой. Способность исполь- зовать язык зависит от наследственности (от генетически детерминированных анатомических особенностей гортани и полости рта), но и несомненно от опыта, т. е. от того, что индивид изучает на протяжении жизни в определенной среде.
Особенно большое значение положение «природа и воспитание» имеет в оценке генетических основ поведения человека.
Установлено, что среда, в которой развивается любой животный организм, очень сильно влияет на некоторые особенности поведения его во взрослом состоянии. Например, женские особи обезьян-резус, отделяемые после рождения от их матерей и лишаемые возможности в раннем возрасте общаться с другими обезьянами, недостаточно активны затем в половом поведении, в социальных играх.
Известно, что особую важность в определении поведения имеет нервная система. Несомненно также значение гормональной системы, ибо у высших животных, включая человека, гормоны принимают участие не только в репродукции, но и в детерминировании индивидуального темперамента, реакций на стресс. Животные, половое поведение которых является «осознанным», к гормонам чувствительны только в определенное время (сезон), в другое время гормональный стимул не вызывает у них никакого ответа. Напротив, у человека способность к репродукции не зависит от сезона, поэтому гормональное влияние в половом поведении является меньшим. Как нервная, так и гормональная система в конечном итоге находятся под генетическим кон-
тролем (природы). Поэтому поведенческий потенциал человека, вероятно, детерминируется генетически, но этот потенциал может быть реализован только в условиях определенной среды (воспитания).
В контексте познания основ поведения человека часто прибегают к рассмотрению биологических основ преступности. В конце прошлого века итальянский психиатр Чезаре Ломброзо (1835-1909) сформулировал биологическую теорию преступности, суть которой заключается в том, что основу преступности составляют биологические факторы, т. е. преступники являются «прирожденными» преступниками. Эта теория, в которой биологическим фактором преступности позднее стали признавать наследственность, оказалась очень живучей до нашего времени. Точно так же известны утверждения о генетических основах проституции и других пороках людей. Между тем еще никто не идентифицировал ни гены преступности, ни гены проституции. Современные представления основаны на том, что наследуются не гены преступности или проституции, а реакции, т. е. способность индивидов реагировать тем или иным образом в тех или иных условиях жизни.
Вопреки достижениям биологических наук, серьезную проблему составляют поиски методов измерения (оценки) интеллекта человека. Еще на заре развития генетики Ф. Гальтон и К. Пирсон в Англии предложили для оценки интеллекта использовать коэффициент корреляции интеллекта у детей и их родителей, а также у детей одной семьи (сибсов). Отстаивая эффективность генеалогического подхода, Ф. Гальтон даже считал, подобно многим людям своего поколения, что женщины по интеллекту стоят ниже мужчин. Однако этот метод не нашел широкого применения.
В
Современная методика определения уровня умственных способностей детей основана на выявлении разных способностей (уровень общих знаний, сообразительность, словарный запас, географические
знания, выполнение арифметических заданий и т. д.) и на суммарной оценке этих способностей.
Определение умственных способностей с помощью IQ-теста вызывает на протяжении многих лет противоречивое отношение со стороны многих ученых, причем эта противоречивость основана на преувеличении либо биологических, либо социальных факторов развития человека, т. е. на признании или непризнании наследуемости коэффициента интеллектуальности.
Между тем многочисленные данные свидетельствуют о том, что коэффициент интеллектуальности характеризуется значительной наследуемостью. В то же время установлено, что вариабельные результаты IQ-теста, часто имеющие место, зависят от влияния нескольких генов и что многие генетические эффекты осуществляются в очень широком диапазоне через среду. Таким образом, вопреки многим недостаткам, IQ-тест может использоваться для определения того направления в поведении людей, которое связано с их особенностью к абстрактному мышлению. Однако его использование по-прежнему осложняется нерешенностью многих вопросов. В частности, в литера- туре нет удовлетворяющего всех точного определения интеллекта, нет согласия в том, что означает, когда говорят, что один индивид «умнее» другого, нет одинакового понимания важности различий в интеллекте или в амбициях, альтруизме, жестокости и успехах в достижении людьми поставленных целей. По-прежнему остаются невыясненными вопросы о различиях по IQ-тесту людей, принадлежащих к разным социальным группам. Но самое главное состоит в том, что вопреки наследуемости IQ все же не совсем ясно, какова степень влияния на показатели IQ-тестов «природы» и «воспитания» раздельно.
Исследование генетических основ поведения еще отстает от изучения других разделов биологии человека. Главная причина заключается в том, что у человека невозможно прямо связать генетику поведения с менделевским отношением, равно как трудно определить степень наследуемости основных признаков. Кроме того, существуют сложности в изучении поведенческих реакций на молекулярном уровне.
ГЕНЕТИЧЕСКАЯ ИНДИВИДУАЛЬНОСТЬ
В рассмотрении нормальной наследственности человека выдающееся значение имеет осознание его генетической индивидуаль- ности.
Все организмы, возникающие в результате перекрестного оплодотворения гамет, представляют собой полигибридные гетерозиготные виды по многим парам генов. Человек не является исключением из этого правила и также имеет полигибридную гетерозиготную природу. В соот- ветствии с закономерностями расщепления и независимого перераспределения генов одиночная половая клетка человека содержит лишь половину (гаплоидное число) хромосом и генов, представленных в соматических клетках организма в диплоидном наборе. Если, например, какой-либо индивидуум является гетерозиготным организмом по трем парам генов Аа, Вв и Сс, то его половые клетки несут лишь половину таких генов. Следовательно, потомству такого индивида будет передана также половина генов родительского организма, например генов А, В и С. Между тем разные половые клетки одного и того же родителя несут разные наборы хромосом, а оплодотворение той или иной клетки является делом случая. Поэтому от одних и тех же родителей дети наследуют разные наборы генов как от одного, так и от другого родителя, вследствие чего братья и сестры всегда имеют разные генотипы.
Учитывая соображения, приведенные выше, а также то, что один эякулят человека содержит около 200 000 000-500 000 000 сперматозоидов, можно допустить, что индивид, гетерозиготный, например, по 30 парам генов, способен дать 230 разных типов гамет. Поскольку в оплодотворении участвует по одной гамете от каждого родителя, то, исходя из способности каждого родителя давать 230 разных типов гамет, можно представить, что в потомстве одной брачной пары количество возможных генотипов зигот, возникающих после объединения разных половых клеток, будет составлять величину порядка 260.
Как видно, расщепление и независимое перераспределение генов (рекомбинация) являются причиной гигантского генетического разнообразия (полиморфизма) людей. В природе нет двух индивидов с одинаковым генотипом. Каждый человек несет специфический набор генов, вследствие чего каждый человек генетически индивидуален и неповторим. Исключение составляют лишь однояйцовые близнецы, для которых характерны одинаковые генотипы.
Однако генетическое разнообразие людей уменьшается в результате родственных браков (браков между двоюродными братьями и сестрами), частота которых, например, в странах Азии и Африки составляет 1:20, а в Англии - 1:200. В отдаленной перспективе родственные браки создают благоприятный эффект, устраняя вредные аллели из генного пула. Но их ближайший эффект представляется
неблагоприятным вследствие повышенной возможности появления наследственной болезни уже в следующем поколении.
На очень большом материале, собранном в Японии, установлено, что дети двоюродных братьев и сестер имеют возможность в одном из 16 случаев стать гомозиготными по локусам, унаследованным от их дедов и контролирующим наследственную глухоту и мышечную дистрофию. Установлено также, что смерть детей в браках двоюродных братьев и сестер на 4,4% выше, чем в неродственных браках.
Подобным образом генетически индивидуальны организмы разных видов животных и растений (кроме клонов бактерий и сомати- ческих клеток животных и растений, а также чистых линий растений и инбредных линий животных).
ПАТОЛОГИЧЕСКАЯ НАСЛЕДСТВЕННОСТЬ
Интерес к наследственной патологии человека восходит еще ко временам Гиппократа (460-370 гг. до н. э.), который, вероятно, первым привлек внимание к повторению в семьях таких аномальных признаков, как косоглазие и облысение. Он наблюдал также, что эпилепсия и некоторые болезни глаз, вызывающие слепоту у людей преклонного возраста, встречались лишь в определенных семьях. Интерес к генетике человека в XVIII в. проявил П. Маупертус, собиравший родословные семей, в которых встречались полидактилия и альбинизм. Анализируя родословные, он предсказывал возможность будущих рождений с этими аномалиями.
Во второй половине XIX в. Ф. Гальтон применил к изучению наследственности человека статистику. Он привлек также внимание к социальному аспекту наследственной патологии, показав, что в основе наследственных болезней лежит отношение «ген-метаболическая реакция». Более того, он даже предположил, что некоторые из таких болезней, как, например, алкаптонурия, контролируются рецессивными генами.
Наследственная
патология приобрела очень большое значение. Например, смертность детей
в раннем возрасте в результате врожденных пороков развития по
приблизительным подсчетам в
Сейчас известно, что от 1/4 до 1/2 всех болезней представляют болезни генетической природы. Считают, что около 3% новорожденных страдают от того или иного генетического дефекта. Одна из каж- дой гамет человека несет ошибочную информацию, введенную новой мутацией, большая часть которых «оплачивается» ранними абортами. Описано свыше 3000 разных наследственных аномалий, которые представляют собой генетический груз современного человечества. Различают сегрегационный и мутационный груз (рис. 71).
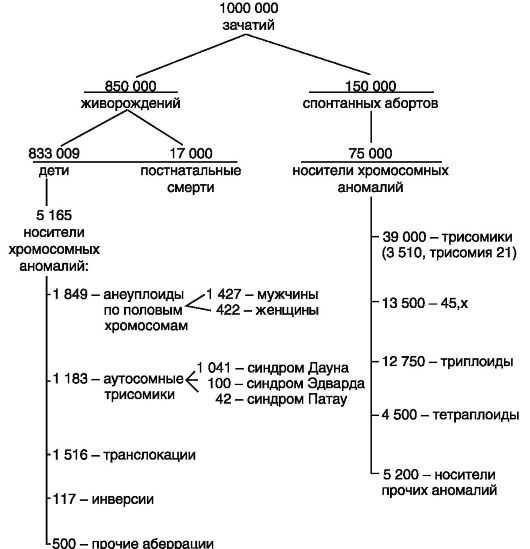
Рис. 71. Хромосомные аномалии у новорожденных и спонтанных абортусов
Сегрегационный груз - это часть генетического груза, унаследованная людьми современных поколений от людей, принадлежащих к поколениям, жившим на протяжении предыдущих веков. В сущности, сегрегационный груз представлен мутациями «старого» порядка. Мутационный груз - это часть генетического груза, которая обусловлена мутациями генов и хромосом, возникающими заново в каждом новом поколении.
Различают наследственную патологию, обусловленную мутациями хромосомных генов и хромосом, патологию, обусловленную мутациями митохондриальных генов, а также патологию, в которой имеет место наследственное предрасположение.
НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ, ОБУСЛОВЛЕННАЯ МУТАЦИЯМИ ГЕНОВ
Известно более 1500 наследственных болезней, являющихся результатом мутаций генов и встречающихся с разной частотой. Различают аутосомно-доминантные и аутосомно-рецессивные генные болезни, а также болезни, детерминируемые генами, локусы которых находятся на половых хромосомах. Важнейшей особенностью генных болезней является их гетерогенность, заключающаяся в том, что разные мутации могут сопровождаться сходным фенотипическим проявлением болезни. Это явление затрудняет клиниче- скую диагностику многих генных болезней.
Аутосомно-доминантный тип наследования характерен для ряда болезней (табл. 21). Такие болезни встречаются в каждом поколении у гетерозиготных носителей, причем среди сибсов в соотношении 1:1. Далее, для этих болезней характерна как полная, так и не всегда полная пенетрантность генов. Не всегда полной является и экспрессивность генов, причем различия затрагивают не только разные семьи, но и членов одной семьи. Например, нейрофиброматоз в одних семьях имеет генерализованный характер, в других проявляется в виде отдельных кожных поражений. Часть аутосомнодоминантных болезней может проявляться лишь через некоторое время после рождения. Например, хорея Хантингтона проявляется примерно между 30-40 годами жизни больных. Наконец, для аутосомно-доминантных болезней характерно протекание с повышенной тяжестью у гомозиготных доминантных индивидуумов.
Таблица 21. Аутосомно-доминантные болезни
Болезнь Централопатическая эпилепсия | Проявление Первичные приступы возникают в возрасте между 4-16 годами, после чего реже и обычно исчезают к 40 годам |
Глаукома | Слепота, вызываемая повышенным давлением жидкости в глазу и дегенерацией нервных клеток (вызывается также аутосомными рецессивными генами) |
Хорея Хантингтона | Непроизвольные движения лица и конечно- стей, позже наступают нарушения психики. Симптомы начинают проявляться примерно между 30-40 годами |
Мышечная дистрофия | Аномалия функций мышц (вызывается также ауто- сомным рецессивным и Х-сцепленным генами) |
Полипоз кишечника | Формирование множественных полипов, обычно ведущих к раку |
Пигментирующий ретинит | Воспаление сетчатки глаза с повышенной пигментацией, ведущее к слепоте |
Брахидактилия | Укорочение концевых костных фаланг (короткопалость) |
Ахондр оплазия | Карликовость, макроцефалия, укорочение конечностей |
Ахондроплазия - результат миссенс-мутации, сопровождаемой заменой глицина на аргинин в белке, ответственном за пролифера- цию хрящей в суставах длинных костей. Например, карлики фертильны, но гомозиготная ахондроплазия является летальной в неонатальный период, поражая 25% потомства гетерозиготных родителей. Частота ахондроплазии 1:15 000.
Аутосомно-рецессивные болезни (табл. 22) встречаются чаще, чем аутосомно-доминантные. и проявляются лишь у гомозигот- ных носителей мутантных аллелей, рождающихся в семьях, где оба родителя гетерозиготны или один является гомозиготой, второй гетерозиготой. Конечно, больные рождаются и в семьях, где оба родителя - гомозиготные носители мутантных аллелей (больными). Для распространения болезней, наследуемых по этому типу, характерна неравномерность. Например, болезни Тея-Сакса, Нимена- Пика и мышечная деформирующая дистония очень часты среди восточно-европейских евреев (ашкенази), достигая частот, превышающих частоты этих болезней среди населения других национальных
групп. В частности, болезнь Тея-Сакса, причиной которой является мутация гена, контролирующего лизосомную гексозоаминидазу А, среди евреев-ашкенази в Австрии составляет 11%, в Чехословакии - 9%, в Венгрии - 7%, в Румынии - 4%, в Польше и России - 3%. Однако среди людей этой группы очень редка фенилкетонурия. Акаталазия, наследственный дисхроматоз и некоторые другие заболевания с высокой частотой встречаются среди японцев. Кровные браки способствуют появлению гомозиготных рецессивных носителей очень редких мутантных аллелей.
Таблица 22. Аутосомно-рецессивные болезни
Болезнь | Проявление |
Серповидноклеточная анемия | Развитие хронической гипоксии и анемии с расстройствами кровообращения и тромбозами |
Цистический фиброз | Нарушения функций поджелудочной и других желез, образование толстого слоя слизи, ведущей к пневмонии и эмфиземе. Смерть наступает обычно в детском возрасте. Представляет собой наиболее частую генетическую аномалию детей (1 на 3700 рождений) |
Галактоземия | Пониженные уровни галактозо-1-фосфатури- дилтрансферазы ведут к увеличению печени, катаракте и нарушениям психики |
Гидроцефалия | Избыточное накопление жидкости в черепной коробке, вызывающее физические и психические нарушения |
Глухота врожденная | Около половины случаев детской глухоты вызывается этим аллелем |
Болезнь Нимена- Пика | Накопление липидов в нервных клетках, вызываю- щее нарушение психики, увеличение печени, замедленный рост. Смерть наступает обычно в первые три года жизни |
Пернициозная анемия | Нарушения в метаболизме витамина В2, что ведет к симптомам, связанным с уменьшением в крови количества эритроцитов |
Болезнь Тея-Сакса | Прогрессивно развивающиеся паралитические явления, нарушения психики, слепота. Смерть наступает в первые три года жизни. В 27-53 случаях встречается в браках двоюродных сестер и братьев (на 10 000) |
Фенилкетонурия | Нарушения в тонусе мышц, уменьшение пигментации кожи, волос, радужной оболочки глаз, микроцефалия, умственная отсталость |
Еще в начале XX в. было показано, что эритроциты многих индивидуумов агглютинируются сывороткой кроликов, иммунизированных кровью обезьян-резус. Эритроцитарный антиген, который ответствен за эту реакцию, получил название резус-фактора, а ген, который детерминирует это свойство, был назван Rr (или Rh rh). Следовательно, индивидуумы, эритроциты которых содержат Rh-фактор, являются резус-положительными (Rh+). У резусположительных индивидов (Rh+) отец также является Rh+ (генотип RR), но мать Rh- (генотип rr). Когда резус положительный, плод находится в матке резус-негативной женщины, то он вызывает образование в крови матери Rh-антител, которые реагируют с эритроцитами плода. Эта реакция сопровождается развитием анемии плода и его абортом или смертью после рождения (рис. 72).
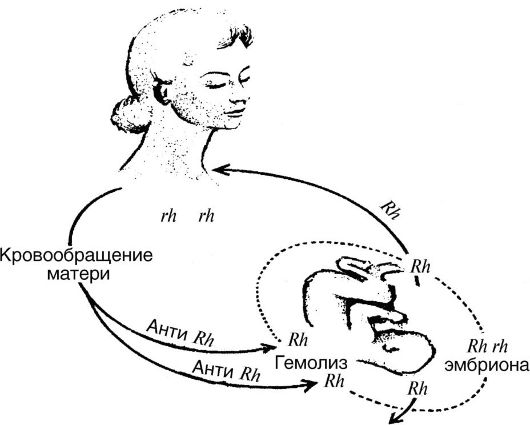
Рис. 72. Индукция Rh-антигеном
Такая аутосомно-рецессивная аномалия, как альбинизм, который обусловлен рецессивным геном, вследствие чего альбинизм рецессивен по отношению к нормальной пигментации, также встречается в разных странах с неодинаковой частотой. В Европе и США один альбинос приходится на 20 000 жителей, тогда как в Южной Америке и Африке - значительно чаще.
Анализ родословных многих семей, в которых обнаружены альбиносы, свидетельствует, что дети-альбиносы обычно рождаются от родителей-неальбиносов наряду с детьми-неальбиносами, при-
чем отношение между детьми-неальбиносами и детьми-альбиносами является менделевским (3:1). Решающие доказательства того, что альбинизм - это наследственный признак, получены при изучении идентичных близнецов. Как правило, если один из них альбинос, другой также является альбиносом, тогда как из двух неидентичных близнецов лишь один может быть альбиносом. Неальбиносы бывают или гомозиготными доминантными организмами (АА), или гетерозиготными организмами (Аа). Альбиносы являются гомозиготными рецессивными организмами (аа). Брак между альбиносом и неальбиносом не дает детей-альбиносов, так как рецессивный аллель а редок, и большинство людей не являются носителями такого аллеля. В браке между альбиносом и неальбиносом дети-альбиносы могут быть только тогда, когда неальбиносы являются гетерозиготным организмом, причем среди детей соотношение между альбиносами и неальбиносами будет составлять 1:1. В браках между альбиносами рождающиеся дети всегда альбиносы.
Среди наследственных болезней, передающихся в качестве аутосомно-рецессивных признаков и приуроченных к отдельным гео- графическим районам, интерес представляет серповидно-клеточная анемия. Известно, что нормальные эритроциты обладают гемоглобином А, каждая молекула которого состоит из двух полипептидных цепей α и двух цепей β. Каждый α-полипептид представлен специфическими последовательностями, состоящими из 141 аминокислоты, а каждый β-полипептид - специфическими последовательностями, состоящими из 149 аминокислот. Напротив, у больных серповидноклеточной анемией были найдены эритроциты серповидной формы, которые обладают гемоглобином S, являющимся результатом мутации (замены пары А-Т на пару Т-А) гена Hb^, контролирующего синтез гемоглобина А, к аллелю НЬ|, контролирующему синтез гемоглобина S. Данная мутация сопровождается тем, что в эритроцитах больных одна половина гемоглобина оказывается гемоглобином А, другая - гемоглобином S. Химические различия между гемоглобинами А и S заключаются в том, что β-полипептидные цепи в их глобинах (белковых частях) различаются одной аминокислотной заменой: в β -цепи гемоглобина А шестой аминокислотой является глутаминовая кислота, а в β-цепи гемоглобина S-валин. Генотип индивидов, гомозиготных по S-аллелю, будет Hb|/Hb|. Удлиненные серповидные эритроциты не способны обеспечивать транспорт кислорода к тканям, поэтому во многих случаях болезнь заканчивается смертью
еще в детском возрасте. В результате мутация гена НЪ^ в виде замены в нем пар азотистых основании происходит ооразование и других вариантов гемоглобина (помимо гемоглобина S), которых известно более 100 и которые обнаружены в разных популяциях людей. Эти варианты различаются между собой электрофоретически, а образование некоторых из них также сопровождается неблагоприятными фенотипическими эффектами разной тяжести.
Свыше 200 генов локализовано на половых хромосомах. Перечень некоторых наследственных болезней, которые контролируются генами, локализованными на Х- или Y-хромосомах, приведен в табл. 23. На хромосоме X идентифицировано свыше 70 генов, контролирующих гемофилию, мышечную дистрофию, цветовую слепоту (дальтонизм), ювенильную глаукому, оптическую атрофию (дегенерацию зрительного нерва) и др. Большинство из этих болезней наследуются по рецессивному типу. Доминантный тип наследования болезней, которые детерминируются генами, сцепленными с Х-хромосомой, очень редок.
Наиболее известной из генных Х-сцепленных болезней является гемофилия (несвертывание крови на воздухе), детерминируемая рецессивным геном и характеризующаяся довольно высокой смертностью, особенно в детском возрасте. Гемофилией болеют почти только мужчины, причем те, которые являются сыновьями женщин - носителей рецессивного гена гемофилии. Если мужчиныгемофилики вступают в брак со здоровыми женщинами, то их сыновья будут здоровы и не будут нести ген гемофилии, так как наследуют хромосому Y, свободную от этого гена. Что касается дочерей мужчингемофиликов, то все они будут внешне здоровыми, но будут гетерозиготными по гену гемофилии, т. е. носителями этого гена. Более того, сыновья последних в 50% случаев также унаследуют ген гемофилии (равным образом гетерозиготными окажутся и 50% дочерей материгемофилика). Поскольку у мальчиков нет второй Х-хромосомы, то унаследованный ими рецессивный мутантный ген гемофилии проявляет свое действие, и они страдают гемофилией. Поскольку у девочек две хромосомы X, то вследствие того, что на второй хромосоме X локализован доминантный (нормальный) ген, унаследованный рецессивный ген не проявляет своего действия и девочки не болеют гемофилией. Таким образом, в рассмотренном случае 50% мальчиков будут поражены гемофилией и 50% девочек окажутся гетерозиготными носителями гена гемофилии.
Таблица 23. Наследственные болезни, контролируемые генами, локализованными на Х- или Y-хромосоме
Болезнь Гемофилия | Проявление Недостаток фактора крови VIII, сопровождаю- щийся несвертыванием крови, в результате чего гемофилия даже при малейшем повреждении может стать смертельной |
Гипофосфатемия | Потеря организмом фосфора и недостаток в кальции, что сопровождается размягчением и мальформацией костей |
МукополисахаридозII | Нарушение в метаболизме белково-углеродистых соединений, отставание в росте, умственная отсталость, глухота |
Мышечная дистрофия | Нарушение структуры и функции мышц, часто начинающиеся в 20-30-летнем возрасте |
Ночная слепота | Неспособность видеть в темноте |
Синдром Рейфенстейна | Форма мужского псевдогермафродитизма, характеризующаяся недостаточным развитием тестисов и повышенным развитием грудных желез |
Тестикулярная феминизация | Нарушение в развитии мужчин, в результате чего они имеют внешние признаки женщин, включая влагалище, но не имеют матки. Гонады, локализованные в брюшной полости, являются дегенеративными тестисами |
Y-сцепленная волосатость ушей | Длинные волосы, растущие с внутреннего края ушей |
Синдактилия | Перепончатообразное сращение второго и тре- тьего пальцев на ноге |
Гемофилией могут страдать и женщины, но лишь тогда, когда они рождаются от родителей, один из которых является гемофиликом (отец), другой - гетерозиготным носителем (мать). Однако это встречается очень редко.
Передача рецессивного гена, детерминирующего гемофилию, от гетерозиготных носительниц к их дочерям, внукам и т. д., которые становятся гетерозиготными носителями и сыновья которых в 50% случаев болеют гемофилией, хорошо прослеживается при ознакомлении с генеалогией некоторых царствовавших семей в Европе, которая восходит к английской королеве Виктории, бывшей гетерозиготной
по полулетальному гену гемофилии в Х-хромосоме. От гемофилии умерли три правнука королевы Виктории - испанские инфанты Альфонсо, Гонсалес и Хуан, являющиеся сыновьями Альфонса XIII и Виктории-Евгении Баттенбергской. Гемофиликом был также сын последнего русского царя Николая II, который унаследовал ген гемофилии от своей матери, бывшей царицы Александры Федоровны (Алисы). Последняя унаследовала этот ген через мать от своей бабки королевы Виктории (рис. 73).
Давно идет обсуждение вопроса о происхождении гена гемофилии у королевы Виктории. Достоверно известно, что ни ближайшие, ни дальние родственники ее родителей не обладали таким геном. Поэтому предполагают, что Виктория оказалась носителем свежей мутации гена или же она была дочерью фаворита ее матери, который болел гемофилией.
Другим примером наследования генов, сцепленных Х-хромосомой, является наследование дальтонизма, который, например, в США встречается у 8% мужчин и у 0,5% женщин. Наследование дальтонизма похоже на наследование гемофилии, так как рецессивный ген локализован на хромосоме X. Отец передает Х-хромосому ко всем дочерям, но ни к одному из сыновей, а мать передает одну из ее хромосом X ко всем детям. В связи с этим все сыновья матери-дальтоника тоже являются дальтониками, причем независимо от состояния зрения отца. Однако если отец имеет нормальное зрение, то нормальное зрение будут иметь все дочери от этого брака, хотя они и будут являться гетерозиготными носителями. В браке последних с мужчинами, зрение которых нормально, будут рождаться девочки с нормальным зрением, а мальчики - наполовину дальтоники и наполовину с нормальным зрением. Девочка-дальтоник может родиться лишь в браке мужчины-дальтоника с женщиной-дальтоником или гетерозиготной носительницей. В литературе описан случай, когда мужчина был одновременно гемофиликом и дальтоником, что является доказательством локализации этих генов на одной и той же хромосоме X.
Синдром Леша-Найяна - это заболевание почек мальчиков, сопровождающееся умственной отсталостью и связанное с недоста- точностью гипоксантин-фосфорибозилтрансферазы, необходимой для синтеза ДНК. Наследуется через рецессивный ген, сцепленный с Х-хромосомой. Следовательно, мать передает дефектный ген мальчикам через Х-хромосому. Предполагают, что 1/2 мальчиков, родившихся от матери-носителя, будут наследовать дефектный ген.
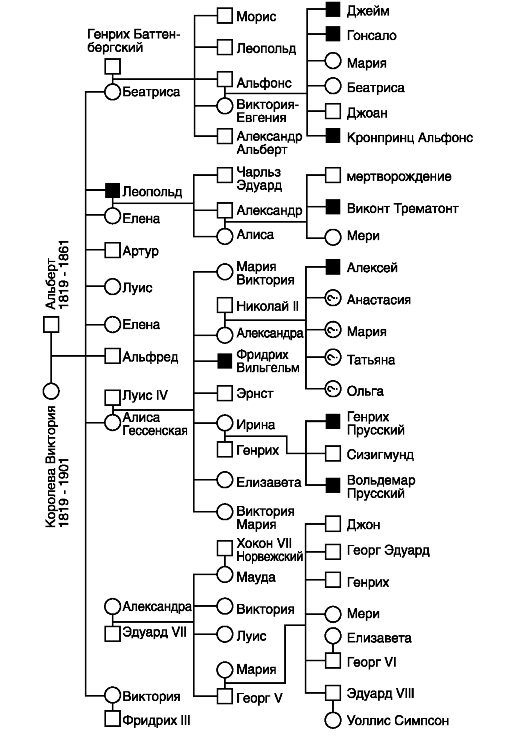
Рис. 73. Родословная потомков королевы Виктории, в которой прослеживается наследование гена гемофилии: □ - нормальный мужчина; О - нормальная женщина; ■ - мужчина-гемофилик
Мышечная дистрофия ювенильного возраста также зависит от рецессивного гена, сцепленного с Х-хромосомой. Эта болезнь про- является в мышечном истощении, которое начинается в раннем подростковом возрасте и заканчивается параличами конечностей в среднем и позднем подростковом периоде. Умирают обычно, не достигнув 21 года. Болезнь наследуется по тому же типу, что и синдром Леша- Найяна.
Примерами наследственных аномалий, контролируемых генами, локализованными на Y-хромосомах, являются синдактилия (перепончатообразное сращение 2-го и 3-го пальцев на ноге) и гипертрихиоз (волосатость края ушной раковины). Поскольку Y-хромосома встречается только у мужчин, эти гены передаются к потомству только по мужской линии.
Характерной особенностью ряда генных болезней является то, что их географическая приуроченность приходится на «старые» популяции людей, что объясняют так называемым компенсационным преимуществом. Классическим примером такой компенсации является преимущество гетерозиготных носителей гена серповидноклеточной анемии относительно малярии. Гомозиготы по этому гену погибают, но гетерозиготы более устойчивы к малярии, чем нормаль- ные. При болезни Тея-Сакса у евреев-ашкенази компенсационное преимущество обеспечивается большей устойчивостью их к туберкулезу по сравнению с другим населением в том же самом регионе. Поэтому смертность их от болезни Тея-Сакса является «платой» гомозигот за выживание гетерозигот.
Характерной особенностью генных болезней является также и то, что многие из них, помимо специфических клинических черт, сопро- вождаются также умственной отсталостью больных. В частности, умственная отсталость установлена в случае около 60 нозоологических форм аутосомно-доминантных и аутосомно-рецессивных аномалий (включая фенилкетонурию). По данным американских авторов, в США около 1/3 населения, возможно, имеет один или несколько аутосомных рецессивных генов, которые в гомозиготном состоянии могут сопровождаться умственной отсталостью индивидуумов. Считают также, что 23% всех случаев умственной отсталости контролируется генами, локализованными на половых хромосомах и детерминирующими разные наследственные болезни.
Исследования молекулярных механизмов в этиологии генных болезней позволили установить существование таких болезней, воз-
никновение которых связано с нарушениями регуляции действия генов. Например, синдром Рубенштейна-Тайби, который характе- ризуется нарушениями в строении концевых фаланг конечностей и умственной отсталостью больных, является результатом мутации гена, детерминирующего синтез белка, действующего в качестве коактиватора ц-АМФ-регулируемой экспрессии этого гена.
НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ, ОБУСЛОВЛЕННАЯ
МУТАЦИЯМИ ХРОМОСОМ
Наследственные болезни, связанные с мутациями хромосом, имеют широкое распространение. Считают, что хромосомные болезни живорожденных составляют 35-40% от всех наследственных болезней.
Для всех или почти всех хромосомных аномалий характерной особенностью является то, что они начинают проявляться еще во внутриутробном периоде развития, оказываясь летальными. Установлено, что гибель зародышей начинается уже на стадиях зиготы и бластулы (в первые две недели развития). Хромосомные мутации ответственны примерно за 50% спонтанных абортов и 6% мертворождений. Считают, что те или иные хромосомные дефекты имеются у 0,6-0,7% всех новорожденных. Смертность от хромосомных болезней в перинатальный период составляет приблизительно 1:1000.
Различают хромосомные наследственные болезни, детерминируемые мутациями количества хромосом, и хромосомные наследственные болезни, детерминируемые мутациями структуры хромосом.
Мутационные нарушения плоидности хромосом человека в сторону гаплоидии неизвестны. Что касается полиплоидии, то она описана в виде триплоидии и тетраплоидии при исследовании спонтанно абортированных плодов и эмбрионов, а также мертворожденных. По некоторым данным, 1% всех зачатий человека представлен триплоидными зиготами. Аборты триплоидов, как правило, происходят к концу третьего месяца беременности. Полиплоидия является результатом извращений митотического деления зародышей или нерасхождения полного генома в течение мейоза, ведущего к диплоидным гаметам.
Наиболее часто встречаются болезни, являющиеся результатом анеуплоидии аутосом и половых хромосом. «Клинически значимые»
аномалии составляют половину всех хромосомных аномалий новорожденных. В табл. 24 представлены данные о некоторых из таких наследственных болезней.
Наиболее частой формой анеуплодии (гетероплодии) являются полисомии по тем или иным хромосомам, частота которых составляет 5,7 ? 10-4 на гамету или генерацию. К наиболее известным аутосомным полисомиям относятся трисомии, когда одна из пар в хромосомном наборе имеет добавочную хромосому, т. е. представлена тремя хромосомами (2п + 1). Например, болезнь Дауна, характеризующаяся серьезными нарушениями здоровья, включая нарушения психической деятельности, обусловлена трисомией по 21-й паре хромосом. У шимпанзе синдром Дауна бывает по 22-й паре, которая гомологична 21-й паре человека (рис. 74).
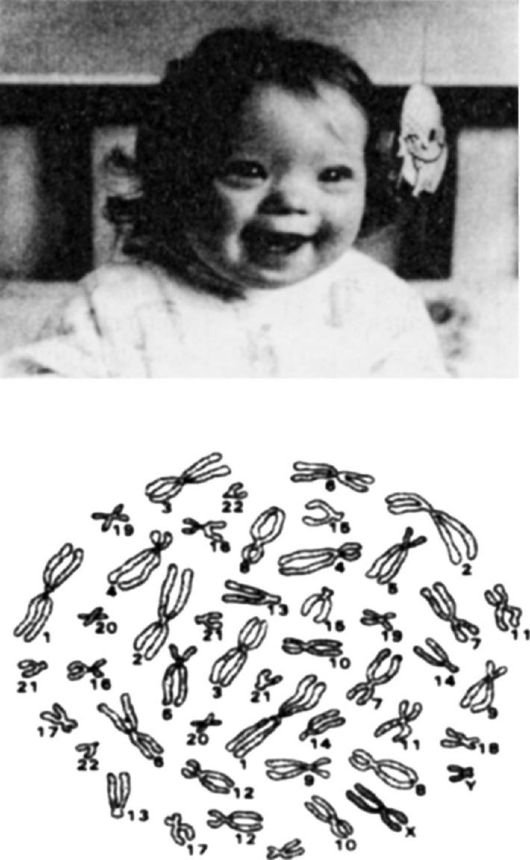
Рис. 74. Синдром Дауна (кариотип)
310 Глава VIII. Нормальная и патологическая наследственность человека Таблица 24. Некоторые наследственные болезни, детерминируемые
Хромосомная формула | Клинический синдром | Частота при рождении | Главные фенотипические характеристики |
47,ХХ+21 или 47,XY+21 | Болезнь Дауна | 1:700 (у европейцев) | Монголоидность, открытый рот с большим языком, умственная отсталость. 1/6 часть больных умирает в первый год после рождения |
47,ХХ+13 или 47,XY+13 | Трисомия 13 (синдром Патау) | 1:20 000 | Глухота, аномалии сердца, полидактилия, умственная дефективность |
47,ХХ+18 или 47,XY+18 | Трисомия 18 (синдром Эдварда) | 1:8000 | Множественные врожденные пороки многих органов. Умственная отсталость. 90% больных умирают в первые 6 месяцев. Известен случай, когда один ребенок дожил до 5 лет |
45,Х | Синдром Шерешев- ского- Тернера | 1:3000 | Женщины с недостаточным половым развитием, короткой фигурой, нарушениями сердечнососудистой системы |
47,XXY | Синдром Клайнфельтера | 1:500 | Мужчины с недоразвитыми тестисами, но развитой грудью, с женским голосом, длинными конечностями |
47,ХХХ | Синдром Трипло-Х | 1:700 | Больные женщины внешне не отличаются от нормальных, но их фолликулы недостаточно развиты. Менструации нерегулярны |
48,ХХХХ 48,XXXY 48,XXVY 49,XXXXY 50,ХХХХХХ | Различные полисомии | Данных нет | Аномалии скелета, умственная отсталость и другие симптомы |
Трисомии бывают и в случае малых хромосом. Например, трисомия по 13-й паре обусловливает синдром Патау, 18-й паре - синдром Эдвардса. Известны такие трисомии по 22-й, 8-й и другим парам хромосом. Описаны также случаи аутосомных тетрасомий и пентасо-
мий, но они летальны. Что касается аутосомных моносомий, то они тоже летальны.
Более частой формой анеуплоидии является анеуплоидия по половым хромосомам, частота которой составляет 9,3?10-4 на гаме- ту или генерацию. Различают полисомии по половым хромосомам и Х-моносомии. Наиболее известными примерами наследственных заболеваний, в основе которых лежит анеуплоидия по половым хромосомам, являются синдромы Клайнфельтера и Трипло-Х, которыми страдают женщины, а также синдром 47,XYY, который характеризуется агрессивностью, умственной отсталостью и антиобщественным поведением больных мужчин. Причинами трисомии считают нерасхождение хромосом при гаметогенезе (мейозе) у одного из родителей. Примером наследственного заболевания, в основе которого лежит Х-моносомия, является синдром Шерешевского-Тернера. Х-моносомии служат, вероятно, главной причиной полового недоразвития женщин (рис. 75, 76, 77).
Частота многих хромосомных аномалий зависит от возраста матерей, что хорошо показано в случае синдрома Дауна (табл. 25).
Как правило, мутации количества хромосом происходят в гаметах одного из родителей. Поэтому все клетки организма, в зачатии кото-
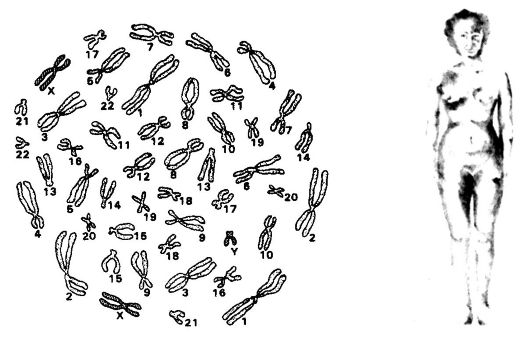
Рис. 75. Кариотип синдрома Рис. 76. Синдром Трипло-Х
Клайнфельтера (47,XXY; 48XXXY; (47,ХХХ)
49XXXXY или 48XXYY)
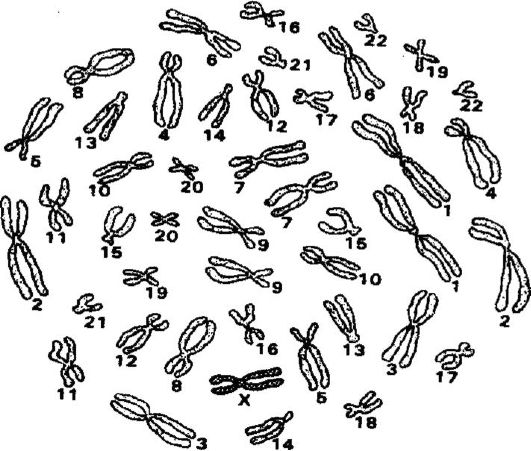
Рис. 77. Кариотип синдрома Шерешевского-Тернера (46,Х)
Таблица 25. Возраст матерей и частота синдрома Дауна
Возраст матери, лет | Частота синдрома |
до 20 | - |
до 30 | 1:1000 |
до 30-34 | 1,6:1000 |
до 35-39 | 4:1000 |
до 40-44 | 14:1000 |
до 45 и выше | 25:1000 |
рого принимала участие одна из мутантных гамет, будут содержать аномальный хромосомный набор. Однако иногда количественные хромосомные мутации могут случаться в процессе первых делений зиготы, образованной нормальными гаметами. Из такой зиготы разовьется организм, часть клеток которого будет иметь нормальный диплоидный набор, другая же часть - аномальный (рис. 78). Это явление называют хромосомным мозаицизмом, а индивидов, обладающих мозаицизмом, - хромосомными мозаиками. Мозаицизм чаще всего проявляется по половым хромосомам. Такие мозаики имеют генотипы Х/ ХХ, X/ XY, XX/XY, XXY/XX.
Для хромосомных болезней, причиной которых являются мутации количества хромосом, так же как и для генных, характерна в качестве «сопутствующего» симптома умственная отсталость. Она
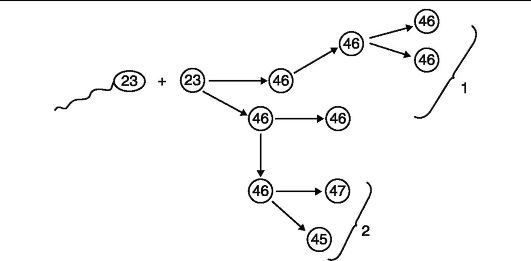
Рис. 78. Формирование хромосомного мозаицизма: 1 - линия клеток с нормальным числом хромосом; 2 - линия клеток с аномальным
числом хромосом
часта в случае аутосомных синдромов. Что касается половых хромосом, то около 1% умственно отсталых женщин имеют одну или более лишнюю хромосому X.
Умственная отсталость имеет важное социальное значение, но ее изучение очень ограничено тем, что человеческие психические черты еще не сведены до количественных механизмов. Кроме того, отсутствуют адекватные тесты. IQ-тест не полностью оправдал возлагавшихся на него надежд, поскольку невозможно определить, что измеряется с помощью этого теста.
Хромосомные болезни, детерминируемые мутациями структуры хромосом, более редки, а их клиническое проявление менее выражено или неспецифическое по сравнению с аномалиями, вызываемыми мутациями количества хромосом. Мутации структуры хромосом выявлены во всех парах аутосом. Часто многие из этих мутаций называют частичными трисомиями и моносомиями. Например, делеция по хромосомной паре 5 вызывает синдром «кошачий крик», связанный с анатомическими изменениями лицевого скелета, синдром «дупликации - деления 11» в хромосоме 3 заключается в спонтанных абортах. Описаны, однако, два рождения, при которых ребенок не способен сидеть, не может есть твердую пишу, имеет очень короткий нос. Предполагают, что при синдроме Дауна в некоторых случаях происходит транслокация сегмента хромосомы 21 на другую
хромосому. По данным японских авторов, 14 из 321 пациента болезнью Дауна характеризовались транслокацией части хромосомы 21 на одну из нескольких других хромосом.
НАСЛЕДСТВЕННАЯ ПАТОЛОГИЯ, ОБУСЛОВЛЕННАЯ МИТОХОНДРИАЛЬНЫМИ ГЕНАМИ
Известно небольшое количество наследственных болезней, получивших название митохондриальных из-за того, что в их основе лежат изменения в ДНК митохондрий, т. е. митохондриальных генов.
Установлено свыше 80 различных генов, которые необходимы для продукции компонентов дыхательной цепи митохондрий и мутации которых повреждают окисление в клетках.
Одной наиболее известной болезнью является наследственная оптическая нейропатия Лебера, связанная с поражением мышц глаза и проявляющаяся в виде внезапной потери зрения в период между юноше- ским и взрослым возрастом из-за инактивации зрительного нерва.
Описаны также хроническая прогрессирующая офтальмоплегия, синдромы Кернса-Сэйра и Пирсона, митохондриальная энце- фаломиопатия и молочный ацидоз.
Известны данные, в соответствии с которыми перестройки в митохондриальной ДНК фенотипически проявляются в развитии диабета - Diabetus melitus.
Известно, что митохондриальная ДНК передается по материнской линии, что прочно установлено у дрозофил и мышей. Но у морских голубых двухстворчатых раковин рода Mytilus она передается как по материнской, так и по мужской линии, но передача зависит от пола потомства. Мужские особи получают материнские митохондрии от отцов, женские - от матерей. Напротив, дочери обычно получают лишь материнскую митохондриальную ДНК. Поскольку в спермиях очень низкий уровень женских митохондрий, то считают, что все или почти все дочерние митохондрии должны иметь материнское происхождение. У этой раковины женские митохондрии передаются матерями сыновьям и дочерям, а мужские митохондрии - отцами сыновьям. Могут быть также ограничения (передача женских митохондрий отцами дочерям).
Каким образом наследуются митохондриальные гены у человека? Поскольку митохондрии из сперматозоидов не проникают в яйце-
клетку, то индивид наследует митохондрии (митохондриальные гены) от матери. Мутации митохондриальных генов могут передаваться в яйцеклетки, поэтому риск передачи велик. Итак, это пример мате- ринской наследственности.
БОЛЕЗНИ С НАСЛЕДСТВЕННЫМ ПРЕДРАСПОЛОЖЕНИЕМ
Болезни, в патогенезе которых играет роль наследственность и проявление которых зависит от действия факторов внешней среды, являются болезнями с наследственным предрасположением. К таким болезням относятся атеросклероз, гипертоническая болезнь, ишемическая болезнь сердца, ревматизм, язвенная болезнь, дерматиты, некоторые формы диабета, шизофрения и др. Доводами в пользу наследственной предрасположенности служат существенно большее накопление повторных случаев болезни среди родственников больных по сравнению с популяционной частотой данного заболевания, а также значительное повышение показателей конкордантности в парах монозиготных близнецов по сравнению с такими показателями в парах дизиготных близнецов (табл. 26).
Генетическая природа болезней с наследственными предрасположениями неодинакова. Различают моногенные и полигенные болезни с наследственным предрасположением.
Моногенные болезни с наследственным предрасположением - это болезни, при которых предрасположенность определяется одним геном во взаимодействии с точно известным фактором среды.
Полигенные болезни с наследственным предрасположением - это болезни, при которых предрасположенность определяется многими генами во взаимодействии с многими факторами среды. По этой причине такие болезни еще называют мультифакториальными (рак, гипертония, шизофрения и др.).
Изучение мультифакториальных болезней затруднено по многим причинам, одна из которых заключается в поисках генетических маркеров предрасположения. Например, в случае атеросклероза повышенный уровень холестерина контролируется несколькими генами, один из которых ответствен за повышение уровня холестерина (семейная гиперхолестеринемия), второй - за повышение в сыворотке концентрации триглицеридов (семейная глицеридемия), третий -
за повышение уровня обоих липидов (комбинированная гиперлипидемия). Поскольку холестерин и триглицериды имеют значение в развитии раннего атеросклероза коронарных сосудов, то считают, что эти гены в сочетании с фактором среды создают механизм предрасположения к атеросклерозу и развитию инфаркта миокарда.
Таблица 26. Частота встречаемости ряда болезней среди родственников и в популяции
Болезнь | Частота, % | Конкордантность у близнецов, % | ||
среди родственников | в популяции | монозиготных | дизиготных | |
Ишемическая болезнь сердца | 30-60 | 19 | 67 | 43 |
Язвенная болезнь | 8 | 0,6 | 50 | 14 |
Шизофрения | 14 | 1 | 67 | 18 |
Ревматизм | 10 | 2 | 37 | 7 |
Болезни с наследственной предрасположенностью характеризуются различной степенью выраженности, что является отражением разных уровней накопления факторов предрасположения и комбинации их с неодинаковыми по степени и направлению взаимодействия факторами среды (стресс, климатические условия, инфекции и др.).
ПРИНЦИПЫ ДИАГНОСТИКИ, ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ
Различают пренатальную и постнатальную диагностику. В пренатальной диагностике используют ряд методов, среди которых наиболее эффективным является амниоцентез (рис. 79).
Суть этого метода заключается в трансабдоминальном получении околоплодной жидкости и исследовании ее непосредственно путем микроскопии содержащихся в ней клеток или после культивирования последних. С помощью метода амниоцентеза возможно опреде- ление пола и кариотипа плода. Широкое распространение получил метод ультразвуковой диагностики (УЗИ). Пренатальную диагностику обычно используют в семьях, где риск рождения наследственно больного ребенка составляет около 3-5%.
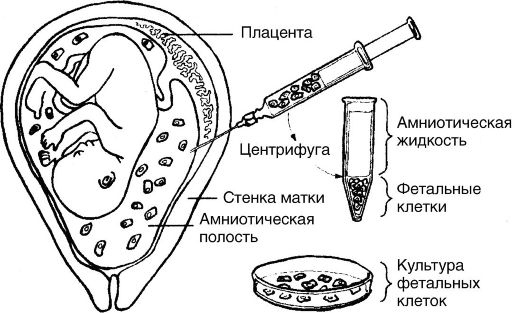
Рис. 79. Амниоцентез
Постнатальная диагностика наследственных болезней основывается на результатах клинического, параклинического и генетического обследования пациентов. Генетическое обследование основывается в первую очередь на результатах генеалогического анализа. В зависимости от показаний привлекают также цитогенетические, биохимические, иммунологические и другие методы.
Большое значение имеют методы массовой («просеивающей») диагностики с целью проверки населения на возможность скрытых форм наследственных аномалий. «Просеивающую» диагностику применяют к новорожденным с целью выявления галактоземии, муковисцидоза и других наследственных болезней и к определенным группам населения с целью выявления гетерозиготного носительства болезни Тея-Сакса, серповидноклеточной анемии и талассемии.
Во многих случаях диагностика наследственных болезней является успешной. Однако при гетерозиготных наследственных аномалиях (разных аномалий, но сходных по проявлению) она очень затруднена. Особенно сложной является диагностика умственной отсталости, так как она осложнена разнообразием факторов среды.
Подходы к лечению наследственных болезней являются теми же, что и при болезнях другой природы, причем врач должен руководствоваться установкой о возможности лечения многих наследственных болезней. Как и в случае других болезней, различают симптоматическое (воздействие на симптомы болезни), патогенетическое
(воздействие на патогенез болезни) и этиологическое (воздействие на причину болезни) лечение наследственных болезней.
Симптоматическое и патогенетическое лечение наследственных болезней заключается в равных формах терапии. При некоторых наследственных болезнях прибегают к диетотерапии. Поскольку, например, патогенез такого наследственного заболевания, как галактоземия, связан с накоплением в клетках галактозы из-за отсутствия фермента ф-Д-галакто-1-фосфат-уридилтрансферазы, вследствие чего развиваются изменения в печени и головном мозге и, наконец, ослабление умственной деятельности, то лечение болезни обычно проводят исключением материнского молока и назначением диеты, не содержащей галактозы. Другой пример диетотерапии представляет лечение фенилкетонурии, которая может быть обнаружена простым исследованием мочи новорожденных. Новорожденному с этой болезнью назначается диета с пониженным содержанием фенилаланина в период раннего детства, что предупреждает умственную отсталость и другие симптомы фенилкетонурии. Когда «вылеченные» становятся матерями, они вновь должны вернуться к диете с пони- женным содержанием фенилаланина и таким образом обеспечить подходящую среду для плода. Их потомство (девочки) тоже может нуждаться в низкой по фенилаланину диете в раннем детстве, чтобы предупредить симптомы болезни.
С целью лечения часто прибегают к введению в организм недостающего фактора. Например, при пернициозной анемии, гемофилии и антигемофильной глобулиновой недостаточности прибегают к периодическим инъекциям недостающего в организме белка, что временно улучшает состояние больных. Для лечения ряда болезней используют переливание крови или удаление из организма токсических веществ с помощью лекарственных средств. Лекарственную терапию часто используют с целью индукции недостающих ферментов. Например, отдельные наследственные болезни лечат фенобарбиталами, которые стимулируют индукцию недостающих ферментов. При гемолитической болезни новорожденных (К/г+-дети) матерям вводят глобулин Rh, который разрушает Rh-позитивные клетки плодового происхождения и предупреждает образование Rh-антител, которые могут повредить другие плоды в последующих беременностях.
Важное место в лечении наследственных болезней занимает хирургическое лечение путем удаления органов или частей орга- нов, коррекция повреждений или трансплантаций. Например, часто
прибегают к удалению толстой кишки при полипозе, селезенки при сфероцитозе, почек при цистинозе. Многие аномалии (незаращение верхней губы, врожденные пороки сердца, полисиндактилия, болезнь Нимана-Пика и др.) исправляют хирургической коррекцией или с помощью трансплантаций. Однако важно подчеркнуть, что при симптоматическом и патогенетическом лечении генетический дефект у больных сохраняется. Вступая в детородный возраст, наследственно больные передают неблагоприятные гены своему потомству.
В широком плане в рамках методов симптоматического и патогенетического лечения можно рассматривать и евфенические мероприятия, под которыми понимают компенсацию естественных недостатков человека в фенотипе, т. е. улучшение здоровья человека через фенотип. Часто евфенические мероприятия называют лечением адаптивной средой. Они известны давно, и к ним относят пренатальную и постнатальную заботу о потомстве, иммунизации, переливания крови, трансплантации органов, пластическую хирургию, обогащение диет белками, витаминами и микроэлементами, исключение из диет отдельных углеводов, физическую культуру, лекарственную терапию при тех наследственных болезнях, проявление которых зависит от факторов среды. Однако евфенические мероприятия, как и симптоматическое и патогенетическое лечение, тоже не приводят к радикальным результатам, ибо благодаря евфенике нельзя кардинально преодолеть наследственные дефекты. Кроме того, эффекты, достигнутые в результате евфенических мероприятий, не передаются по наследству и не сопровождаются уменьшением количества небла- гоприятно действующих генов в популяциях человека.
Этиологическое лечение наследственных болезней, которое должно приводить к кардинальному исправлению наследственных аномалий, еще не разработано. Однако в связи с достижениями физико-химической биологии сформулированы и разрабатываются программы изменения в желаемом направлении фрагментов генетического материала, детерминирующих наследственные аномалии человека. Они исходят в основном из двух идей генной инженерии. В соответствии с одной из этих идей предполагают, что исправление дефекта может быть осуществлено изменениями в нужном русле «испорченного» фрагмента (группы нуклеотидов или отдельного нуклеотида) генетического материала направленными мутациями. Такой способ исправления наследственных аномалий может быть
реализован путем индукции обратных мутаций в случае открытия сложных мутагенов, которые соответствуют единицам хромо- сомы, подвергаемой изменению, и несут химические группы соответствующей реактивности, обеспечивающие их комплементарность исправленной хромосоме. В соответствии с другой идеей исправление генетического дефекта может быть обеспечено заменой в клетке неудовлетворительного фрагмента хромосомы «лучшим» хромосомным фрагментом. Предполагают, что вводимый фрагмент должен быть синтезированным или иметь естественное происхождение, причем в любом случае он должен быть гомологичным заменяемому фрагменту хромосомы, за исключением тех его участков, которые являются замещающими и имеют структуру желаемого характера. Разработка этих идей представляет сейчас важное направление в ген- ной инженерии.
Таким образом, это есть случай генной терапии, под которой понимают замену мутантного (болезнетворного) белка в клетках организма на нормальный белок, восстанавливающий нормальные клеточные функции. Разработки методов генной терапии обеспечиваются различными методами клеточной и генной инженерии.
Профилактика наследственных болезней сводится к профилактике болезней, унаследованных от предыдущих поколений, и к про- филактике болезней, вновь возникающих в результате мутаций в зародышевых клетках родителей.
Профилактика болезней, унаследованных от предыдущих поколений (сегрегационный груз), осуществляется главным образом с помощью медико-генетической консультации. Основные задачи медико- генетического консультирования сводятся к тому, чтобы определить степень риска рождения ребенка с наследственной патологией в данной семье и помочь родителям принять правильное решение, а также насторожить врача на риск рождения наследственно отягощенного ребенка и облегчить возможность постановки раннего диагноза и принятия лечебных мер. В глобальном плане медико-генетическая консультация должна понижать частоту неблагоприятных генов в популяциях человека. Результативность медико-генетической консультации зависит от знания родословной и точности определения скрытого носительства патологических генов, диагностики гетерозиготного состояния. Медико-генетическая консультация связана с исключительной морально-этической ответственностью консультирующего генетика или врача в познании тайн при анализе родос-
ловных и в определении степени риска. От медико-генетической консультации как врачебного заключения следует отличать консультацию как учреждение. В СССР первая медико-генетическая консультация была организована еще в конце 20-х гг. XX в. (С.Н. Давиденков). В настоящее время медико-генетические консультации существуют во многих странах мира и в России.
Профилактика наследственных болезней, вновь возникающих в результате мутаций, сводится к предупреждению загрязнения среды обитания человека факторами, которые могут действовать как мутагены. В условиях научно-технической революции серьезное значение приобретает организация генетического мониторинга (слежение за генетическими процессами в популяции человека).
ВОПРОСЫ ДЛЯ ОБСУЖДЕНИЯ
1. Применимы ли законы Г. Менделя к человеку? Если да, то покажите, на чем основана их применимость.
2. Чем отличаются методы изучения наследственности человека от классического генетического анализа?
3. Каким образом можно установить принадлежность группы сцепления к той или иной паре хромосом человека?
4. Какова ценность метода клонирования генов в генетике человека?
5. Назовите нормальные признаки человека, детерминируемые аутосомно-доминантными и рецессивными генами.
6. Приведите примеры наследственности человека, сцепленной с полом.
7. Что вы знаете о генетической индивидуальности людей?
8. Что такое генетический груз и чем он определяется?
9. Как классифицируют наследственные болезни?
10. Назовите генные наследственные болезни, наследуемые как аутосомно-доминантные и рецессивные признаки, а также при- знаки, сцепленные с полом.
11. Что вы знаете о хромосомных болезнях, их частоте, особенностях распространения и этиологии?
12. Что вы знаете о болезнях с наследственным предрасположением и об их отличиях от наследственных болезней?
13. Аномалия «заячья губа» встречается с частотой 0,1%. Конкордантность по этому признаку между идентичными близнецами
составляет 50%, между сибсами - 3,5%, между двоюродными братьями и сестрами - 0,7%, а между троюродными братьями и сестрами - 0,3%. На степень конкордантности не влияет пол. Можете ли вы на основе этих данных определить тип наследования «заячьей губы»?
14. В эритроцитах человека найдено три электрофоретически различных формы кислой фосфатазы А, В и С. При исследовании 178 англичан оказалось, что их эритроциты содержат разные фосфа- тазы, а именно:
• фосфатаза А - 17 человек, фосфатазы В + С - 9;
• фосфатаза В - 61 человек, фосфатазы А + С - 5;
• фосфатаза С - 0, фосфатазы А, В и С - 0;
• фосфатазы А + В - 86 человек.
Объясните эти результаты. Почему ни у кого не найдена фосфатаза С?
15. Какими могут быть заключения генетического консультанта в следующих трех случаях:
а) Дядя женщины (брат ее матери) болел гемофилией. Женщина забеременела и желает знать, каков риск рождения у нее ребенкагемофилика. Как могла бы измениться ситуация, если бы дядей- гемофиликом был брат ее отца?
б) Женщина имела двоюродного брата, который умер от фиброцистита. Имеется ли риск рождения у этой женщины больного ребенка?
в) Старшая сестра мужчины страдала хореей Хантингтона. Мужчине 40 лет, и он хочет знать о риске для себя и детей, которых он желает иметь. Что вы скажете в ответ на его вопросы?
16. Какова, по вашему мнению, результативность лечения наследственных болезней?
17. В чем состоит профилактика наследственных болезней? Каковы ограничения генетической консультации?
18. Что вы знаете о научных направлениях в разработке способов радикального лечения наследственных болезней?
19. Что такое генетический мониторинг и каково его значение в профилактике наследственных болезней?
20. Каковы, по вашему мнению, эффективные методы в разработке способов генной терапии?