Клиническая биохимия : учебное пособие. 3-е издание / под ред. В.А. Ткачука. - 2008
|
|
|
|
ГЛАВА 2 СИСТЕМА ГЕМОСТАЗА
Система гемостаза - одна из защитных систем организма, обеспечивающая сохранение крови в жидком состоянии в пределах кровеносных сосудов и образование тромбов в области повреждения стенки сосудов. Гемостатический процесс включает пять стадий: локальную вазоконстрикцию, формирование тромбоцитарного тромба, стабилизацию его фибрином, ретракцию тромба и растворение после восстановления поврежденной стенки сосуда. Свертывание крови обеспечивается взаимодействием белков плазмы и клеток крови с поврежденным эндотелием или субэндотелиальными структурами. Условно система гемостаза подразделяется на три системы: свертывания, противосвертывания и фибринолиза, которые тесно взаимосвязаны.
2.1. КОМПОНЕНТЫ СИСТЕМЫ СВЕРТЫВАНИЯ
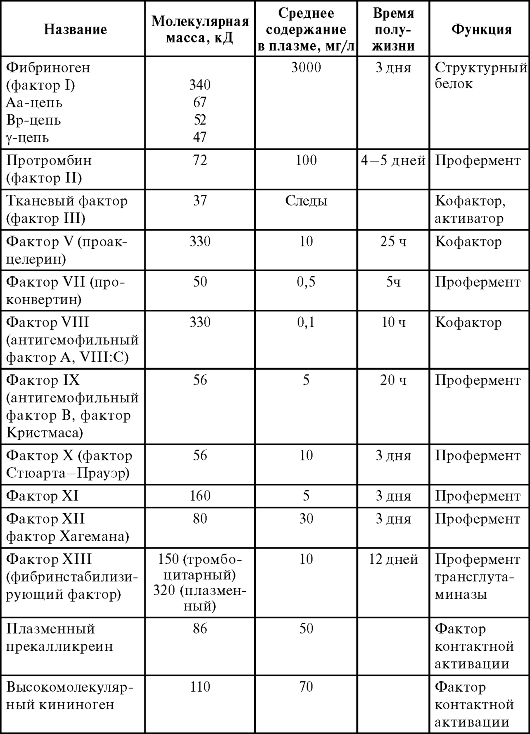
Основные компоненты системы свертывания крови были идентифицированы к середине 1950-х годов (табл. 2.1). Большинство факторов свертывания были открыты при исследовании крови больных с нарушениями свертываемости, их назвали по фамилиям больных или по предполагаемому характеру выполняемой функции. Кроме этого, ряд факторов был открыт практически одновременно в разных лабораториях, они получали разные названия, что затрудняло систематизацию данных.
В 1957 г. Международный комитет по номенклатуре факторов свертывания крови ввел цифровые обозначения факторов. Порядковый номер факторам присваивали примерно в той последовательности, в которой они были открыты. Плазменные факторы свертывания были пронумерованы римскими цифрами, а тромбоцитарные - арабскими. Активированные формы факторов обозначаются добавлением к цифре буквы а. Факторам Флетчера и Фитцжеральда- Вильямса-Фложак цифровые обозначения не были присвоены, так как вскоре после обнаружения этих факторов было установлено, что это соответственно плазменные прекалликреин и высокомолекулярный кининоген.
Таблица 2.1. Факторы системы свертывания крови
 ФАКТОРЫ СИСТЕМЫ СВЕРТЫВАНИЯ КРОВИ
ФАКТОРЫ СИСТЕМЫ СВЕРТЫВАНИЯ КРОВИ
В активированной форме шесть плазменных факторов системы свертывания крови и калликреин - специализированные сериновые протеазы. То есть основной механизм активации свертывания крови - ограниченный протеолиз. Протеазные домены происходят из С-концевых частей молекул проферментов. Аминокислотные последовательности этих участков факторов свертывания крови гомологичны таковым трипсина и химотрипсина. Селективность действия протеаз системы гемостаза обеспечивается специфическими структурами N-концевых частей молекул.
Gla-домены. В N-концевой части протромбина, факторов X, IX и VII, а также протеинов C и S содержится до 12 остатков γ-карбоксиглутаминовой кислоты (Gla), образующейся в результате посттрансляционного γ-карбоксилирования остатков Glu под действием карбоксилазы и витамин К-эпоксидредуктазы (рис. 2.1). Эти остатки связывают ионы Са2+, что стабилизирует структуру белков и необходимо для эффективного взаимодействия факторов с фосфолипидами. При дефиците витамина К или приеме его антагонистов степень γ-карбоксилирования снижается и гепатоциты секретируют факторы, обладающие низкой функциональной активностью. Их называют PIVKA-белками, индуцированными антагонистами витамина К.
Крингл-домены. Протромбин, фактор XII и ферменты системы фибринолиза содержат участки, состоящие из ~80 аминокислотных остатков, которые образуют петлю, свернутую в форме сплющенного тора. В стабилизации структуры важная роль принадлежит трем дисульфидным мостикам и боковым аминокислотным цепям, образующим внутреннюю, наиболее консервативную часть петли. Значительная вариабельность участков, образующих наружную часть петли, обеспечивает специфичность взаимодействия с другими компонентами системы гемостаза.
EGF-домены. Факторы VII, X, IX и XII содержат последовательность из ~50 аминокислот, гомологичную таковой фактора роста эпидермиса. EGF-домены обеспечивают связывание факторов со специфическими рецепторами на поверхности клеток или рецепторными доменами факторов V и VIII.
Последовательность ароматических аминокислот. Протромбин, факторы VII, X и IX содержат 5-членную последовательность Phe-Trp-X-X-Tyr, в которой ароматические боковые цепи аминокислот
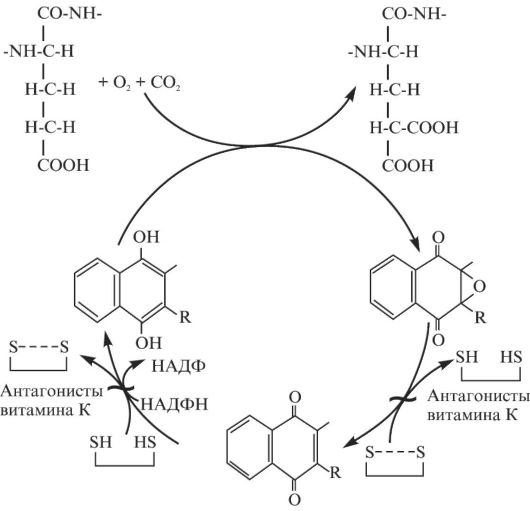 Рис. 2.1. Витамин
К-зависимое образование γ-карбоксиглутаминовой кислоты. Антагонисты
витамина К (пероральные антикоагулянты) ингибируют восстановление
эпоксидной и хиноновой форм витамина К, образующихся в ходе реакции
карбоксилирования остатков глутаминовой кислоты
Рис. 2.1. Витамин
К-зависимое образование γ-карбоксиглутаминовой кислоты. Антагонисты
витамина К (пероральные антикоагулянты) ингибируют восстановление
эпоксидной и хиноновой форм витамина К, образующихся в ходе реакции
карбоксилирования остатков глутаминовой кислоты
экспонированы наружу, что создает гидрофобный участок, взаимодействующий с мембранами клеток.
Домены фибронектина. Фактор XII содержит два участка, гомологичных фрагментам фибронектина типа I и II. Предполагается, что фрагмент типа II фибронектина принимает участие во взаимодействии с коллагеном.
Фактор XIII представлен двумя формами: плазменной и тромбоцитарной. Плазменная форма - тетрамер, состоящий из двух пар цепей (α и β), тромбоцитарная содержит только α-цепи. Активация плазменной формы фактора XIII протекает в два этапа. Вначале под действием тромбина или фактора Ха происходит расщепление α-цепей с освобождением пептида активации, затем тетрамер распадается с образованием активного фермента, состоящего из двух α'-цепей.
Фактор ХШа катализирует образование ε-(γ-глутамил)-лизиновых ковалентных связей между полипептидами в комплексах белков (рис. 2.2). Под действием фактора ХШа происходят сшивание смежных молекул фибрин-мономеров в полимере, а также сшивание с фибрином α2-антиплазмина, коллагена и фибронектина.
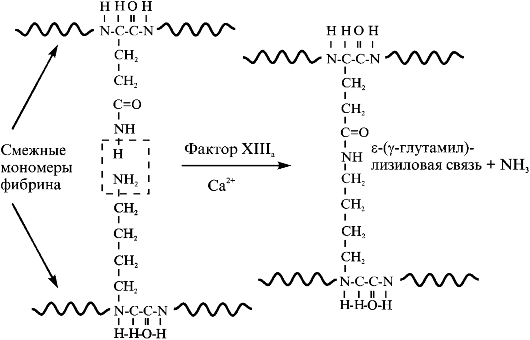 Рис. 2.2. Образование ковалентных связей между боковыми цепями остатков глутамина и лизина смежных молекул фибрин-мономеров в полимере
Рис. 2.2. Образование ковалентных связей между боковыми цепями остатков глутамина и лизина смежных молекул фибрин-мономеров в полимере
БЕЛКИ-РЕГУЛЯТОРЫ АКТИВНОСТИ ПРОТЕАЗ СВЕРТЫВАНИЯ
КРОВИ
Факторы V и VIII - гомологичные белки, выполняющие сходную роль в процессе свертывания крови. Они обеспечивают связывание витамин К-зависимых факторов IXa и Xa и их субстратов - фактора Х и протромбина с фосфолипидами. Наличие множественных связей в этих комплексах обеспечивает эффективное взаимодействие ферментов с субстратами и увеличение эффективности реакций активации в сотни тысяч раз.
Факторы V и VIII синтезируются в виде полипептидов, состоящих соответственно из 2196 и 2332 аминокислотных остатков. Оба белка содержат по три А-домена, гомологичных церулоплазмину,
по два гомологичных С-домена и один большой центральный В-домен, который отщепляется при активации факторов. Фактор VIII легко подвергается протеолизу в С-концевой области В-домена, а в плазме обнаруживается в виде набора гетеродимеров, взаимодействующих при участии Са2+. Активируются факторы V и VIII под действием тромбина или фактора Ха. Тромбин расщепляет фактор V по остаткам Arg в положениях 709, 1018 и 1545, а фактор VIII по остаткам Arg 372, 740 и 1689. В активированной форме фактор V - гетеродимер (А1-А2/А3-С1-С2), а фактор VIII - гетеротример (А1/А2/А3-С1-С2), субъединицы которых взаимодействуют при участии ионов Са2+. Участки связывания активированных факторов V и VIII с фосфолипидами находятся во фрагментах, происходящих из C-концевых частей исходных молекул.
В плазме фактор VIII циркулирует в виде комплекса с фактором фон Виллебранда (фФВ) - адгезивным белком, обеспечивающим связывание тромбоцитов с субэндотелиальными структурами и склеивание тромбоцитов при их агрегации. Другая функция фФВ - повышение стабильности фактора VIII и защита его от инактивации протеином С. фФВ синтезируется только в мегакариоцитах и клетках эндотелия как полипептид, состоящий из 2050 аминокислотных остатков, который вначале димеризуется, а затем мультимеризуется с образованием комплексов, достигающих молекулярной массы 20000 кД. Димеры фФВ постоянно секретируются в плазму. Мультимеры находятся в α-гранулах тромбоцитов и тельцах Вайбеля-Паладе эндотелия и секретируются из них при активации тромбоцитов или повреждении эндотелия. Мультимерам фФВ принадлежит ведущая роль в адгезии тромбоцитов и образовании тромбоцитарных агрегатов в условиях высокого напряжения сдвига, которое характерно для мелких сосудов и стенозированных участков артерий.
Тканевый фактор (ТФ) - трансмембранный гликопротеин, выполняющий функции рецептора фактора VII и модулятора его активности. Кофакторная активность тканевого фактора определяется как апобелком, так и фосфолипидами мембраны клеток. Связывание одноцепочечной формы фактора VII с ТФ изменяет структуру фактора VII таким образом, что становится возможным его расщепление на двухцепочечную форму, которая в комплексе с ТФ активирует факторы свертывания крови X и IX, что инициирует каскад реакций гемокоагуляции. ТФ - конститутивный компонент мембран ряда типов клеток, в норме не контактирующих с плазмой крови. Высокое содержание ТФ характерно для коры головного мозга, миокарда, клеток эпидермиса
и эпителия, выстилающего слизистые оболочки органов. Клетки эндотелия и моноциты могут экспрессировать ТФ под воздействием медиаторов воспаления, эндотоксина, окисленных липопротеинов низкой плотности и иммунных комплексов, что может быть одной из причин активации внутрисосудистого свертывания крови при ряде заболеваний.
ТФ состоит из 263 аминокислотных остатков; это отдаленный представитель суперсемейства белков, включающего рецепторы гормона роста, ИЛ-1-7 и ИФН (α и β). Исследования последних лет показали, что функция ТФ не ограничивается только активацией свертывания крови. Связывание фактора VIIa с ТФ индуцирует Са2+-сигналы в клетках, стимулирует миграцию ГМК, рост и метастазирование опухолей. В эмбриогенезе экспрессия ТФ начинается раньше, чем экспрессия фактора VII. Селективное повреждение гена ТФ приводит к аномалиям в развитии кровеносных сосудов и гибели эмбрионов мышей.
ФИБРИНОГЕН
Фибриноген состоит из трех пар неидентичных полипептидных цепей, обозначаемых как Аα, Вβ и γ; его молекула симметрична, вытянута, слегка изогнута и имеет размер 7 × 48 нм. Основные черты пространственной организации молекулы фибриногена как структуры, состоящей из трех модулей, были определены Холлом и Слейтером в 1959 г. с помощью электронной микроскопии. N-концевые части всех трех полипептидов образуют центральную область взаимодействия двух половин молекулы фибриногена (Е-домен), которые ковалентно связаны между собой тремя дисульфидными мостиками. Далее следует область, в которой все три субъединицы закручены в суперспираль длиной ~16 нм. Примерно посередине суперспирали имеется короткая область нарушения регулярности структуры. Она служит одним из участков специфического расщепления плазмином. За суперспиралью каждая из полипептидных цепей фибриногена образует свою структуру. С-концевые фрагменты β- и γ-цепей образуют на концах молекулы фибриногена глобулы, которые латерально смещены от оси суперспирали (D-домен). После спирального участка α-цепи загибаются, и их С-концевые участки взаимодействуют друг с другом вблизи центра молекулы (рис. 2.3).
В агрегации тромбоцитов фибриноген выполняет роль мостиков, связывающих между собой активированные клетки. При активации на поверхности тромбоцитов открываются специфические рецепторы, состоящие из гликопротеинов IIь-IIIа, взаимодействующих
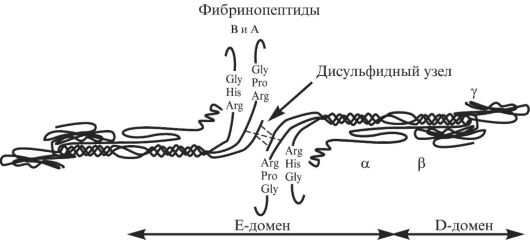 Рис. 2.3. Структура фибриногена
Рис. 2.3. Структура фибриногена
с аминокислотными последовательностями RGD и HHLGGAKQAGDV, имеющиеся в С-концевых областях α-цепи (остатки 572-574) и γ-цепи (остатки 400-411) фибриногена. Данные о значении остатков 572-574 в связывании с тромбоцитами противоречивы. По-видимому, ведущая роль во взаимодействии с тромбоцитами принадлежит γ-цепям фибриногена.
Полимеризация фибриногена начинается после того, как в результате отщепления тромбином N-концевого 16-членного фибринопептида А в α-цепи открывается участок Gly-Pro-Arg, взаимодействующий с комплементарным участком в С-концевой части γ-цепи. Этого достаточно для того, чтобы инициировать процесс самосборки протофибрилл, в которых периферический D-домен одной молекулы взаимодействует с центральным E-доменом другой молекулы (рис. 2.4). Отщепление от N-конца Ββ-цепи 14-членного фибринопептида B и экспозиция дополнительного участка взаимодействия, содержащего последовательность Gly-His-Arg, значительно ускоряют процесс сборки и латеральную ассоциацию протофибрилл с образованием больших узлов фибриновых волокон. Ветвление волокон с образованием трехмерной фибриновой сетки происходит с участием С-концевых доменов α-цепей и служит необходимым этапом формирования прочного геля. Механическая стабильность фибрина и его устойчивость к лизису повышаются, когда под действием фактора ХШа образуются ковалентные связи между смежными мономерами фибрина в полимере. Вначале между собой попарно сшиваются γ-цепи, а затем сшиваются α-цепи фибриногена с образованием длинных полимеров.
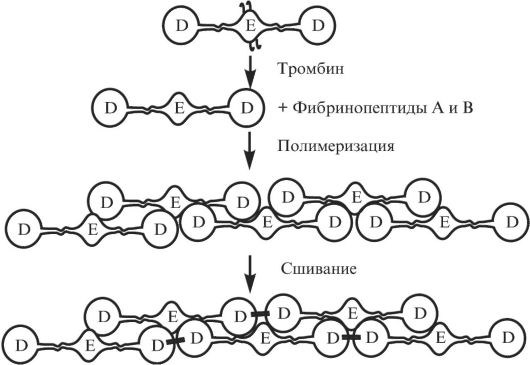 Рис. 2.4. Схема взаимодействия молекул фибрин-мономеров в процессе полимеризации и сшивания
Рис. 2.4. Схема взаимодействия молекул фибрин-мономеров в процессе полимеризации и сшивания
Последовательность каждой из цепей фибриногена кодируется соответствующим геном, однако их значительная гомология свидетельствует о том, что возникли они в результате дупликации одного гена-предшественника. Экспрессия трех генов фибриногена координируется, а сборка белка начинается с Ββ-цепи. Аα-и Ββ-цепи фибриногена состоят из 610 и 461 аминокислотных остатков соответственно. γ-Цепь фибриногена имеет два варианта. В плазме кроме основной формы, состоящей из 411 остатков, присутствует минорная (~10%) форма, в которой в результате альтернативного сплайсинга С-концевой тетрапептид заменен 20-членным пептидом. Фибриноген, содержащий удлиненную γ'-цепь, менее эффективно взаимодействует с тромбоцитами. Обнаружены полиморфные участки в Аα-цепи - Thr/Ala312 и в Ββ-цепи - Arg/Lys448. У мужчин, гомозиготных по Arg448, уровень фибриногена в крови ниже, чем у гетерозиготных и гомозиготных по Lys448. Обнаружен полиморфизм и в промоторной области Ββ-гена, контролируемой ИЛ-6. Это свидетельствует о том, что уровень фибриногена частично определяется генетически. Кроме этого, различные варианты фибриногена могут различаться по их роли в патогенезе атеротромбоза.
Фибриноген подвергается множественной посттрансляционной модификации, заключающейся в гликозилировании, фосфорилировании, сульфатировании Tyr418,422 минорных γ'-цепей и гидроксилировании Pro31 Вβ-цепей, что значительно увеличивает число возможных вариантов этого белка в организме.
В Аα-цепи фибриногена обнаружено два участка фосфорилирования - Ser3 и Ser345. Физиологическое значение фосфорилирования Ser3, который находится в фибринопептиде А, неизвестно. Фосфорилирование по этому участку не влияет на скорость отщепления фибринопептида А in vitro. Другой участок фосфорилирования находится вблизи области, участвующей во взаимодействии молекул при полимеризации и сшивании. По данным экспериментов in vitro, степень фосфорилирования этого участка влияет на структуру фибринового геля. Дефосфорилированный белок образует более толстые нити при полимеризации. Степень фосфорилирования возрастает в условиях, когда стимулируется синтез фибриногена. Это может свидетельствовать о том, что исходно фибриноген секретируется в фосфорилированной форме и постепенно дефосфорилируется фосфатазой крови. Фосфорилированный фибриноген более устойчив к протеолизу, чем дефосфорилированный.
Фибриноген связывает три иона Са2+ с высоким (Kd ~1 цМ) и около 10 с низким (Kd ~l мМ) сродством. Участки высокого сродства, гомологичные Са2+-связывающим центрам кальмодулина, находятся в С-концевой области γ-цепи фибриногена. Аминокислотные замены в этой области приводят к нарушению полимеризации фибрина. Са2+ связывается остатками сиаловых кислот с низким сродством.
Молекула фибриногена содержит четыре раздваивающиеся углеводные цепи, образующие N-гликозидную связь с Asn52 и Asn364 γ- и β-цепей соответственно. На концах углеводных цепей могут находиться остатки сиаловых кислот, играющие важную роль в полимеризации фибрина. При отщеплении сиаловых кислот скорость полимеризации фибрина увеличивается, а удаление всех углеводных компонентов устраняет влияние Са2+ на полимеризацию и приводит к увеличению латеральной агрегации фибриновых нитей. Увеличение содержания сиаловых кислот при заболеваниях, сопровождающихся повышением активности сиалилтрансферазы, - одна из причин приобретенной дисфибриногенемии.
Дисфибриногенемия характеризуется тем, что содержание белка в плазме может быть близким к норме, но некоторые из его функций
нарушены. Дефектные фибриногены обозначают по наименованию города, в котором они впервые были обнаружены. К настоящему времени описано более 80 вариантов аминокислотных замен, которые влияют на отщепление фибринопептидов, полимеризацию, взаимодействие с тромбоцитами и эритроцитами, сшивание и разрушение фибрина.
КЛИНИЧЕСКОЕ ЗНАЧЕНИЕ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ
ФИБРИНОГЕНА
Фибриноген - белок острой фазы; его уровень в плазме может значительно повышаться при многих заболеваниях. Синтез фибриногена стимулируется ИЛ-6. Концентрация фибриногена повышается с возрастом, при нарушениях метаболизма (например, при гиперлипидемии), курении и приеме пероральных контрацептивов. Для фибриногена характерны сезонные изменения концентрации с максимумом в зимние и минимумом в летние месяцы.
В проспективных исследованиях показано, что с повышением уровня фибриногена увеличивается риск заболеваний ССС. Хотя уровень фибриногена коррелирует с многими известными факторами риска заболеваний ССС, данные ряда проспективных исследований свидетельствуют о том, что фибриноген может рассматриваться как независимый фактор риска, значимость которого увеличивается при сочетании с гиперхолестеринемией и повышением систолического АД. Значимость фибриногена в патогенезе заболеваний ССС может быть обусловлена тем, что этот белок определяет вязкость плазмы крови, повышает агрегацию тромбоцитов и эритроцитов. Фибриноген накапливается в области атеросклеротических бляшек, где после превращения в фибрин стабилизирует тромбоцитарные агрегаты. Фибрин и продукты его разрушения стимулируют пролиферацию ГМК и моноцитов, обеспечивая матрикс для роста клеток и связывая тромбин, обладающий митогенной активностью.
АКТИВАЦИЯ СВЕРТЫВАНИЯ КРОВИ
Свертывание крови происходит в результате серии реакций, в которых путем ограниченного протеолиза образуются активные формы компонентов системы. Многоступенчатость и ферментативная природа реакций активации обеспечивают возможность резкого усиления начального сигнала и эффективного контроля процесса свертывания крови, который должен протекать локально в области повреждения стенки сосуда. В основе современной схемы механизма активации свертывания крови лежит каскад последовательных реакций.
Два типа взаимодействий могут приводить к инициации свертывания крови, и каскад реакций гемокоагуляции принято представлять в виде двух возможных путей активации - внутреннего и внешнего (рис. 2.5). Внешний путь получил такое название, поскольку для его инициации необходим тканевый фактор (тромбопластин), который в норме
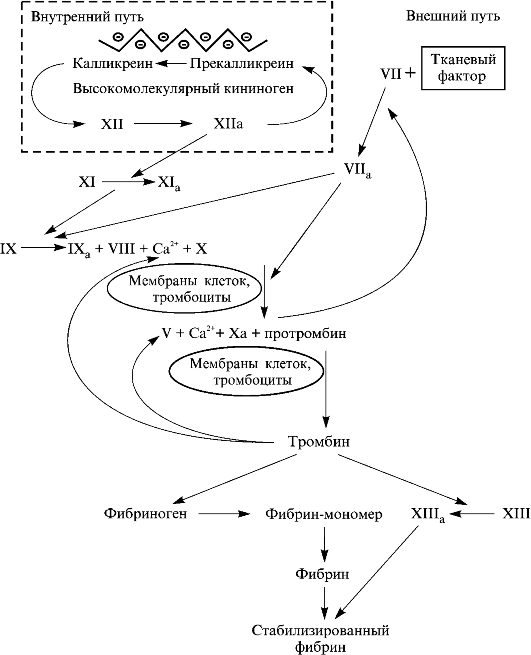 Рис. 2.5. Схема основных реакций активации свертывания крови
Рис. 2.5. Схема основных реакций активации свертывания крови
в крови и клетках, контактирующих с кровью, отсутствует и появляется при повреждении ткани. Все компоненты внутреннего пути присутствуют в крови и начинается он с контактной активации фактора XII и прекалликреина.
Внутренний путь
Связывание фактора XII с компонентами субэндотелиального слоя, активированными тромбоцитами, мицеллами фосфолипидов или бактериальными липополисахаридами, изменяет его конформацию таким образом, что он становится высокочувствительным к протеолитической активации калликреином; в комплексе с высокомолекулярным кининогеном повышает активацию прекалликреина, который затем может обеспечить дополнительную активацию фактора XII.
Начальный этап активации фактора XII - расщепление по остатку Arg353 с образованием фермента, состоящего из двух полипептидных цепей, соединенных дисульфидным мостиком. Протеазной активностью обладает легкая цепь, а тяжелая цепь содержит домены, обеспечивающие взаимодействие с другими компонентами. Калликреин плазмы может далее расщеплять еще две связи по остаткам Arg334 и Arg343, что приводит к образованию Р-формы фактора ХIIа, которая обладает ферментативной активностью, но в результате потери основной массы тяжелой цепи не способна взаимодействовать с поверхностью и активировать фактор XI.
Детали контактной активации свертывания крови и ее физиологическое значение не выяснены. Наследственный дефицит фактора XII, прекалликреина или высокомолекулярного кининогена не проявляется повышенной кровоточивостью у больных.
На следующей стадии фактор ХПа активирует фактор XI, структура которого имеет много общего со структурой прекалликреина. Высокомолекулярный кининоген служит кофактором активации фактора XI, протекающей на поверхности. Расщепление связи Arg369~He370 в каждой из субъединиц приводит к образованию фермента, состоящего из двух легких и двух тяжелых цепей, соединенных дисульфидными мостиками. Тяжелые цепи содержат участки, обеспечивающие взаимодействие с высокомолекулярным кининогеном, и необходимы для Са2+-зависимой активации фактора IX.
Активация фактора IX происходит в результате последовательного расщепления двух пептидных связей Arg145-Ala146 и Arg180-Val181 c высвобождением пептида активации и может протекать под
действием как фактора XIa, так и комплекса фактора VII с тканевым фактором в присутствии Са2+. Исследование кинетики реакции активации фактора IX под действием фактора XIa или комплекса тканевый фактор-фактор VIIa показало, что оба пути активации примерно одинаково эффективны. Нормальный уровень пептида активации фактора IX у больных с дефицитом фактора XI и сниженный уровень при дефиците фактора VII свидетельствуют в пользу того, что по крайней мере стационарная активация фактора IX протекает под действием фактора VIIa. О физиологической значимости активации фактора IX фактором VIIa свидетельствует также и то, что симптомы кровоточивости у больных с дефицитом фактора XI менее выражены, чем у больных с дефицитом факторов IX или VIII.
Активация фактора Х под действием фактора IXa протекает на поверхности фосфолипидов при участии ионов Са2+ и фактора VIIIa, выполняющего роль матрицы, обеспечивающей связывание и оптимальное взаимодействие факторов IXa и X, что увеличивает скорость реакции в сотни тысяч раз. Фактор Х состоит из двух цепочек, образующихся в результате расщепления связей Arg139-Arg140 и/или Arg142-Ser143 и соединенных дисульфидным мостиком. Активация фактора Х происходит после расщепления связи Arg194-Ile195 в N-концевой части тяжелой цепи с освобождением пептида активации. Другой физиологически важный активатор фактора Х - комплекс тканевый фактор-фактор VIIa.
Внешний путь
При контакте крови с клетками, экспрессирующими тканевый фактор, фактор VII плазмы связывается с высоким сродством 3 нМ) с тканевым фактором. Образование комплекса резко увеличивает чувствительность фактора VII к протеолитической активации, заключающейся в расщеплении связи Arg152-Ile153. Активация может происходить под действием следов активных форм факторов X и IX, а также в результате аутоактивации. В плазме около 1% фактора VII постоянно находится в двухцепочечной форме, которая в отсутствие тканевого фактора не ингибируется антитромбином III и не может активировать свои естественные субстраты - факторы X и IX.
Фактор VII может активироваться и под действием фактора ХПа. Данная реакция вряд ли играет существенную роль в обеспечении гемостаза в норме, но она может лежать в основе холодовой активации фактора VII при хранении плазмы при 4 °С. Эту возможность необходимо учитывать при подготовке образцов для исследования функциональной активности фактора VII.
ОБРАЗОВАНИЕ ТРОМБИНА
Фактор Х, активированный на поверхности фосфолипидов комплексом фактор IXa-VIII., или комплексом фактор VIIa-тканевый фактор, формирует комплекс с фактором V и протромбином. Тромбин образуется в результате последовательного расщепления двух связей в молекуле протромбина. После гидролиза связи Arg320-Ile321 образуется мейзотромбин, в котором две половины исходной молекулы связаны дисульфидным мостиком. Мейзотромбин обладает протеолитической активностью, сходной с тромбином в активации ПрС и служит мощным вазоконстриктором, но не обладает прокоагулянтными активностями тромбина и не инактивируется антитромбином III. После гидролиза связи Arg271-Thr272 из комплекса высвобождаются N-концевой фрагмент активации протромбина 1+2, содержащий все регуляторные домены, и молекула α-тромбина, состоящая из двух цепочек, соединенных дисульфидным мостиком. Активный центр и субстратсвязывающий участок находятся в B-цепи, состоящей из 259 аминокислотных остатков. A-цепь исходно состоит из 49 аминокислотных остатков, но в тромбине человека от ее N-конца может дополнительно отщепляться 13-членный пептид без существенного изменения ферментативных свойств.
В-цепь α-тромбина человека легко подвергается аутолизу. Наиболее чувствительные связи Lys154-Gly155, Arg70-Tyr71 и Arg73- Asn74. В результате их расщепления разрушается структура участков, обеспечивающих взаимодействие тромбина с макромолекулярными субстратами. Образующиеся при этом β-, β'-и γ-тромбины обладают сходной с α-тромбином активностью в отношении низкомолекулярных субстратов, но значительно меньшей (< 1% от исходной) свертывающей активностью.
После образования тромбин диссоциирует из протромбиназного комплекса и может участвовать в регуляции многих физиологических процессов. Наиболее известные функции тромбина прокоагулянтная, антикоагулянтная, вазоактивная и митогенная. На завершающей стадии свертывания крови тромбин обеспечивает превращение фибриногена в фибрин и активирует фактор XIII, стабилизирующий фибрин. Кроме этого, тромбин оказывает прокоагулянтное действие, активируя свое образование.
ПОЛОЖИТЕЛЬНЫЕ ОБРАТНЫЕ СВЯЗИ
Положительные обратные связи (см. рис. 2.5) имеют очень важное значение в инициации свертывания крови. Их наличие обеспечивает возможность преодоления ингибиторного потенциала крови и образования тромбина в количестве, достаточном для активации тромбоцитов и образования фибрина. Тромбин может ускорять свое образование, активируя тромбоциты, факторы V и VIII, а также, возможно, факторы VII и XI.
Тромбин - наиболее мощный из известных активаторов тромбоцитов, этот путь активации тромбоцитов служит ведущим механизмом образования артериальных тромбов. Активация осуществляется через специфический рецептор, принадлежащий к семейству рецепторов, содержащих семь трансмембранных доменов, и заключается в отщеплении тромбином N-концевого пептида от внеклеточного участка рецептора. Открывающийся при этом новый N-концевой участок служит связанным лигандом, который активирует рецептор. Согласно этому механизму тромбин в отличие от других агонистов действует каталитически и одна молекула тромбина может активировать много рецепторов.
В двух ключевых реакциях свертывания крови важную роль играют факторы Va и VIIIa, обеспечивающие образование на поверхности фосфолипидов комплексов, в которых каталитическая эффективность (kcat/Km) реакций активации фактора Х и протромбина увеличивается в сотни тысяч раз. Способность к образованию фосфолипидзависимых комплексов с соответствующими ферментами и субстратами у факторов V и VIII появляется после активации тромбином, заключающейся в расщеплении как минимум трех связей. Активация фактора VIII необходима также для его диссоциации из комплекса с фФВ. Происходит это после расщепления по Arg1689, находящемуся в области участка взаимодействия с фФВ. Кроме тромбина, эти факторы могут активироваться и под действием фактора Ха, но активация протекает с меньшей скоростью и фактор Ха не может активировать фактор VIII, находящийся в комплексе с фФВ.
Основная положительная обратная реакция факторов Ха и IXa - активация фактора VII.
ФУНКЦИЯ ФОСФОЛИПИДОВ
Наличие многоступенчатого каскада ферментативных реакций может обеспечить значительное усиление начального стимула, необходимое для быстрого тромбирования поврежденных участков сосудов, по которым под давлением и с высокой скоростью циркулирует кровь. В то же время свертывание крови должно быть ограничено участком повреждения стенки сосуда. Один из механизмов ограничения - локализация активации свертывания на поверхности поврежденных или стимулированных клеток. Причем необходимо участие клеток, экспрессирующих тканевый фактор, и активированных тромбоцитов, на поверхности которых могут протекать зависимые от прокоагулянтных фосфолипидов реакции активации свертывания крови.
Для мембран клеток характерно асимметричное распределение фосфолипидов. Наружный слой мембраны формируется преимущественно из холиновых фосфолипидов, а обладающие про-коагулянтными свойствами аминофосфолипиды фосфатидилсерин и фосфатидилэтаноламин находятся на цитоплазматической стороне мембраны. Асимметричное распределение фосфолипидов поддерживается АТФ-зависимыми транслоказой, перемещающей аминофосфолипиды с наружной стороны мембраны на внутреннюю, и флоппазой, осуществляющей медленный транспорт аминофосфолипидов и холиновых фосфолипидов с внутреннего на наружный слой мембраны. Повышение внутриклеточной концентрации Са2+ ингибирует транслоказу, активирует скремблазу, осуществляющую перераспределение фосфолипидов, и калпаин, участвующий в формировании и отшнуровывании микровезикул, содержащих фосфатидилсерин.
2.2. СИСТЕМА ПРОТИВОСВЕРТЫВАНИЯ
С фосфолипидами связано антикоагулянтное действие аннексина V. Этот белок впервые был выделен из сосудов пуповины, а затем - и из плаценты и назван соответственно сосудистым и плацентарным антикоагулянтом. Содержание аннексина V в плазме очень низкое (~5 нг/мл), но этот белок может секретироваться из эндотелиальных клеток и оказывать локальное антикоагулянтное действие, заключающееся в конкурентном ингибировании связывания фактора X (Xa) с фосфолипидами.
ИНГИБИТОРЫ СЕРИНОВЫХ ПРОТЕАЗ
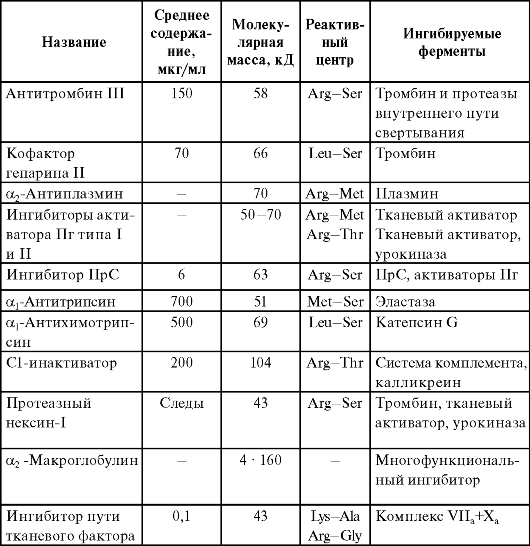
Большинство компонентов системы гемостаза в активированной форме - сериновые протеазы. В регуляции их активности играют важную роль белки - ингибиторы протеаз (табл. 2.2). Все они, за исключением ингибитора пути тканевого фактора (ИПТФ) и α2-мaкроглобулинa, - представители одного семейства, получившего название «серпины», которое образовалось в ходе эволюции
Таблица 2.2. Белковые ингибиторы протеаз крови
 из
одного белка-предшественника параллельно с возникновением
специализированных протеаз. Название - аббревиатура английского термина
(от англ. serpins, serine protease inhibitors - ингибиторы
сериновых протеаз). Кроме ингибиторов, в это семейство входят такие
белки, как овальбумин, ангиотен-зиноген и тироксинсвязывающий глобулин.
Серпины состоят из ~400 аминокислотных остатков, а различия молекулярной
массы в основном обусловлены различной степенью гликозилирования.
Гомология наиболее выражена в С-концевой области, содержащей реактивный
центр (участок, взаимодействующий с активным центром фермента), и
наименее выражена в N-концевой области, легко подвергающейся протеолизу.
Расщепление связей в N-концевом участке серпинов играет важную
физиологическую роль. В случае ангиотензиногена это приводит к
высвобождению ангиотензина, а в случае антитромбина III - к изменению
его гепаринсвязывающих свойств.
из
одного белка-предшественника параллельно с возникновением
специализированных протеаз. Название - аббревиатура английского термина
(от англ. serpins, serine protease inhibitors - ингибиторы
сериновых протеаз). Кроме ингибиторов, в это семейство входят такие
белки, как овальбумин, ангиотен-зиноген и тироксинсвязывающий глобулин.
Серпины состоят из ~400 аминокислотных остатков, а различия молекулярной
массы в основном обусловлены различной степенью гликозилирования.
Гомология наиболее выражена в С-концевой области, содержащей реактивный
центр (участок, взаимодействующий с активным центром фермента), и
наименее выражена в N-концевой области, легко подвергающейся протеолизу.
Расщепление связей в N-концевом участке серпинов играет важную
физиологическую роль. В случае ангиотензиногена это приводит к
высвобождению ангиотензина, а в случае антитромбина III - к изменению
его гепаринсвязывающих свойств.
Механизм действия серпинов состоит в образовании стехиометрического комплекса с протеазой, в котором после расщепления по реактивному центру ингибитор остается ковалентно связанным с серином активного центра фермента. Комплекс протеаза-ингибитор выводится из кровотока и разрушается. Таким образом, серпины - белки, выполняющие свою функцию один раз в жизни.
Исследования, выполненные in vitro, показали, что в близких к физиологическим условиях комплекс протеаза-ингибитор может диссоциировать, но с очень низкой скоростью (t1/2 около трех дней), возрастающей при повышении рН или под действием нуклеофильных реагентов. При этом высвобождаются активный фермент и ингибитор, расщепленный в С-концевой области.
Рентгеноструктурные исследования кристаллов α1-антитрипсина показали, что пептидная связь реактивного центра находится в напряженном интермедиатном состоянии. Это обеспечивает эффективное взаимодействие с активным центром протеазы и в то же время высокую чувствительность серпинов к инактивации путем окисления остатков Met вблизи реактивного центра или протеолитического расщепления петли, содержащей реактивный центр. Локальная инактивация серпинов путем окисления и расщепления эластазой может способствовать ограничению участков воспаления, а системная инактивация может быть одним из механизмов развития ДВС-синдрома при сепсисе или змеиных укусах.
АНТИТРОМБИН III
Антитромбин III - один из наиболее подробно исследованных ингибиторов плазмы крови. Он может ингибировать все протеазы системы свертывания крови, а также плазмин, трипсин и Сls-компонент системы комплемента. Концентрация антитромбина III в плазме составляет 0,15-0,18 мг/мл, t1/2 - около трех дней. У младенцев после рождения концентрация антитромбина III примерно в два раза ниже и постепенно повышается в течение первых 6 мес жизни. Основное место синтеза антитромбина III - клетки паренхимы печени, но некоторое его количество синтезируется также и эндотелием. При инфузии антитромбина III восстановление его уровня в плазме происходит медленнее, чем можно было бы ожидать, исходя из объема крови и количества введенного белка. Это свидетельствует о том, что некоторое количество антитромбина III может депонироваться в тканях и выполнять роль буфера, поддерживающего стабильный уровень антитромбина III в плазме.
Антитромбин III состоит из 432 аминокислотных остатков, имеет три дисульфидных мостика и четыре участка гликозилирования. Реактивный центр образует пара Arg393-Ser394, но для специфичности действия важное значение имеет также и то, что в положении 392 находится глицин. Взаимодействие антитромбина III с тромбином протекает в две стадии. На первой образуется обратимый комплекс, в котором затем происходят расщепление ингибитора и образование ковалентной связи между ферментом и ингибитором.
Скорость ингибирования протеаз антитромбином III значительно повышается в присутствии сульфатированных олигосахаридов (один из представителей - гепарин). Минимальная структура, обеспечивающая активацию антитромбина III, - пентасахарид (рис. 2.6), содержащий четыре специфически расположенные сульфогруппы. Активирующее действие такого пентасахарида обусловлено влиянием на положение петли, содержащей реактивный центр. В антитромбине III, в отличие
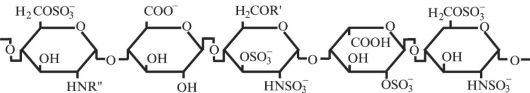 Рис. 2.6. Структура фрагмента гепарина, обеспечивающего активацию антитромбина III
Рис. 2.6. Структура фрагмента гепарина, обеспечивающего активацию антитромбина III
от неактивируемых ингибиторов, она частично погружена в основную структуру белка и экспонируется наружу после взаимодействия с кофактором. Такая мобильность реактивного центра обеспечивает возможность регуляции активности ингибитора и предохраняет чувствительный участок от неспецифической инактивации.
В физиологических условиях гепарин связывается с антитромбином III с высоким сродством и стехиометрически. Ведущая роль во взаимодействии с гепарином принадлежит остаткам Arg47, Lys125, Arg129, Arg132, Lys133. Они образуют сферу положительных зарядов антитромбина III, изменения пространственной локализации которых в результате модификации близлежащих аминокислот (Trp49, Lys53) или расщепления дисульфидной связи в С-концевой части молекулы приводят к снижению способности связывать гепарин.
Специфическое связывание гепарина зависит от третичной структуры антитромбина III и в то же время приводит к ее изменению. Одно из следствий изменения структуры антитромбина III - значительное увеличение скорости ингибирования тромбина. Кажущаяся константа скорости реакции возрастает от ~1 до ~107 М-1с-1. Другое следствие изменения структуры - увеличение сродства к гепарину. После начального связывания гепарина с относительно низким сродством (Kd ~4?10-5 М) происходят изменения структуры антитромбина III и сродство к гепарину повышается (Kd ~10-8 M). После образования комплекса с протеазой и расщепления по реактивному центру происходят новые изменения конформации антитромбина III, приводящие уже к снижению сродства к гепарину. К следствиям снижения сродства к гепарину после образования комплекса протеаза-антитромбин III относят то, что в ингибировании протеаз гепарин действует как бы каталитически: после образования комплекса протеаза-антитром- бин III он может высвобождаться из комплекса и участвовать в новом акте. Снижение сродства к гепарину после образования комплекса протеаза-антитромбин III может способствовать диссоциации комплекса с поверхности эндотелия и удалению его из крови.
Активация антитромбина III, обеспечиваемая пентасахаридным фрагментом, содержащим в определенных положениях сульфогруппы, - очень важный, но не единственный механизм, определяющий антикоагулянтное действие гепарина. Высокая скорость ингибирования тромбина, примерно на порядок превышающая скорость ингибирования фактора Ха, обусловлена тем, что в этой реакции гепарин не только активирует антитромбин III, но и связывается с тромбином,
выполняя роль матрицы, обеспечивающей эффективное взаимодействие протеазы с ингибитором (см. рис. 2.6; рис. 2.7). Кроме тромбина, это взаимодействие реализуется при ингибировании факторов IXa и XIa, но не играет существенной роли при ингибировании факторов X.,, ХПа и калликреина. Роль матрицы могут выполнять только те молекулы гепарина, которые содержат не менее 18 сахаридных остатков.
В кровеносных сосудах функцию кофактора антитромбина III могут выполнять гликозаминогликаны и гликопротеины люминальной поверхности эндотелия, которые имеют гепариноподобные структуры. На взаимодействие антитромбина III и тромбина с гликопротеинами эндотелия и стенки сосудов влияет ряд белков плазмы. Богатый
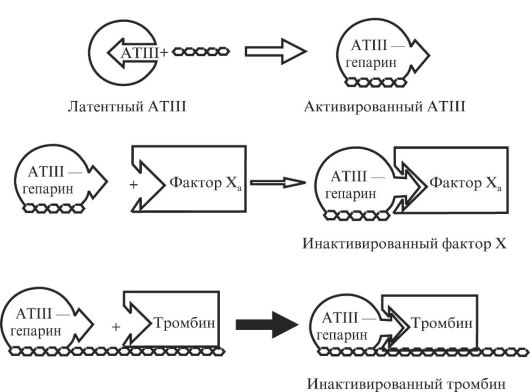 Рис. 2.7. Роль
гепарина в инактивации факторов свертывания. При связывании АТШ с
пентасахаридом, содержащим четыре сульфогруппы, происходят изменения
структуры ингибитора, вследствие которых экспонируется его реактивный
центр. Этого достаточно для ингибирования фактора Xa. Βысокая
скорость ингибирования тромбина комплексом АТШ - гепарин обусловлена
тем, что в этой реакции гепарин выполняет роль не только активатора АТШ,
но и матрицы, обеспечивающей эффективное взаимодействие ингибитора с
ферментом. Роль матрицы могут выполнять структуры, содержащие не менее
18 углеводных остатков. АТШ - антитромбин III
Рис. 2.7. Роль
гепарина в инактивации факторов свертывания. При связывании АТШ с
пентасахаридом, содержащим четыре сульфогруппы, происходят изменения
структуры ингибитора, вследствие которых экспонируется его реактивный
центр. Этого достаточно для ингибирования фактора Xa. Βысокая
скорость ингибирования тромбина комплексом АТШ - гепарин обусловлена
тем, что в этой реакции гепарин выполняет роль не только активатора АТШ,
но и матрицы, обеспечивающей эффективное взаимодействие ингибитора с
ферментом. Роль матрицы могут выполнять структуры, содержащие не менее
18 углеводных остатков. АТШ - антитромбин III
гистидином гликопротеин и секретируемый из α-гранул тромбоцитов фактор IV, а также витронектин могут связываться с гепариноподобными структурами и препятствовать инактивации тромбина и фактора Ха. Активированные нейтрофилы могут в значительной степени подавлять антикоагулянтные свойства поверхностных гликопротеинов эндотелиальных клеток, что может быть одним из механизмов развития тромбозов при сепсисе и воспалениях.
Таким образом, когда тромбин вымывается потоком крови из места образования и контактирует с неповрежденным эндотелием, он может быть нейтрализован антитромбином III, связанным с гликопротеинами стенки сосудов. Очевидно, что этот механизм инактивации тромбина более эффективно действует в микроциркуляции, где соотношение между объемом протекающей крови и поверхностью сосуда значительно выше, чем в магистральных сосудах.
НАСЛЕДСТВЕННЫЙ И ПРИОБРЕТЕННЫЙ ДЕФИЦИТ
АНТИТРОМБИНА III
Частота наследственного дефицита антитромбина III в популяции, передаваемого как аутосомно-доминантный признак, составляет один случай на 2000-5000 человек. Наиболее частые клинические проявления дефицита антитромбина III - тромбозы глубоких вен и тромбоэмболии легочной артерии. Вероятность развития тромботических осложнений у больных с наследственным дефицитом антитромбина III увеличивается с возрастом и составляет менее 10% в возрасте до 15 лет и более 85% - старше 50 лет. Частота выявления дефицита антитромбина III у больных с венозными тромбозами составляет около 3%.
В плазме больных с наследственным дефицитом антитромбина III даже при отсутствии клинических признаков тромбозов значительно повышено по сравнению с нормой содержание фрагмента F1+2, отщепляющегося при активации от протромбина. Повышенный уровень фоновой активации свертывания крови при дефиците антитромбина III значительно увеличивает вероятность тромбообразования при наличии таких факторов риска, как травма, операция, инфекция, беременность и роды, прием пероральных контрацептивов.
В зависимости от того, какая из функций антитромбина III нарушена, выделяют несколько типов наследственного дефицита антитромбина III. Наиболее часто отмечают тип I дефицита, при котором примерно в одинаковой степени снижены функциональная активность антитромбина III и его концентрация. Этот тип дефицита
подразделяют на две подгруппы. При типе Ia молекулярные свойства антитромбина III и t1/2 в циркуляции не изменены, снижена только его концентрация. При типе Ib в дополнение к сниженной концентрации часть синтезирующихся молекул антитромбина III видоизменена.
Тип II характеризуется тем, что у больных концентрация антитромбина III находится в пределах нормы, но его свойства изменены. В зависимости от того, какие свойства нарушены, выделяют три подгруппы. В подгруппу IIа включены аномалии антитромбина III, когда наблюдают изменение нескольких функций. Например, в антитромбине III «Будапешт» в результате точечной замены Pro429 Leu не образуется дисульфидная связь между Cys430 и Cys247, что отражается в значительном изменении третичной структуры ингибитора и его способности взаимодействовать с протеазами и гепарином. В две другие подгруппы включены аномалии антитромбина III, при которых нарушено взаимодействие с протеазой (IIb) и гепарином (IIc). В популяции дефицит типа IIс в гетерозиготной форме встречается чаще, чем другие типы дефицитов, но тромбозы развиваются в редких случаях гомозиготной формы.
Приобретенный дефицит антитромбина III может быть обусловлен сниженным синтезом, повышенным потреблением или потерей белка при нефротическом синдроме и заболеваниях ЖКТ. Во всех этих случаях отмечают параллельное снижение концентрации и активности антитромбина III. Основное место синтеза антитромбина III - клетки паренхимы печени. Поэтому заболевания, сопровождающиеся снижением белково-синтетической функции печени или транскапиллярного тока, приводят к снижению уровня антитромбина III. Снижение концентрации антитромбина III часто наблюдают в ходе развития ДВС-синдрома при разнообразных заболеваниях. На начальных этапах, когда снижение уровня антитромбина III еще не достигло критического уровня, терапия гепарином может быть достаточно эффективной. Однако антикоагулянтное действие гепарина определяется уровнем антитромбина III. При снижении уровня антитромбина III для достижения сопоставимого действия на свертывание крови требуется введение большего количества гепарина, чем при нормальном уровне антитромбина III. В то же время интенсивная гепаринизация может приводить к дальнейшему снижению уровня антитромбина III, обусловленному увеличением его потребления.
Снижение синтеза антитромбина III наблюдается при лечении L-аспарагиназой, эстрогенами и синтетическими препаратами, обладающими эстрогенным свойством.
КОФАКТОР II ГЕПАРИНА
Селективный ингибитор тромбина, получивший название кофактора II гепарина (ГК-II), принадлежит к семейству серпинов и характеризуется значительной структурной гомологией с антитромбином III, которая наиболее выражена вблизи реактивного центра и в положении аминокислот, принимающих участие во взаимодействии с гепарином. Сродство к гепарину у ГК-II на порядок ниже, чем у антитромбина III, но в отличие от последнего ГК-II активируется дерматансульфатом и многими другими синтетическими и природными полианионами. Минимальная структура, обеспечивающая активацию ГК-II, - сульфатированный гексасахарид.
Важная характеристика ГК-II - его селективность. Из протеаз крови с высокой скоростью он ингибирует только тромбин и значительно медленнее - лейкоцитарный катепсин G. При оптимальной концентрации дерматансульфата кажущиеся константы скорости второго порядка составляют 6,4 ? 108 М-1с-1 для тромбина и 8,4 ? 104 М-1с-1 для катепсина G. При этом реактивный центр ГК-II представлен парой аминокислот Leu444-Ser445, связь между которыми тромбин в отличие от катепсина G в других белках не гидролизует. Замена Leu444 Arg в рекомбинантном ГК-II приводит к увеличению на два порядка скорости ингибирования тромбина.
В N-концевой части молекулы ГК-II имеется фрагмент (остатки 54-75), содержащий два сульфатированных тирозина и характерную для С-концевой части гирудина последовательность кислых аминокислот, взаимодействующую с анионсвязывающим участком тромбина. Высокая специфичность ГК-II в ингибировании тромбина может быть обусловлена тем, что во взаимодействии с тромбином принимает участие не только реактивный центр ингибитора, но и специфическая последовательность кислых аминокислот N-концевой части молекулы.
Существенное различие между антитромбином III и ГК-II по тому, какой из гликозаминогликанов требуется им для проявления максимальной активности, свидетельствует о том, что эти два ингибитора могут взаимодополнять друг друга, контролируя активность тромбина в разных участках сосуда. Антитромбин III обеспечивает нейтрализацию тромбина при участии гепарино-подобных структур на поверхности эндотелия, а ГК-II - во внутренней оболочке артерий и средней оболочке сосудов, где на долю дерматансульфата приходится до 70% от всех гликозаминогликанов.
ПРОТЕАЗНЫЙ НЕКСИН-I
Протеазный нексин-I (ПН-I) - белок с молекулярной массой 43 000 Д, принадлежащий к семейству серпинов, ингибирует протеазы системы фибринолиза, ПрС и тромбин. Гепарин повышает скорость ингибирования протеаз, и в его присутствии константа скорости ингибирования тромбина ПН-I достигает величины 4,8 ? 108 М-1с-1, что на порядок превышает значение, определенное для антитромбина III. В плазме концентрация ПН-I много ниже (~1 нг/мл), чем антитромбина III или ГК-II, однако ПН-I обнаружен на поверхности тромбоцитов, где его локальная концентрация может достигать величины, достаточной для регуляции активности тромбина и ПрС.
Другим местом действия ПН-I может быть межклеточное пространство. Связывание ПН-I с белками внеклеточного матрикса повышает его специфичность как ингибитора тромбина. В наиболее высокой концентрации ПН-I присутствует в головном мозге в структурах, окружающих кровеносные сосуды и астроциты. Возможно, ПН-I - основной регулятор митогенной активности тромбина в межклеточном пространстве.
ИНГИБИТОР ТКАНЕВОГО ФАКТОРА
В плазме крови присутствует белок, при участии которого фактор Ха может ингибировать активацию фактора Х комплексом фактор VIIa-тканевый фактор. Этот ингибитор получил несколько наименований: антиконвертин, ингибитор внешнего пути и ЛП-связанный ингибитор. XIII Конгресс международного общества по тромбозам и гемостазу рекомендовал использование термина ИПТФ.
Основное место синтеза и локализации ИПТФ - клетки эндотелия капилляров и мегакариоциты. Некоторое количество ИПТФ может синтезироваться также и моноцитами. Гепатоциты не содержат иРНК, кодирующую ИПТФ, и уровень этого ингибитора не снижается при поражениях печени.
В плазме основная часть ИПТФ находится в виде комплекса с ЛП: ~50% его связано с ЛПНП и ТГ, 40-45% - c ЛПВП, где он ковалентно связан дисульфидным мостиком с апобелком А-II, и только 5-10% приходится на свободную форму, обладающую наибольшей антикоагулянтной активностью. Некоторое количество ИПТФ находится в тромбоцитах и секретируется при их активации, что может повышать локальную концентрацию этого ингибитора в области тромбообразования.
У больных с нарушениями липидного обмена концентрация ИПТФ в плазме повышена. Наиболее высокое содержание ИПТФ характерно для гиперлипидемии типа Па и Пь, что, по-видимому, обусловлено перераспределением ингибитора между эндотелием и ЛП. Концентрация ИПТФ в плазме значительно повышается при инъекции гепарина, но длительная гепаринотерапия может приводить к заметному снижению уровня свободного ИПТФ в плазме и его антикоагулянтного действия.
ИПТФ синтезируется как полипептид, состоящий из 276 аминокислотных остатков. Он может подвергаться посттрансляционному N-гликозилированию по остаткам аспарагина в положениях 117, 167 и 228, сульфатированию олигосахаридной цепочки и фосфорилированию по Ser2. Физиологическое значение этих модификаций не установлено. В N-концевой области белка имеется последовательность кислых аминокислот, за которой следуют три гомологичных участка, образующих домены ингибитора Кунитца, а С-концевая область представлена участком, содержащим большое количество щелочных аминокислот. Ряд связей этого участка легко подвергается протеолизу и в плазме ИПТФ присутствует в виде нескольких форм с молекулярной массой от 34 до 41 кД. Протеолиз С-концевой области ИПТФ приводит к потере его способности взаимодействовать с ЛП.
Гепарин увеличивает эффективность ингибирования протеаз. В С-концевой части ИПТФ обнаружено два участка связывания гепарина. Один из них - последовательность щелочных аминокислот, а другой - фрагмент Gly212-Phe243, входящий в структуру третьего ингибиторного домена. Наибольшим сродством обладают молекулы гепарина, имеющие молекулярную массу более 10 кД и/или высокую плотность зарядов. Специфическая пентасахаридная последовательность, обеспечивающая активацию антитромбина III, не участвует во взаимодействии с ИПТФ.
Каждый из трех ингибиторных доменов ИПТФ обладает потенциальным реактивным центром, находящимся в области жесткой третичной структуры, поддерживаемой тремя дисульфидными мостиками на протяжении ~50 аминокислотных остатков. Эффективная инактивация протеаз ингибиторами Кунитца обеспечивается тем, что их структура хорошо соответствует структуре субстрат-связывающего участка активного центра протеазы. В отличие от серпинов, ИПТФ не образует ковалентной связи с протеазой и может быть вытеснен из активного центра конкурентным ингибитором.
Три ингибиторных домена ИПТФ характеризуются значительной гомологией структуры, но только два из них непосредственно участвуют в ингибировании, и каждый обладает специфичностью в отношении ингибируемой протеазы. Первый домен связывается с активным центром фактора VIIa, а второй - фактора Xa. Таким образом, одна молекула ИПТФ может инактивировать два фактора свертывания, но при этом эффективное ингибирование фактора VIIa наблюдают лишь в присутствии фактора Ха и тканевого фактора. Для описания ингибирования активации свертывания крови под действием ИПТФ был предложен двухстадийный механизм. Вначале ИПТФ образует обратимый комплекс с фактором Ха, который затем связывается с комплексом фактор VIIa-тканевый фактор. В результате образуется четырехкомпонентный комплекс, стабильность которого обеспечивается множественными взаимодействиями: ингибиторных доменов с активными центрами, Gla-доменов с тканевым фактором, гидрофобных участков с фосфолипидами.
Исследования влияния ИПТФ и антитромбина III на активацию свертывания in vitro показали, что продолжительность лаг-фазы в образовании тромбина определяется концентрацией тканевого фактора и ИПТФ. Эти данные свидетельствуют о том, что функция ИПТФ состоит в снижении скорости образования факторов IXa и Xa до величины, при которой их активность может контролироваться антитромбином III. Антитромбин III и ИПТФ, действуя на разные стадии активации, обеспечивают наличие определенного порога, только после преодоления которого может начаться массивная генерация тромбина, необходимая для свертывания крови.
Введение рекомбинантного ИПТФ в дозах, не оказывающих выраженного влияния на изменение основных коагулологических тестов, значительно замедляет тромбирование механически поврежденных участков вен кроликов и снижает частоту реокклю-зий после тромболизиса тканевым активатором Пг артериальных и венозных тромбов. Локальное введение ИПТФ в область повреждения внутренней оболочки артерий при баллонной ангиопластике ингибирует образование тромбоцитарных агрегатов и тромбов на поврежденной поверхности, а также развитие рестенозов после операции.
2.3. СИСТЕМА ПРОТЕИНА С
ПРОТЕИН С
В 1976 г. во фракции витамин К-зависимых факторов свертывания был обнаружен белок, который был назван протеином С (ПрС). Вскоре оказалось, что этот белок идентичен описанному ранее аутопротромбину II-А и служит проферментом, который после активации тромбином специфически расщепляет факторы Va и VIIIa, что приводит к прерыванию каскада реакций активации свертывания крови.
ПрС синтезируется в печени как полипептид, состоящий из 461 аминокислотного остатка, включая 42-членный сигнальный пептид, необходимый для посттрансляционного у-карбоксилирования девяти остатков Gln. ПрС подвергается N-гликозилированию по четырем участкам, его молекулярная масса составляет 62 кД. В плазме ПрС находится в 2-цепочечной форме, образующейся в результате расщепления связи Arg157-Thr158 в молекуле предшественника и удаления двух С-концевых аминокислот легкой N-концевой цепи. Концентрация ПрС в плазме составляет ~4 мг/л, a t1/2 - 6-8 ч.
Легкая цепь ПрС состоит из Gla-домена и двух EGF-доменов, которые обеспечивают связывание Са2+ с высоким сродством и взаимодействие ПрС с фосфолипидами, комплексом тромбин-тромбомодулин, протеином S и фактором Va. Тяжелая цепь ПрС содержит протеолитический домен, который гомологичен другим протеазам свертывания и трипсину. Активный центр образуют остатки His211, Asp257 и Ser360. ПрС обладает узкой субстратной специфичностью и с высокой скоростью расщепляет только активированные формы факторов Va и VIIIa.
Активация ПрС происходит в результате расщепления связи Arg169-Leu170 в N-концевой части тяжелой цепи и сопровождается освобождением пептида активации. Впервые ПрС был активирован тромбином и протеазой из яда змеи Рассела. Однако свободный тромбин активирует ПрС с очень низкой скоростью и только в отсутствие ионов Са2+. Эндотелий сосудов в тысячи раз ускоряет активацию ПрС благодаря тромбомодулину, обеспечивающему активацию ПрС тромбином.
ТРОМБОМОДУЛИН
Тромбомодулин (ТМ) - трансмембранный гликопротеин, состоящий из 557 аминокислотных остатков. На N-конце молекулы ТМ находится домен, гомологичный лектинам, за которым следуют шесть повторяющихся EGF-доменов, а также фрагмент, обогащенный Ser/Thr, содержащий хондроитинсульфатную боковую цепь и участки О-гликозилирования, затем трансмембранный домен и короткий С-концевой цитоплазматический участок.
ТМ связывает тромбин с высоким сродством (Kd ~0,5 нМ). Связывание тромбина и изменение его субстратной специфичности обеспечиваются структурами 5-го и 6-го EGF-доменов, которые взаимодействуют с субстратсвязывающим участком активного центра тромбина, и хондроитинсульфатной цепью, которая связывается с последовательностью щелочных аминокислот тромбина. Наличие на ТМ двух участков взаимодействия с тромбином подтверждается связыванием двух молекул тромбина с одной молекулой ТМ. Во взаимодействии с ПрС и его активации важная роль принадлежит Asp349 4-го EGF-домена и области, связывающей 4-й и 5-й EGF-домены ТМ.
Связывание тромбина с ТМ увеличивает каталитическую эффективность реакции активации ПрС в ~20000 раз и ингибирует прокоагулянтные функции тромбина: активацию тромбоцитов, фактора V и свертывание фибриногена. ТМ, кроме изменения направленности каталитического действия тромбина, может выполнять и функцию удаления этого белка. Связывание с ТМ стимулирует эндоцитоз комплекса и разрушение тромбина лизосомальными ферментами, а также инактивацию тромбина антитромбином III.
ТМ экспрессируется в эндотелиальных клетках вен, артерий, капилляров и лимфатической системы большинства органов, за исключением головного мозга. Содержание иРНК, кодирующей ТМ, убывает в последовательности: сердце > поджелудочная железа > легкие > скелетные мышцы > почки > печень > плацента. Концентрация ТМ в микроциркуляции может достигать ~10 нМ. В следовых количествах ТМ обнаруживается также в тромбоцитах, плазме и моче.
Синтез ТМ может стимулироваться тромбином, цАМФ, ретиноевой кислотой, пентоксифиллином и ИЛ-4. Экспрессия ТМ может снижаться под воздействием эндотоксина, ИЛ-1, TNF, гипоксии и ряда инфекций. Поскольку большинство из перечисленных воздействий стимулирует синтез тканевого фактора, то в результате может
происходить значительное изменение соотношения между про- и антикоагулянтными активностями эндотелия.
Приемлемых для клинических исследований методов определения функциональной активности ТМ пока не предложено. В экспериментах на мышах показано, что полная инактивация гена ТМ приводит к гибели эмбрионов на стадии, предшествующей развитию ССС. Это свидетельствует о том, что функция ТМ, по-видимому, не ограничивается только контролем свертывания крови. Обнаружено, что описанный ранее поверхностный белок плода фетомодулин по структуре и свойствам идентичен ТМ. ТМ может конкурировать с клеточным рецептором за связывание с тромбином и таким образом подавлять митогенное действие тромбина и его влияние на экспрессию молекул адгезии на эндотелии. При гетерозиготной форме дефицита ТМ тромбозы у животных развивались лишь при наличии дополнительных повреждающих стимулов.
ПРОТЕИН S
Протеин S (ПрS) - витамин К-зависимый белок, увеличивающий скорость инактивации протеином С факторов Va и VIIIa в составе протромбиназного и Х-азного комплексов. Потенцирующий эффект наблюдают только в присутствии фосфолипидов, и функция может заключаться в повышении сродства ПрСа к мембранам.
синтезируется в печени, эндотелии, интерстициальных клетках семенников и мегакариоцитах. Секретируется как обладающий активностью одноцепочечный гликопротеин с молекулярной массой ~80 кД, состоящий из 635 аминокислотных остатков. N-кон- цевая область характеризуется высокой степенью гомологии с другими витамин К-зависимыми белками. Она состоит из Gla-домена и 4 EGF-доменов. Между Gla-доменом и 1-м EGF-доменом аминокислотная последовательность образует стабилизированную дисульфидным мостиком петлю, две связи которой легко расщепляются тромбином. Расщепление приводит к инактивации В отличие от других витамин К-зависимых белков С-концевая часть молекулы не содержит протеазного домена. Аминокислотная последовательность в области остатков 243-635 гомологична таковой глобулина, связывающего половые гормоны, но специфического связывания стероидов с
ПрS не обнаружено.
ПрS связывается с высоким сродством (Kd ~0,5 нМ) с С4Ь-свя- зывающим белком (С4Ь-СБ) системы комплемента. В плазме около
половины находится в виде комплекса с С4Ь-СБ, который не обладает антикоагулянтной активностью. В образовании комплекса принимают участие С-концевая область и N-концевая область β-цепи С4Ь-СБ. Участки связывания С4Ь-компонента находятся на α-цепях С4Ь-СБ, и связывание ПрS не влияет на взаимодействие и расщепление этого компонента протеазным фактором I системы комплемента. Это свидетельствует о том, что обладающий высоким сродством к фосфолипидам, может участвовать также и в локальной регуляции воспалительного процесса.
СИСТЕМА ПРОТЕИНА С В РЕГУЛЯЦИИ
Факторы IXa и Xa в составе протромбиназного и теназного комплексов на поверхности фосфолипидов частично защищены от действия антитромбина III, во-первых, из-за препятствий, создаваемых компонентами комплексов. Кроме того, антитромбин III активируется на поверхности неповрежденных эндотелиальных клеток, а активированные факторы свертывания образуются на фрагментах мембран клеток или тромбоцитах, т.е. пространственно разобщены. Когда тромбин вымывается потоком крови из места образования и контактирует с неповрежденным эндотелием, то он может либо инактивироваться антитромбином III, либо связаться с ТМ и активировать ПрС. Последний, действуя каталитически, расщепляет активированные формы факторов V и VIII, что приводит к распаду комплексов и подавлению свертывания, а свободные формы факторов IXa и Xa могут ингибироваться антитромбином III при контакте с эндотелием (рис. 2.8).
Функция системы ПрС, по-видимому, не ограничивается только остановкой процесса свертывания крови. Инфузия ПрСа оказывала более выраженное защитное действие по сравнению с другими антикоагулянтами на выживаемость обезьян при экспериментальном ДВС. В экспериментах in vitro показано, что ПрСа может снижать экспрессию молекул адгезии и синтез цитокинов макрофагами.
Инактивация протеина С
t1/2 ПрСа в кровотоке составляет около 10 мин, что обусловлено значительно меньшей по сравнению с другими протеазами скоростью его инактивации белковыми ингибиторами плазмы. По кинетическим параметрам наиболее эффективные ингибиторы ПрСа относят к семейству серпинов: ингибитор ПрС, α1-антитрипсин и протеазный нексин-1. Константы скорости инактивации ПрСа
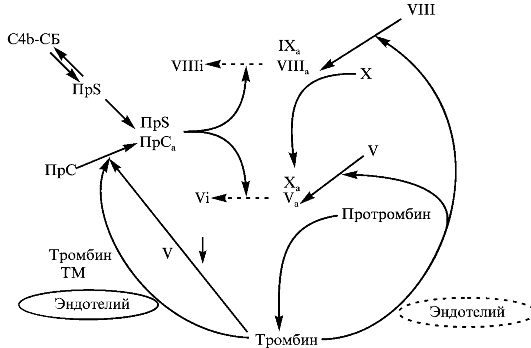 Рис. 2.8. Схема
антикоагулянтного действия ПрС. Тромбин при контакте с неповрежденным
эндотелием связывается с ТМ и активирует ПрС, расщепляющий факторы VIIIa и Va. Это приводит к остановке реакций активации свертывания крови
Рис. 2.8. Схема
антикоагулянтного действия ПрС. Тромбин при контакте с неповрежденным
эндотелием связывается с ТМ и активирует ПрС, расщепляющий факторы VIIIa и Va. Это приводит к остановке реакций активации свертывания крови
этими ингибиторами на 2-3 порядка ниже, чем тромбина. Гепарин увеличивает скорость инактивации ПрСа в 20-40 раз, что также значительно меньше, чем увеличение скорости инактивации тромбина. Наличие четко выраженного оптимума концентрации гепарина свидетельствует о том, что он выполняет роль матрицы, на которой происходит взаимодействие ПрСа с ингибитором.
Кроме перечисленных выше ингибиторов в инактивации ПрСа в плазме могут принимать участие α2-антиплазмин и α2-макроглобулин, а также ИПТФ в присутствии гепарина. Наличие конкуренции между фактором Ха и ПрСа за взаимодействие с ИПТФ свидетельствует о том, что инактивация ПрСа обеспечивается вторым ингибиторным доменом ИПТФ.
Наследственный и приобретенный дефицит
Дефицит ПрС или ПрS, как и дефицит антитромбина III, - факторы, предрасполагающие к развитию венозных тромбозов и тромбоэмболий. При гетерозиготной форме дефицита тромбозы редко проявляются в возрасте до 14 лет, но вероятность их развития значительно
увеличивается с возрастом и при наличии других осложняющих факторов, таких, как травмы, операции и иммобилизация больного, беременность и использование пероральных контрацептивов. Значительных отличий в клинической картине тромбозов при дефиците антитромбина III, ПрС или не обнаружено, за исключением несколько более высокой вероятности тромбофлебитов при дефиците ПрС/ПрS и тромбозов при беременности и приеме пероральных контрацептивов у носителей с дефицитом антитромбина III. Дефицит
может предрасполагать и к развитию артериальных тромбозов в молодом возрасте. Гомозиготный дефицит ПрС или встречается крайне редко и связан с тяжелыми тромботическими осложнениями в неонатальном периоде.
Частота гетерозиготного дефицита ПрС в популяции может составлять один случай на 300 человек. У больных с семейными тромбозами частота выявления дефицита ПрС или составляет 2-5%. Классификация врожденного дефицита ПрС включает два, а
- три типа. Чаще всего отмечают тип I дефицита, при котором наблюдают параллельное снижение функциональной активности и концентрации этих белков. Тип II дефицита характеризуется более выраженным снижением функциональной активности по сравнению с концентрацией этих белков. К типу III дефицита относят случаи, когда снижено содержание свободной формы белка при близком к норме содержании общего Всего к настоящему моменту охарактеризовано около 30 нуклеотидных замен в ДНК, кодирующей ПрС. В большинстве случаев это происходит в результате превращения 5-метилцитозина в тимин вследствие дезаминирования в нуклеотидной паре CpG. Сложная организация гена (длина более 80 тысяч пар оснований, 15 экзонов, 14 интронов и наличие псевдогена) ограничивает возможности исследования; молекулярные основы дефицита ПрS установлены лишь у нескольких больных.
Изменение соотношения между свободной и связанной с С4Ь-СБ формами ПрS может лежать в основе приобретенного дефицита при нефротическом синдроме, когда в результате нарушения процесса реабсорбции белков в проксимальных канальцах нефронов свободный
ПрS выходит в мочу, а высокомолекулярный комплекс с С4Ь-СБ не подвергается клубочковой фильтрации. Снижение концентраций
ПрS и ПрС наблюдается при беременности и приеме гормональных контрацептивов, заболеваниях печени и ДВС. Существуют данные о снижении уровня ПрС при гомоцистеинурии.
Для лечения и профилактики повторных тромбозов у больных с дефицитом ПрС или применяют антагонисты витамина К. В начале терапии наблюдают транзиторное состояние гиперкоагуляции, обусловленное более быстрым снижением концентрации ПрС и ПрS, чем витамин К-зависимых факторов свертывания, которое
может привести к развитию некроза кожи у больных с исходно сниженным уровнем или ПрС. Во избежание этого рекомендуют начинать прием антагонистов витамина К на фоне гепаринотерапии и только после достижения требуемого стабильного уровня антикоагуляции отменять гепарин.
Резистентность и тромбозы
Исследование влияния экзогенного ПрСа на свертываемость in vitro крови членов семьи с тромбофилией привело к открытию феномена резистентности к ПрСа. Для достижения такой же степени удлинения АЧТВ, как в плазме доноров, к плазме этих больных необходимо было добавить значительно больше ПрСа. Молекулярный механизм, лежащий в основе обнаруженного феномена, был идентифицирован как замена в факторе V Arg506 на Gln. Такая форма фактора V, обладающая нормальной коагулянтной активностью, но устойчивая к расщеплению под действием ПрСа, была названа фактором VЛейден.
ПрСа расщепляет три пептидные связи в N-концевой цепи фактора Va, причем скорость гидролиза по Arg в положениях 306 и 679 увеличивается после расщепления по Arg506. Замена Arg506 Gln делает невозможным расщепление трипсиноподобной протеазой по этому участку, и в результате инактивация фактора VЛейден под действием ПрСа протекает значительно медленнее, чем нормального.
Замена Arg506 Gln - наиболее часто встречающаяся (но не единственная) причина врожденной резистентности к ПрСа. В экзоне 13-го гена фактора V обнаружены полиморфные участки, которые могут быть связаны с резистентностью к ПрСа.
Резистентность к ПрСа обнаруживают у 10-20% больных, страдающих тромбозом глубоких вен. При селективном анализе семейных венозных тромбозов частота выявления резистентности достигает 50%, а при тромбозах, связанных с беременностью, может доходить до 60%, что в 10 раз выше, чем суммарная частота выявления молекулярных дефектов антитромбина III, ПрС и при этой патологии. В популяции частота гетерозиготных носителей фактора VЛейден составляет 3-5%, а при обследовании 50 больных с резистентностью к ПрСа гомозиготная форма выявлена у шести из 47 носителей. Эти
данные свидетельствуют о том, что замена Arg506 на Gln в факторе V - наиболее часто встречающийся генетический дефект, связанный с заболеванием. Однако статистический анализ связи данной мутации с тромбозами показал, что этот дефект per se менее значим, чем дефекты других компонентов системы противосвертывания.
Риск развития венозных тромбозов у больных с резистентностью к ПрСа многократно возрастает при наличии других предрасполагающих факторов, таких как дефицит компонентов противосвертывающей системы, травма, операция, беременность и использование пероральных контрацептивов. Пероральные контрацептивы per se могут вызывать феномен резистентности к ПрСа, сравнимый по величине с гетерозиготным дефектом в факторе V. У женщин фактор VЛейден - также и фактор риска инфаркта миокарда в молодом возрасте. Риск инфаркта миокарда значительно возрастет при сочетании фактора VЛейден с метаболическими нарушениями. Наиболее значимым следует считать сочетание мутации и курения, при котором отмечали 30-50-кратное увеличение риска инфаркта миокарда.
Чувствительность фактора Va к протеолизу под действием ПрСа может изменяться также в результате посттрансляционных модификаций. In vitro показано, что скорость инактивации фактора Va под действием ПрСа увеличивается после дегли-козилирования или фосфорилирования тромбоцитарной казеин-киназой-II. Феномен резистентности к ПрСа может развиться в результате повышения в плазме концентрации фактора VIII - другого субстрата ПрСа, способного конкурентно снижать скорость протеолиза фактора Va комплексом ПрСа-ПрS. К числу наиболее часто встречающихся причин приобретенной резистентности к ПрСа могут быть отнесены нарушения иммунной системы.
АНТИФОСФОЛИПИДНЫЙ СИНДРОМ
Еще в начале 1950-х годов было обнаружено, что у больных системной красной волчанкой появляется ингибитор свертывания крови, который впоследствии был назван волчаночным антикоагулянтом. Установлению природы ингибитора способствовало выявление нормализации свертываемости крови после добавления избытка фосфолипидов, а не нормальной плазмы, как в случае гемофилии. Методом радиоиммунного анализа было показано, что развитие тромбозов у больных системной красной волчанкой связано с появлением в плазме АТ, взаимодействующих с кардиолипином,
и волчаночный антикоагулянт был первоначально идентифицирован как АТ к фосфолипидам. Широкое использование высокочувствительных методов анализа показало, что АТ к фосфолипидам появляются при многих заболеваниях и могут быть вызваны также приемом лекарств и неблагоприятными экологическими факторами. Это привело к внедрению термина «антикардиолипиновый», а затем «антифосфолипидный синдром».
AT могут быть представлены иммуноглобулинами классов G, М или A и проявлять различные свойства как клинически, так и при исследованиях с использованием разных тест-систем. Например, было отмечено, что тромбозы более характерны для аутоиммунных, чем для инфекционных заболеваний, а применение колонки с фосфолипидами позволило разделить АТ к кардиолипину и волчаночный антикоагулянт. Такое разнообразие AT следовало ожидать, если бы в роли Аг выступали только фосфолипиды, и вскоре выявили, что в действительности АТ образуются к белок-фосфолипидным комплексам. Наиболее частый Аг для образования антикардиолипиновых АТ - комплекс β2-гликопротеин I (апоЛП H) с анионными фосфолипидами, а для волчаночного антикоагулянта - протромбин-фосфолипидный комплекс. У больных с антифосфолипидным синдромом обнаружены также АТ к аннексину-V, ПрС и тромбомодулину и гепарину. Эти АТ могут значительно различаться по сродству, влиянию на активность факторов свертывания и t1/2 AT в кровотоке.
AT к фосфолипидам могут нарушать следующие мембранзависимые реакции системы гемостаза: синтез и секрецию вазоактивных соединений; активацию витамин К-зависимых факторов свертывания; активацию ПрС; инактивацию факторов Va и VIIIa комплексом ПрСa-ПрS; активацию антитромбина III гликопротеинами эндотелия.
Таким образом, АТ к фосфолипидам были первоначально идентифицированы как антикоагулянт, замедляющий свертывание крови in vitro. Однако их способность ингибировать основные реакции системы противосвертывания при наличии прокоагулянтного влияния на тромбоциты и эндотелий приводит к тому, что у больных с антифосфолипидным синдромом риск тромбозов значительно выше риска кровотечений.
Клинически антифосфолипидный синдром характеризуется рецидивирующими тромбозами различной локализации. Тромбы могут развиваться в венозной (70%) и артериальной (30%) системах, а также в системе микроциркуляции. В случаях повторных спонтанных
выкидышей и внутриутробной гибели плода, причиной которых могут быть тромбозы сосудов плаценты, частота обнаружения АТ к фосфолипидам доходит до 50%. Так, при антифосфолипидном синдроме могут быть выявлены тромбофлебит глубоких вен голени, цереброваскулярные нарушения (синдром Снеддона), обусловленные тромбозом сосудов мозга. Livedo reticularis - рисунок на коже, напоминающий кружева или ячейки рыболовной сети, чаще локализованный в области нижних конечностей и обусловленный венулитом. Также возможны легочная гипертензия, синдром Бадда-Киари (сочетание симптомов внутрипеченочной портальной гипертензии, возникающее при нарушении оттока крови из печеночных вен вследствие их тромбоза), инфаркт миокарда, гемолитическая анемия, тромбоцитопения, тромботический неинфекционный эндокардит (чаще с поражением митрального и аортального клапанов), асептические некрозы головок бедренных костей, а также ложноположительная реакция Вассермана.
СИСТЕМА ФИБРИНОЛИЗА
Образование фибрина выполняет важную защитную функцию, обеспечивая остановку кровотечения в местах повреждения сосудов. После выполнения гемостатической функции и регенерации ткани фибрин должен быть удален для восстановления нормального кровотока. Это осуществляется фибринолитической системой, состоящей из профермента (плазминогена), ферментов, превращающих Пг в сериновую протеазу, и сложной системы ингибиторов, контролирующих как активацию Пг, так и сам процесс лизиса фибрина (рис. 2.9, табл. 2.3).
Кроме удаления внутрисосудистых тромбов компоненты системы фибринолиза участвуют также в разрушении внеклеточного матрикса и могут выполнять важную функцию в регуляции таких процессов, как эмбриогенез, регенерация тканей, рост и метастазирование опухолей.
ПЛАЗМИНОГЕН И ПЛАЗМИН
Фермент, непосредственно расщепляющий фибрин, - плазмин (ПЛ), образующийся при активации Пг. Пг синтезируется в печени и секретируется как одноцепочечный белок, состоящий из 790 аминокислотных остатков и содержащий Glu в качестве N-концевой аминокислоты, но в плазме присутствует в виде нескольких изоформ. Наличие двух основных изоформ обусловлено разной степенью
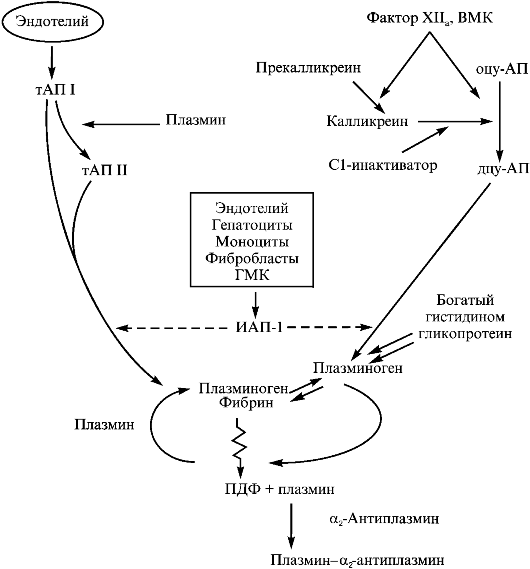 Рис. 2.9. Основные
реакции системы фибринолиза. Сплошными стрелками показаны реакции
секреции и активации компонентов, пунктирными - их ингибирование. тАП
активирует только Пг, связанный с фибрином. Урокиназа может активировать
как свободный, так и связанный с фибрином Пг. Богатый гистидином
гликопротеин - конкурентный ингибитор связывания Пг с фибрином.
Расщепление фибрина до растворимых фрагментов протекает в несколько
этапов. После образования Х-фрагментов фибрина в его структуре
экспонируются дополнительные участки связывания Пг и тАП, что
увеличивает скорость разрушения фибрина. оцу-АП - одноцепочечный
активатор Пг урокиназного типа; дцу-АП - двухцепочечная форма активатора
Пг урокиназного типа; ИАП - ингибитор активаторов Пг
Рис. 2.9. Основные
реакции системы фибринолиза. Сплошными стрелками показаны реакции
секреции и активации компонентов, пунктирными - их ингибирование. тАП
активирует только Пг, связанный с фибрином. Урокиназа может активировать
как свободный, так и связанный с фибрином Пг. Богатый гистидином
гликопротеин - конкурентный ингибитор связывания Пг с фибрином.
Расщепление фибрина до растворимых фрагментов протекает в несколько
этапов. После образования Х-фрагментов фибрина в его структуре
экспонируются дополнительные участки связывания Пг и тАП, что
увеличивает скорость разрушения фибрина. оцу-АП - одноцепочечный
активатор Пг урокиназного типа; дцу-АП - двухцепочечная форма активатора
Пг урокиназного типа; ИАП - ингибитор активаторов Пг
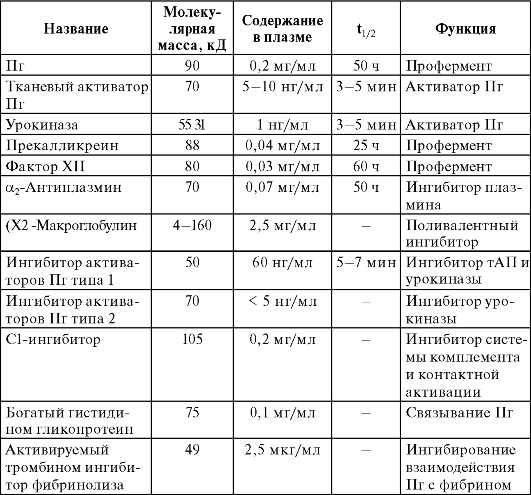
Таблица 2.3. Основные компоненты системы фибринолиза
 гликозилирования. Glu-Пг I содержит две углеводные цепочки, связанные с Asn288 и Thr345, а Glu-Пг II - только одну, связанную с Thr345.
Степень гликозилирования влияет на сродство Пг к остаткам Lys, а также
на скорость его активации и расщепления плазмином. Плазмин легко
расщепляет три связи - Arg67-Met68, Lys76-Lys77 или Lys77-Val78
- в молекуле Пг с высвобождением N-концевого полипептида. Образующиеся
при этом формы Пг обладают сходными свойствами, и их принято называть
Lys-Пг независимо от того, какова в действительности N-концевая
аминокислота. Lys-Пг активируется с большей скоростью, чем Glu-Пг, и
быстрее выводится из кровотока. Различия в скорости активации особенно
заметны в отсутствие
гликозилирования. Glu-Пг I содержит две углеводные цепочки, связанные с Asn288 и Thr345, а Glu-Пг II - только одну, связанную с Thr345.
Степень гликозилирования влияет на сродство Пг к остаткам Lys, а также
на скорость его активации и расщепления плазмином. Плазмин легко
расщепляет три связи - Arg67-Met68, Lys76-Lys77 или Lys77-Val78
- в молекуле Пг с высвобождением N-концевого полипептида. Образующиеся
при этом формы Пг обладают сходными свойствами, и их принято называть
Lys-Пг независимо от того, какова в действительности N-концевая
аминокислота. Lys-Пг активируется с большей скоростью, чем Glu-Пг, и
быстрее выводится из кровотока. Различия в скорости активации особенно
заметны в отсутствие
кофакторов активации, таких, как фибрин или аналоги Lys. Эластаза может селективно расщеплять Пг с образованием мини-Пг (Val354-Пг), не содержащего первых трех крингл-доменов.
Третичная структура Пг характеризуется высокой стабильностью, которая обеспечивается 24 дисульфидными мостиками. Большая часть структуры приходится на пять крингл-доменов, содержащих так называемые лизинсвязывающие участки. На крингл-1 находится участок высокого сродства (Kd ~9 мкМ), на остальных - участки низкого сродства (Kd ~5 мМ). Крингл-доменам принадлежит важная функция в связывании Пг с фибрином и мембранами клеток, в активации Пг, а также в обеспечении специфичности и локализации действия плазмина.
Активация Пг происходит в результате расщепления связи Arg561- Val562 с образованием фермента, состоящего из двух цепей, соединенных двумя дисульфидными мостиками. Активный центр находится в легкой B-цепи (С-концевой фрагмент Пг) и представлен остатками His602, Asp645 и Ser740. Легкая цепь - малоспецифическая трипсиноподобная протеаза, способная расщеплять практически все белки плазмы. Специфичность действия плазмина обеспечивается кринглдоменами тяжелой A-цепи (N-цевой фрагмент Пг).
ИНГИБИТОРЫ ЛИЗИСА ФИБРИНА
α2-Антиплазмин (α2-АП) принадлежит к семейству серпинов. Молярная концентрация α2-АП в плазме в два раза ниже, чем концентрация Пг. α2-АП синтезируется в печени и секретируется как одноцепочечный гликопротеин, состоящий из 464 аминокислотных остатков. В крови а2-АП присутствует в виде двух форм: Меt-α2-АП и Asn-α2-An. Asn-α2-An образуется в результате отщепления 12-член- ного пептида от N-конца Met-α2-AП Обе формы α2-АП ингибируют плазмин в растворе с одинаковой скоростью, но Меt-α2-AП обладает меньшим сродством к фибрину, чем Asn-α2-AП
α2-АП ингибирует свободный плазмин с очень высокой скоростью (t1/2 ~0,1-0,5 с). Реакция протекает в две стадии. Вначале быстро образуется обратимый комплекс α2-АП-плазмин (Kd~2 ? 1010 М), в котором затем происходит медленное расщепление α2-АП по реактивному центру Arg354-Met355-С-концевой фрагмент α2-АП остается нековалентно связанным с крингл-1, а N-концевая часть образует ковалентную связь с Ser активного центра плазмина. Взаимодействие а2-АП с субстратсвязывающим участком обеспечивает селективность действия
плазмина. В присутствии быстродействующего ингибитора этот фермент может проявлять свою активность только в месте образования до тех пор, пока остается связанным с фибрином.
Кроме ингибирования плазмина в кровотоке α2-АП принадлежит важная роль в стабилизации фибрина. При полимеризации в фибрин включаются как Пг, так и α2-АП в примерно равном количестве. Причем α2-АП связывается, а затем и сшивается фактором XIIIa с С-концевой областью а-цепи фибрина, которая в первую очередь расщепляется под действием плазмина. Сшитый с фибрином а2-АП инактивирует плазмин, образовавшийся из связанного при полимеризации Пг, после расщепления С-концевой области α-цепи фибрина, которое еще не приводит к распаду полимера, но открывает новые участки связывания с высоким сродством Пг к тАП. Расщепление фибрина до растворимых фрагментов происходит после активации Пг, связывающегося с частично разрушенным фибрином. Наличие двух стадий в лизисе фибрина - один из механизмов, обеспечивающих смещение во времени процессов образования и лизиса фибрина. Кроме этого, тот факт, что растворение фибрина происходит путем связывания с ним Пг из кровотока, обеспечивает протекание лизиса с поверхности тромба, тем самым предотвращая образование эмболов.
α2-Макроглобулин (α2-МГ) - полифункциональный ингибитор протеаз. Это гликопротеин с молекулярной массой 725 кД, состоящий из четырех идентичных субъединиц. В его структуре имеется последовательность Arg681-Val-Gly-Phe-Tyr-Glu686, пептидные связи которой могут расщепляться многими типами протеаз. Расщепление приводит к конформационным изменениям α2-МГ, в результате которых гидролизуется тиоэфирная связь между Cys949 и Gln952, а карбонильный радикал образует ковалентную связь с протеазой, но в отличие от серпинов не по активному центру. Поэтому комплекс протеаза-α2-МГ сохраняет способность расщеплять низкомолекулярные субстраты, а взаимодействие с белками становится невозможным из-за препятствий, создаваемых макромолекулой ингибитора. Комплекс протеа- за-α2-МГ связывается с рецепторами на фибробластах, макрофагах и клетках РЭС, транспортируется внутрь клеток и расщепляется лизосомальными протеазами.
α2-МГ ингибирует плазмин со скоростью, значительно меньшей, чем α2-АП, но в плазме молярное соотношение Пг/α2-АП составляет 2:1, и α2-МГ может играть важную роль в инактивации плазмина при потреблении а2-АП.
Активируемый тромбином ингибитор фибринолиза. Стимулом к открытию еще одного ингибитора фибринолиза послужило наблюдение зависимости скорости лизиса сгустков плазмы от количества образовавшегося тромбина. Активируемый тромбином ингибитор фибринолиза (АТИФ) был выделен и идентифицирован как карбоксипептидаза U (прокарбоксипептизаза В плазмы), специфически отщепляющая С-концевые остатки Lys, которые экспонируются после частичного расщепления фибрина и формируют новые участки связывания тАП и Пг. Это приводит к снижению скорости лизиса фибрина.
Свободный тромбин активирует АТИФ с низкой скоростью, поэтому антифибринолитический эффект этого ингибитора зависит от количества тромбина, образующегося при активации свертывания крови. Компоненты системы ПрС могут оказывать разнонаправленное действие на АТИФ. Активация ПрС и расщепление фактора Va снижают количество образующегося тромбина, что должно приводить к снижению скорости активации АТИФ. В то же время связывание тромбина с ТМ приводит не только к активации ПрС, но и к 1000-кратному увеличению скорости активации АТИФ комплексом тромбин-ТМ. Результирующая этих двух процессов зависит от многих параметров, определяющих формирование тромба in vivo.
Богатый гистидином гликопротеин. К числу ингибиторов фибринолиза может быть отнесен и богатый гистидином гликопротеин (БГГ). Этот белок с молекулярной массой 75 кД присутствует в плазме и тромбоцитах, из которых секретируется при активации. БГГ обладает высоким сродством к лизинсвязывающему центру 1 Пг (Kd~l мкМ) и может служить конкурентным ингибитором взаимодействия Пг с фибрином. В плазме ~50% Пг находится в виде комплекса с БГГ, обладающего низким сродством к фибрину. В то же время БГГ, секретируемый активированными тромбоцитами, связывается с их поверхностью и может выполнять роль буфера, обеспечивающего накопление Пг в области тромбообразования.
РАСЩЕПЛЕНИЕ ФИБРИНОГЕНА И ФИБРИНА ПЛАЗМИНОМ
Плазмин расщепляет пептидные связи фибриногена в определенном порядке с образованием продуктов разрушения, обозначаемых как X-, Y-, D- и E-фрагменты. Наиболее чувствительна к протеолизу С-концевая часть Аа-цепи, способная расщепляться по остаткам 584, 425 и 207. Затем происходит отщепление N-концевого 42-членного
пептида Вβ-цепи. Фибриноген - симметричная структура, но расщепление по отмеченным остаткам может протекать асимметрично с образованием набора Х-фрагментов, имеющих молекулярную массу от 330 до 240 кД. Х-фрагменты могут полимеризоваться под действием тромбина, хотя и с меньшей скоростью, чем нативный фибриноген.
Полная потеря свертываемости происходит после отщепления одного из D-фрагментов, содержащего периферический участок полимеризации. Далее Y-фрагмент может расщепляться на начальные D- и E-фрагменты, которые после отщепления нескольких коротких пептидов превращаются в конечные фрагменты с молекулярной массой 80 кД (D-фрагмент) и 50 кД (Е-фрагмент).
Расщепление связей в сшитом фибрине происходит примерно в том же порядке, как и при расщеплении фибриногена, а конечные продукты расщепления - E-фрагменты и D-димер. D-димер состоит из двух ковалентно связанных D-фрагментов, принадлежавших смежным молекулам фибрин-мономеров в полимере. В плазме растворимые фрагменты фибрина могут представлять собой набор фрагментов, состоящих из D-димера, связанного ковалентно или нековалентно с Е-фрагментами.
АКТИВАТОРЫ ПЛАЗМИНОГЕНА
Тканевый активатор плазминогена (тАП) синтезируется клетками эндотелия и секретируется как одноцепочечный гликопротеин с молекулярной массой 70 кД. В препаративных количествах тАП может быть выделен из культуры клеток меланомы.
При хроматографии на Lys-сефарозе тАП разделяется на две формы - типы I и II, отличающиеся по содержанию углеводов. Участки гликозилирования - Asn в положениях 117, 184 (только при типе I) и 448. С Asn117 связан линейный олигосахарид, состоящий из остатков маннозы. Два других участка содержат разветвляющиеся до четырех цепочек олигосахариды, содержащие остатки N-ацетил- лактозамина. Степень гликозилирования тАП влияет на кинетику активации Пг. Удельная активность тАП типа I в ~1,5 раза выше, чем активность тАП типа II.
Третичная структура тАП состоит из пальцевидного домена фибронектина, домена фактора роста эпидермиса, двух крингл-доменов и протеазного домена. Домен фибронектина и крингл-2 содержат участки связывания тАП с фибрином. Активный центр образуют остатки His322, Asp371 и Ser478. Плазмин, калликреин и фактор Ха специфи-
чески расщепляют тАП по Arg275~Ile276 с образованием активатора, состоящего из двух цепочек, соединенных дисульфидным мостиком. Физиологическое значение превращения тАП в двухцепочечную форму неизвестно. В отсутствие фибрина двухцепочечный тАП активирует Пг с несколько большей скоростью, чем одноцепочечный, но в присутствии фибрина активаторные свойства двух форм тАП практически не различаются.
Свободный Пг активируется тАП с очень низкой скоростью. Реакция характеризуется низкой величиной kcat и значением Km, более чем на порядок превышающим концентрацию Пг в крови. Фибрин связывает тАП и Пг. Образование комплекса фибрин-тАП-Пг на три порядка увеличивает каталитическую эффективность активации Пг. В процессе разрушения фибрина плазмином на нем экспонируются новые участки связывания Пг с высоким сродством и наблюдается дальнейшее увеличение скорости активации Пг.
тАП обладает коротким t1/2 в кровотоке. Основной орган, обеспечивающий выведение тАП, - печень. При прохождении через печень из крови удаляется 70% активатора. Исследование динамики тАП в крови после внутривенной инфузии показало, что выведение активатора из кровотока характеризуется наличием двух фаз с t1/2 около 3 и 30 мин. Предполагается, что фаза быстрого снижения концентрации тАП обусловлена его клиренсом клетками печени и обратимым связыванием с эндотелием, а фаза медленного снижения - поступлением в кровь активатора, депонированного ранее на эндотелии.
Несколько типов рецепторов могут участвовать в связывании и интернализации тАП. В печени эндотелиальные клетки и клетки Купфера связывают преимущественно свободную форму тАП и взаимодействие с их рецепторами обеспечивается остатками маннозы углеводной цепи, связанной с Аэпц7 первого крингл-домена. Клетки паренхимы содержат Са2+-зависимый рецептор, взаимодействующий с EGF-доменом тАП. Рекомбинантные формы тАП, в которых удалены три N-концевых домена или изменено положение участка гликозилирования, выводятся из кровотока в несколько раз медленнее, чем природная форма тАП.
Гепатоциты содержат рецептор, гомологичный рецептору липопротеинов низкой плотности, который связывает как свободный тАП, так и его комплекс с ингибитором. Этот рецептор участвует в связывании и интернализации также и других протеаз, инактивированных а2-МГ. Он состоит из двух полипептидных цепей с молекулярной
массой 515 и 85 кД. С рецептором взаимодействует еще один белок, имеющий молекулярную массу 39 кД. Лигандсвязывающие участки находятся на субъединице с молекулярной массой 515 кД, располагающейся на поверхности клеток и взаимодействующей с субъединицей с молекулярной массой 85 кД, обладающей трансмембранным и цитоплазматическим доменами. тАП и комплекс тАП-ИАП-1 связываются, по-видимому, с разными участками рецептора, так как белок с молекулярной массой 39 кД ингибирует в большей степени связывание комплекса, чем свободного тАП.
Многие типы клеток специфически связывают Пг со сродством ~1 мкМ. Рецепторы тАП на мембранах клеток, а также такие белки межклеточного матрикса, как тромбоспондин, желатин и коллаген IV типа могут стимулировать активацию связанного с мембранами Пг под действием тАП, но их кофакторная активность значительно меньше, чем у фибрина.
Урокиназа. Трипсиноподобная протеаза, активирующая Пг, впервые была выделена из мочи и названа урокиназой. Выделенный фермент состоял из двух субъединиц, соединенных дисульфидным мостиком. Вскоре было показано, что клетки секретируют урокиназу в виде одноцепочечного белка, который не расщепляет синтетические субстраты и не взаимодействует с ацилирующими реагентами. Это послужило основанием для названия данной формы фермента проурокиназой.
При инкубации Пг с одноцепочечной урокиназой происходит взаимная активация проферментов. Кинетический анализ показал, что образование двухцепочечной урокиназы и плазмина может быть описано моделью, включающей три последовательные реакции: активацию Пг одноцепочечной урокиназой; превращение образующимся плазмином одноцепочечной урокиназы в двухцепочечную; активацию Пг двухцепочечной урокиназой. Связывание Пг с одноцепочечной урокиназой может приводить к конформационным изменениям, в результате которых формируется активный центр, обеспечивающий начальную активацию Пг. В пользу этого свидетельствует то, что способностью активировать Пг обладает рекомбинантная форма активатора, которая в результате замены Lys158 Glu не расщепляется плазмином на двухцепочечную. Таким образом, одноцепочечная урокиназа, по-видимому, - не истинный профермент, и Международный комитет по тромбозу и гемостазу рекомендовал заменить термин «проурокиназа» на «одноцепочечный активатор Пг урокиназного типа» (оцу-АП).
оцу-АП секретируется клетками как белок с молекулярной массой 54 кД, состоящий из 411 аминокислотных остатков. Вторичная структура состоит из трех участков, включающих крингл- и каталитический домен. Активный центр образуют остатки His204, Asp255 и Ser356.
Ряд связей в структуре оцу-АП легко подвергается протеолизу, что обусловливает наличие нескольких изоформ фермента. Из культуры клеток аденокарциномы человека выделена низкомолекулярная (32 кД) форма, которая может образоваться в результате расщепления связи Glu143-Leu144 По ферментативным свойствам эта форма практически не отличается от нативного оцу-АП. Плазмин и калликреин расщепляют связь Lys158-Ile159 с образованием двухцепочечной формы фермента (дцу-АП), в котором легкая (N-концевая) и тяжелая (С-концевая) цепи связаны дисульфидным мостиком. Плазмин может далее расщеплять и связь Lys135-Lys136 с образованием низкомолекулярной формы (33 кД), не содержащей регуляторных доменов. В коммерческих препаратах дцу-АП обнаружена форма, которая может образоваться также в результате расщепления связи Phe157-Lys158.
Тромбин специфически расщепляет Arg156-Phe157 с образованием двухцепочечной формы, не активирующей Пг in vitro, но обладающей заметной тромболитической активностью in vivo. Плазмин может, хотя и с очень низкой скоростью, расщеплять в этой форме связь Lys158-Ile159 с образованием активного дцу-АП.
N-концевая аминокислота тяжелой цепи имеет важное значение в проявлении протеолитической активности дцу-АП. Добавление аминокислот с N-конца или даже консервативная замена Ile159 приводит к резкому снижению способности дцу-АП активировать Пг. Предполагается, что этот остаток после расщепления связи Lys158- Ile159 перемещается в область субстратсвязывающего центра и принимает участие в его формировании.
оцу-АП не обладает высоким сродством к фибрину, но при внутривенном введении вызывает лизис тромбов без выраженной активации Пг в кровотоке. Селективность действия оцу-АП может быть обусловлена: образованием в плазме обратимого комплекса с ингибитором, диссоциирующего при взаимодействии оцу-АП с Пг, связанным с фибрином; локальной активацией оцу-АП на поверхности тромба калликреином, фактором ХПа или плазмином; включением в тромб ряда белков плазмы и клеток крови, взаимодействие которых может приводить к формированию уникальной структуры, обеспечивающей высокое сродство к оцу-АП и увеличение его активности даже без
превращения в дцу-АП. Подтверждением этой точки зрения может служить то, что резистентный к расщеплению плазмином оцу-АП и форма, расщепленная тромбином, обладают намного большей активностью в лизисе тромбов, чем можно было бы ожидать, исходя из кинетики активации Пг in vitro.
Урокиназа синтезируется различными типами клеток: фибробластами, эпителиальными клетками, пневмоцитами, децидуальными клетками плаценты, макрофагами, а также эндотелиальными клетками под воздействием различных стимулов. Многие типы клеток обладают специфическим рецептором к урокиназе (у-АПР). у-АПР - богатый цистеином гликопротеин, не имеющий трансмембранного и внутриклеточного доменов. Взаимодействие с мембраной клетки обеспечивается ковалентной связью С-конца с гликозилфосфатидилинозитолом. Связывание урокиназы с у-АПР обеспечивается N-концевым EGF-доменом и характеризуется высоким сродством (Kd ~10-9-10-10 M) и специфичностью. Другие EGF-содержащие компоненты системы гемостаза с этим рецептором не взаимодействуют. Связывание урокиназы с у-АПР активирует сигналпроводящую систему, дифференциацию клеток, хемотаксис нейтрофилов и миграцию клеток эндотелия. Связанная с рецептором урокиназа активирует Пг и латентные металлопротеазы в межклеточном пространстве, что обеспечивает разрушение внеклеточного матрикса, необходимое для роста, деления и миграции клеток. Повышенное содержание урокиназы, ее рецепторов и ИАП-1 в клетках опухолей ассоциируется с ростом и метастазированием опухолей и плохим прогнозом заболевания.
ИНГИБИТОРЫ АКТИВАТОРОВ ПЛАЗМИНОГЕНА
тАП непрерывно секретируется эндотелием как активный фермент, но в крови он быстро инактивируется. Наиболее эффективный ингибитор тАП и урокиназы - белок из семейства серпинов, получивший название ингибитора активаторов Пг типа 1 (ИАП-1). Ингибитор активаторов Пг типа 2 (ИАП-2), инактивирующий с высокой скоростью урокиназу, синтезируется плацентой, моноцитами и макрофагами. В норме в крови он не обнаруживается, но при беременности и ряде заболеваний его содержание в плазме может значительно возрастать. Ингибиторы, первоначально описанные как ИАП-3 и ИАП-4, впоследствии были идентифицированы как ингибитор ПрСа и протеазный нексин-I соответственно.
ИАП-1 - гликопротеин с молекулярной массой 52 кД, состоящий из 379 аминокислотных остатков. Основное место синтеза ИАП-1 - эндотелий сосудов. ИАП-1 может синтезироваться также гепатоцитами, моноцитами, фибробластами, клетками меланомы и ГМК. Значительное количество ИАП-1 (~90% от общего пула крови) содержится в α-гранулах тромбоцитов и секретируется из них при активации, что может создавать высокую локальную концентрацию ИАП-1 и играть важную роль в стабилизации фибриновой матрицы тромба.
ИАП-1 может находиться в активной и латентной формах, переход между которыми не связан с протеолизом и обратим. In vitro латентный ИАП-1 активируется обработкой хаотропными реагентами. Физиологический механизм активации латентного ИАП-1 неизвестен. Предполагается, что негативно заряженные фосфолипиды, экспонирующиеся на поверхности активированных тромбоцитов, могут активировать латентный ИАП-1. В плазме активная форма ИАП-1 связывается с витронектином, стабилизирующим активную конформацию ИАП-1. Витронектин обеспечивает также связывание ИАП-1 с внеклеточным матриксом.
Взаимодействие ИАП-1 с активаторами протекает в две стадии. Вначале быстро (kcat ~107 М-1-с-1) образуется обратимый комплекс, в котором происходит расщепление связи Arg346-Met347 реактивного центра с освобождением С-концевого пептида ингибитора и образованием ковалентного комплекса активатор-ИАП-1. Комплекс тАП-ИАП-1 может связываться с фибрином и быть конкурентным ингибитором взаимодействия тАП с фибрином.
Повышение активности ИАП-1 - наиболее часто встречающаяся причина снижения фибринолитической активности крови. Анализ соотношения между содержанием в плазме тАП, его активностью и активностью ИАП-1 показал, что доля активного тАП экспоненциально снижается при линейном увеличении активности ИАП-1. При активности ИАП-1 менее 7 МЕ/мл около 33% секретируемого эндотелием тАП находится в активном состоянии, а при активности ИАП-1 более 22 МЕ/мл на долю активного тАП приходится лишь 1,5%. Прямое доказательство того, что повышение ИАП-1 может приводить к патологическому тромбообразованию, получено в экспериментах с трансгенными мышами, у которых венозные тромбозы развивались при повышении экспрессии гена ИАП-1 и исчезали после ее подавления. Гетерозиготная форма врожденного дефицита ИАП-1 протекает
бессимптомно, а у гомозигот наблюдаются медленное заживление ран и повышенная кровоточивость после травм.
В промоторном участке гена ИАП-1 обнаружен 4G/5G-поли- морфизм, который влияет на скорость транскрипции. В популяции встречаемость генотипов 4G/4G, 4G/5G и 5G/5G соответствует 0,25, 0,5 и 0,25. Уровень ИАП-1 в плазме у 4G/4G-гомозигот на ~30% выше, чем у 5G/5G. Активность ИАП-1 в плазме подвержена циркадным изменениям в течение дня с максимумом в утренние часы и в течение года с максимумом в зимние месяцы. ИАП-1 - один из компонентов системы гемостаза, определяющих утреннюю гиперкоагуляцию.
ИАП-1 - один из наиболее быстрореагирующих белков острой фазы. Его активность в плазме может значительно повышаться при многих заболеваниях. Синтез ИАП-1 стимулируется эндотоксином, цитокинами, факторами роста, а также тромбином, тАП и ангиотензином II.
Данные проспективных исследований свидетельствуют о том, что повышение активности ИАП-1 и/или снижение активности тАП связано с риском развития инфаркта миокарда у больных с нестабильной стенокардией, повторного инфаркта миокарда в молодом возрасте и рестенозов после ангиопластики. В то же время необходимо отметить, что при многофакторном анализе значимость ИАП-1 как фактора риска снижается, что отражает его взаимосвязь с другими факторами риска заболеваний ССС. Уровень ИАП-1 в плазме коррелирует с концентрацией инсулина, индексом массы тела, повышением уровня ТГ и снижением содержания ЛПВП, которые характерны для синдрома резистентности к инсулину.
Непосредственное влияние инсулина на синтез ИАП-1, которое показано в экспериментах с культурами клеток, - лишь один из возможных механизмов, связывающих гиперинсулинемию с повышением ИАП-1 в плазме. Другой механизм может заключаться в развитии андроидного типа ожирения. Сравнительный анализ синтеза ИАП-1 в образцах жировой ткани показал, что клетки сальника синтезируют значительно больше ИАП-1, чем подкожный жир. Наконец, третий механизм может заключаться во влиянии инсулина на метаболизм ЛП. В эксперименте показано, что ЛПНП и ЛПОНП увеличивают секрецию ИАП-1 культурой эндотелиальных клеток, причем ЛПОНП, выделенные из плазмы больных с гипертриглицеридемией, обладают более выраженным эффектом, чем ЛПОНП доноров. Большая эффективность ЛПОНП больных обусловлена более высоким сродством этих частиц к клеточному рецептору.
АТ к рецептору апоВ/E устраняли стимулирующее влияние ЛПОНП на синтез ИАП-1. Окисление ЛНП повышает их способность стимулировать синтез ИАП-1 эндотелием вен.
Инсулин и ЛПОНП могут действовать синергично в стимуляции синтеза ИАП-1. Добавление физиологических концентраций инсулина или ЛПОНП, выделенных из плазмы доноров, к культуре клеток HepG2 приводит к двукратному увеличению уровней ИАП-1 в культуральной среде и иРНК в клетках, а при совместном действии наблюдалось 6-кратное увеличение количества иРНК в клетках и концентрации Аг ИАП-1 в культуральной среде.
В связи с тем, что в плазме значительно ниже концентрация тАП, чем других протеаз и его быстродействующего ингибитора, точное измерение активности тАП в крови - методически сложная задача. Изучение содержания тАП с помощью ИФА показало, что повышение Аг тАП позволяет предвидеть клинические осложнения атеросклероза, в основе которых лежит тромбоз. У больных стенокардией с ангиографически подтвержденным поражением коронарных артерий повышение уровня соотношения тАП и Аг коррелировало с частотой развития инфаркта миокарда, инсультов и смертностью. Аналогичные результаты были получены в американском исследовании, в котором наблюдались практически здоровые мужчины. В большом популяционном исследовании факторов риска атеросклероза повышение соотношения тАП и Аг ассоциировалось с появлением его ранних признаков.
В ряде исследований были обнаружены повышение уровня Аг и тАП у больных с тромбозами и наличие отрицательной корреляции между концентрацией Аг и активностью тАП в плазме больных. Предполагается, что повышение уровня Аг и тАП может быть обусловлено увеличением скорости ингибирования тАП в кровотоке и накоплением комплекса тАП-ИАП-1, клиренс которого клетками печени осуществляется медленнее, чем свободного тАП.
СТРЕПТОКИНАЗА
В 1933 г. было обнаружено, что культура β-гемолитического стрептококка продуцирует соединение, вызывающее лизис сгустков крови. Это соединение получило первоначальное название «фибринолизин». Впоследствии оказалось, что для лизиса фибрина под действием стрептококков необходим еще и плазменный фактор. Этот фактор был назван Пг, а фибринолизин переименован в стрептокиназу (СК). В конце 1950-х годов были получены высокоочищенные препараты
СК и началось ее применение для растворения внутрисосудистых тромбов, открывшее эру тромболизиса.
СК - одноцепочечный белок с молекулярной массой 47 кД и состоящий из 386 аминокислотных остатков. СК не имеет собственной ферментативной активности*, а ее эффективность как стимулятора фибринолиза значительно варьирует у животных разных видов. Механизм стимуляции фибринолиза в плазме человека заключается в образовании с высокой скоростью (kass~105 M-1c-1) стехиометрического комплекса СК-Пг (Kd~109 M-1), в котором в результате конформационных изменений в Пг формируется активный центр протеазы. Ферментативные свойства комплекса СК-Пг существенно отличаются от таковых плазмина: комплекс служит активатором Пг, а плазмин - нет; комплекс СК-Пг инактивируется α2-АП и α2-ΜΓ с очень низкой скоростью; комплекс СК-Пг не обладает способностью прямо расщеплять фибрин. После образования комплекса СК-Пг в нем происходит расщепление как Пг с образованием Lys-Пг и плазмина, так и укороченных форм СК:
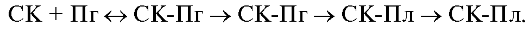 Βсе
эти комплексы обладают способностью активировать Пг, однако по мере
накопления расщепленных форм снижается потенцирующее влияние фибрина на
активацию Пг и увеличивается скорость инактивации ингибиторами плазмы.
Βсе
эти комплексы обладают способностью активировать Пг, однако по мере
накопления расщепленных форм снижается потенцирующее влияние фибрина на
активацию Пг и увеличивается скорость инактивации ингибиторами плазмы.
Повышения тромболитической эффективности СК удалось достичь путем создания препаратов (APSAC, Eminase), представляющих собой эквимолярные комплексы СК-Пг/Пл, в которых Ser активного центра протеазы ацилирован. Эта модификация не влияет на структуру крингл-доменов Пг, и ацилированный комплекс связывается с достаточно высоким сродством (Kd ~10 мкМ) с фибрином. Скорость деацилирования определяется химической структурой ацила и для р-анизоиловых производных t1/2 составляет ~100 мин. Фармакокинетические исследования показали, что t1/2 APSAC в кровотоке больше, чем СК, и составляет ~90 мин. Большее t1/2 и наличие сродства к фибрину способствуют накоплению активатора в области тромба, а относительно медленное восстановление ферментативной активности позволяет применять болюсное введение препарата больным.
* Характерное для ферментов название было дано задолго до того, как был выяснен механизм действия этого белка
2.4. РОЛЬ ТРОМБОЦИТОВ И СОСУДИСТОЙ СТЕНКИ В ПАТОГЕНЕЗЕ АРТЕРИАЛЬНОГО ТРОМБОЗА
Тромботические осложнения заболеваний ССС - наиболее частая причина смерти в современном обществе. В частности, различные проявления острой коронарной патологии в странах западной Европы и США ежегодно уносят почти 1 млн жизней, поэтому неудивительно, что классические представления о патогенезе артериального тромбоза получены на основании экспериментального, морфологического и клинического изучения тромбоза коронарных артерий.
Причинаразвивающегосяинфарктамиокарда-внутрикоронарный тромбоз, возникающий, как правило, на месте имеющейся атеросклеротической бляшки с поврежденной поверхностью. Доказательствами этому положению послужили исследования английских морфологов Дэвиса и Томаса (1984), обнаруживших в 74 из 100 вскрытий умерших от ИБС в первые 6 ч от начала симптомов внутрипросветный тромбоз. Причем все тромбы наблюдались в местах разрывов богатых липидами атеросклеротических бляшек, в значительном проценте случаев тромботические массы проникали через трещину внутрь бляшки и тем самым увеличивали ее размеры.
Фальк (1983), изучая патологоанатомический материал внезапно умерших больных нестабильной стенокардией, практически во всех случаях обнаружил внутрикоронарный тромбоз, причем в 81% случаев тромбы располагались в местах разрывов атеросклеротических бляшек, имели слоистую структуру, что указывало на разный «возраст» тромботических масс, постепенно суживавших просвет коронарной артерии.
Совершенствование ангиографического оборудования, многочисленные ангиографические исследования больных инфарктом миокарда и нестабильной стенокардией без лечения и с введением тромболитических препаратов и наконец создание коронароангиоскопических катетеров позволили визуализировать внутреннюю оболочку коронарных артерий и подтвердить патогномоничность внутрикоронарного тромбоза при инфаркте миокарда и нестабильной стенокардии.
Артериальные тромбы состоят в основном из тромбоцитов с небольшим содержанием фибрина и эритроцитов, поэтому их условно называют белыми тромбами, в отличие от венозных, так называемых красных тромбов, состоящих преимущественно из эритроцитов
и фибриновых нитей. Различия в венозных и артериальных тромбах относительны. Состав тромба определяется его «возрастом».
Атеросклеротическая бляшка - основной элемент атеросклероза. Начальные стадии ее образования связаны с накоплением липидов в макрофагах и в свободном состоянии в тканях с пролиферацией ГМК и образованием коллагена. Вследствие этих процессов образуется атеросклеротическая бляшка с ядром, в котором содержатся внеклеточные липиды. Ядро заключено в фиброзную коллагеновую капсулу. Помимо свободного холестерина, в ядре содержатся пенистые клетки - «нафаршированные» липидами макрофаги. Макрофаги разрушаются, и их липидное содержимое увеличивает ядро бляшки. Небольшие атеросклеротические бляшки часто не видны при коронароангиографии, так как коронарные сосуды способны к ремоделированию. Ишемия миокарда и ее клиническое проявление (стенокардия) появляются, когда бляшка увеличивается настолько, что артерия теряет способность к ремоделированию и возникает хронический стеноз коронарной артерии. Второй возможный вариант развития процесса - быстрое образование тромба на бляшке, и именно это приводит к развитию так называемого острого коронарного синдрома, к клиническим проявлениям которого относят нестабильную стенокардию и инфаркт миокарда. Выделение острого коронарного синдрома связано с тем, что при указанных клинических состояниях отмечают общие морфологические признаки в виде поврежденной атеросклеротической бляшки с разрывами ее поверхности и формированием внутрикоронарного тромбоза.
Тромбообразование на бляшке начинается вследствие истончения и повреждения покрывающего ее эндотелия, при этом тромб располагается на поверхности бляшки. Вторая причина - разрыв покрышки бляшки с обнажением липидного ядра, при этом тромб формируется внутри бляшки, приводя впоследствии к сужению сосуда. При анализе летальных исходов, причиной которых был тромбоз коронарных артерий, Дэвис в 1990 г. показал, что только в 25% случаев тромбоз связан с эрозией эндотелия, а в остальных - с разрывом бляшек. Есть данные, что эрозии эндотелия чаще возникают у женщин.
Таким образом, разрыв атеросклеротической бляшки можно считать составной частью атеросклеротического процесса и важным стимулом для развития тромбоза. Множество факторов контролируют процесс тромбообразования: местный кровоток, врожденные особенности организма, состояние фибринолиза и свертывающей системы
крови больного, стадия развития атеросклеротической бляшки.
Участие тромбоцитов в развитии атеросклеротического процесса подтверждается тем, что фибрин и компоненты тромбоцитов находят внутри бляшки. Смит и Степлз, используя иммуногистохимические методы, обнаружили в бляшке поперечно-сшитый фибрин, протромбин и продукт секреции тромбоцитов - β-тромбоглобулин. Кроме того, похоже, что тромбоциты могут стимулировать рост и пролиферацию компонентов атеросклеротической бляшки, выделяя так называемый тромбоцитарный фактор роста (PDGF), трансформирующий фактор роста (TGF-β) и другие низкомолекулярные субстанции. Особенно важным представляется роль PDGF, так как известно, что он стимулирует рост и пролиферацию ГМК и фибробластов - основу фиброзной ткани внутри атеросклеротической бляшки. Взаимодействие между тромбоцитами и сосудистым эндотелием влияет на рост атеросклеротической бляшки, определяющим фактором которого служит повреждение эндотелия. В 1995 г. Кровелло и соавт. установили, что адгезивная молекула Р-селектина принимает участие в адгезии тромбоцитов, поврежденного эндотелия и лейкоцитов. Установлено, что взаимодействие молекулы Р-селектина с рецептором моноцита инициирует образование цитокинов и тканевого фактора, т. е. моноциты могут принимать участие в развитии атеросклеротической бляшки, не только накапливая липиды и превращаясь в пенистые клетки, но и образуя цитокины, а также активируя каскад коагуляции путем тромбинообразования.
Разрывы мелких бляшек приводят к проникновению тромботических масс внутрь бляшки, стимуляции пролиферации ГМК и дальнейшему росту бляшки. Данный механизм лежит в основе развития хронических стенозов и приводит к формированию стабильной стенокардии.
Выраженность стеноза в коронарной артерии - важный фактор, определяющий клинические проявления разрывов бляшек. Дэвис в 1990 г. показал, что у 81% больных, умерших от тромбоза, развившегося в месте эрозированного эндотелия, были гемодинамически значимые стенозы (60%). С другой стороны, среди лиц, умерших от тромбоза коронарной артерии, развившегося на месте лопнувшей бляшки или изъязвленного поражения, больше половины (60%) имели гемодинамически незначимые (менее 60%) стенозы в коронарных артериях. Эти морфологические данные соответствуют клинико-ангиографическим наблюдениям, подтверждающим,
что возникновение нестабильной стенокардии связано с ростом бляшек в местах умеренных стенозов (Амброуз, 1988). Разрыв бляшек в местах выраженных стенозов не сказывается на коронарном кровотоке, так как длительно существующий стеноз в коронарной артерии способствует развитию коллатерального кровообращения. С другой стороны, разрыв бляшек, умеренно стенозирующих коронарные артерии, чаще проявляется симптомами острого коронарного синдрома из-за отсутствия развитого коллатерального русла.
Форма бляшки также определяет прогрессирование ИБС. Бляшки бывают концентрическими, вызывающими фиксированную степень стеноза коронарной артерии, и эксцентрическими, при которых степень стенозирования может варьировать. Большинство больных ИБС имеют бляшки разной формы. При исследовании коронарного русла у больных стабильной стенокардией соотношение концентрических и эксцентрических бляшек составляет соответственно 76 и 24%. Общепризнано, что при остром коронарном синдроме эксцентрические стенозы отмечают чаще.
Атеросклеротические бляшки отличаются и по составу. Фиброзные (стабильные) состоят в основном из коллагена и протеогликанов. Они составляют до 60% у больных стабильной стенокардией.
Богатые липидами или уязвимые (ранимые) бляшки содержат липидное ядро, отделенное от кровотока тонкой фиброзной капсулой. Скопление липидов часто содержит коллаген, придающий стабильность фиброзной капсуле. У больных, умерших от острого коронарного синдрома, анализ лопнувших бляшек показал наличие в них внеклеточных липидов и сниженное содержание коллагена. В бляшках, где липидное ядро не было нарушено, разрывы наблюдались между слоями коллагена фиброзной капсулы. Бляшки с высоким содержанием липидов предрасполагают к более агрессивному течению ИБС.
Прочность фиброзной капсулы, отделяющей насыщенное липидами ядро от потока крови, зависит от ее толщины. Бляшки с толстой капсулой более устойчивы к разрывам и соответственно к переходу болезни в острый коронарный синдром. Основные составляющие фиброзной капсулы - коллаген и эластин. Нарушение синтеза коллагена или усиление его распада может способствовать истончению фиброзной капсулы.
Таким образом, разрыв капсулы или нарушение целостности эндотелия, покрывающего уязвимую, насыщенную липидами, незначительно стенозирующую просвет коронарной артерии атеросклеротическую
бляшку, - общий первый шаг, приводящий к дестабилизации ИБС. Разрыв бляшки - триггерный механизм для образования богатого тромбоцитами тромба, играющего важную роль в патогенезе острого коронарного синдрома.
Разрыв бляшки приводит к экспонированию тромбогенных субстанций субэндотелиальных слоев. В нагруженных липидами макрофагах образуется большое количество тканевого фактора (ТФ) - мощного стимулятора тромбинообразования. ТФ - низкомолекулярный гликопротеин. На нормальном эндотелии он не экспрессируется, но его большое количество содержится в клетках, представляющих собой содержимое атеросклеротической бляшки, включая макрофаги, ГМК и поврежденные эндотелиальные клетки. ТФ находят также и в адвентиции артерий. Фибриновые и тромбоцитарные скопления в большей степени выражены в богатом липидами ядре бляшки и в местах изъязвлений и разрывов бляшек. ТФ образует комплекс с фактором VII свертывания крови, создавая так называемый комплекс активированный фактор VII-ТФ. Этот комплекс, экспонированный на липидном ядре, активирует фактор Х свертывания крови, который в свою очередь инициирует каскад коагуляции, в результате чего образуется тромбин - ключевой фермент свертывания, превращающий фибриноген в фибрин. Но функции тромбина не ограничиваются ключевой реакцией системы свертывания; он активирует противосвертывающую систему, связываясь с тромбомодулином, экспонированном на эндотелии, а также активирует тромбоциты, связываясь с рецептором Ib. Тромбин - мощнейший индуктор агрегации тромбоцитов. Еще одним сильным стимулятором адгезии и агрегации тромбоцитов, содержащимся как в субэндотелиальных слоях, так и внутри бляшки, служит коллаген. Функцию адгезивного белка выполняет фФВ, присутствующий как в плазме, так и в субэндотелиальных структурах, однако неактивированные тромбоциты могут взаимодействовать только с субэндотелиальной формой фФВ. Разрыв бляшки приводит к экспонированию субэндотелиального фФВ, что способствует первому этапу образования тромбоцитарного тромба, - адгезии тромбоцитов. Адгезия тромбоцитов происходит вследствие связывания фФВ с рецептором мембраны тромбоцитов гликопротеином Ib.
Фактор VIII (фФВ) - многомерный белок плазмы, индуцирующий прилипание тромбоцитов к субэндотелиальным структурам. фФВ, помимо ключевой роли в адгезии и агрегации тромбоцитов, участвует в образовании тромба и процессах коагуляции и служит
защитным белком для фактора VIII свертывания крови. За последние годы достигнуты большие успехи в изучении фФВ. Расшифрована структура белка фФВ, идентифицирован его функциональный домен, определены мутации, приводящие к вариантам фФВ. Ген фФВ расположен в 12-й паре хромосом. фФВ имеет в своей структуре цепочку Arg-Gly-Asp; это соединение находят в структуре многих адгезивных белков, которые способны взаимодействовать с так называемыми интегриновыми рецепторами. фФВ синтезируется исключительно в мегакарио-цитах, в эндотелиальных клетках, находят его и в α-гранулах тромбоцитов, в плазме и субэндотелиальном пространстве. В эндотелиальных клетках фФВ находится в тельцах Вайбеля-Паладе и выделяется из них под воздействием различных физиологических стимулов. Основное количество фФВ находится в эндотелии, субэндотелии и тромбоцитах, поэтому его содержание в плазме непостоянно. фФВ выделяется в кровоток в месте и в момент повреждения эндотелия и, таким образом, участвует в регуляции гемостаза.
фФВ имеет две основные функции. Первая - связывание и стабилизация фактора VIII in vivo и in vitro (защита фактора VIII от инактивации протеином С и фактором Ха); вторая - обеспечение связей между тромбоцитами и сосудистой стенкой (адгезия тромбоцитов) и тромбоцитами (агрегация тромбоцитов).
фФВ взаимодействует с компонентами субэндотелия и клеточными рецепторами при высоких скоростях сдвига, т. е. в мелких сосудах и стенозированных артериях. В этих местах это единственный белок, осуществляющий адгезию.
Установлено, что у больных атеросклерозом и такими заболеваниями, как артериальная гипертензия, сахарный диабет, ИБС по сравнению со здоровыми отмечают повышенный уровень фФВ. Исследование, проведенное Янссоном и соавт. в 1991 г., показало, что повышение уровня фФВ связано с высоким риском развития окклюзии коронарных артерий у больных ИБС, в том числе и после перенесенного инфаркта миокарда. Все это позволяет рассматривать повышенное содержание фФВ как возможный фактор риска развития атеросклероза и тромбоза, предполагая, что повышенное содержание фФВ стимулирует усиленную адгезию и агрегацию тромбоцитов и тем самым усиливает тромбообразование.
После прикрепления тромбоцитов к поверхности поврежденного эндотелия происходит их склеивание друг с другом - так называемый процесс агрегации тромбоцитов. Стимулы к агрегации -
многочисленные агонисты, циркулирующие в кровотоке, содержащиеся в атеросклеротической бляшке, субэндотелии, выделяющиеся из тромбоцитов при адгезии и агрегации, а также нарушение текучести крови в стенозированных участках коронарных артерий.
Мощнейшие стимуляторы адгезии тромбоцитов - коллаген субэндотелиального матрикса, а также тромбин. При активации тромбоцитов происходят изменение их формы и активация рецептора IIb/IIIa- Среди множества стимуляторов активации тромбоцитов (агонистов) следует отметить тромбин, тромбоксан А2, фактор активации тромбоцитов, серотонин, АДФ, норадреналин. Помимо тромбоцитов, АДФ содержится в эритроцитах, которые и служат основным его источником. АДФ освобождается из эритроцитов при их разрушении в турбулентных потоках, возникающих в суженных атеросклеротическими бляшками артериях.
Каждый агонист, взаимодействуя со специфическим рецептором, образует комплекс, и сигнал передается внутрь тромбоцитов при помощи так называемых вторичных посредников. Считается, что большинство агонистов имеют общий путь передачи сигнала - гидролиз фосфотидилинозитол-4,5-дифосфата в диацилглицерол и инозитолтрифосфат, что в последующем вызывает мобилизацию вторичного посредника - ионов кальция. Внутритромбоцитарное увеличение содержания кальция приводит к уменьшению количества тромбоцитов, сокращению секреции АДФ и серотонина, активации мембранной фосфолипазы и арахидонового каскада с образованием тромбоксана А2 - мощного индуктора активации тромбоцитов. Секреция из гранул тромбоцитов биологически активных веществ получила название реакции освобождения, в результате которой в процесс агрегации вовлекаются новые тромбоциты; процесс становится самоподдерживающимся и заканчивается формированием первичного тромбоцитарного тромба. Агрегация тромбоцитов завершается путем формирования мостиков между адгезивными белками (фибриноген, фФВ) и активированными рецепторами IIb/IIIa тромбоцитов. Этот конечный этап агрегации тромбоцитов одинаков при всех возможных видах их стимуляции.
СТРУКТУРА И ФУНКЦИЯ РЕЦЕПТОРОВ ТРОМБОЦИТОВ Рецепторы тромбоцитов - гликопротеины мембраны, большинство из которых относят к семейству так называемых интегринов. Интегрины - гетеродимерные молекулы, состоящие из семейства α- и β-субъединиц, различные комбинации которых и служат
специфическимирецепторамидляразличныхлигандов.Интегрины находят на поверхностях практически всех клеток, они участвуют во многих физиологических реакциях. Известно несколько интегринов, участвующих в адгезии тромбоцитов: гликопротеин Ia/IIa (α2β1) - основной рецептор для коллагена, гликопротеин !С/На (α2β1) - для фибронектина, α6β1 - рецептор для ламинина, α5β3 - рецептор для витронектина. Последний рецептор способен узнавать и другие лиганды: фибриноген, фФВ, связывающиеся и с рецептором IIb/IIIa. Известно несколько рецепторов, по структуре не относящихся к интегринам, участвующих в адгезии тромбоцитов: гликопротеин IV - рецептор для коллагена и тромбоспондина, а также гликопротеин Ib, связывающий фФВ.
Таким образом, за процесс адгезии тромбоцитов ответственно несколько рецепторов мембраны тромбоцитов, среди которых есть представители семейства интегринов и других семейств. Однако основной рецептор, узнающий наибольшее количество лигандов, а именно фибриноген, фибронектин, фФВ и витронектин и участвующий в процессе агрегации, - гликопротеин IIb/IIIa (α2β3) поверхностной мембраны тромбоцитов.
IIb/IIIa-peцептop тромбоцитов - типичный представитель семейства интегринов. Его α-субъединица, или гликопротеин Щ (молекулярная масса 36 кД), состоит из тяжелой и легкой цепей. Легкая цепь имеет короткий цитоплазматический хвост, трансмембранную часть и короткий внеклеточный домен. Тяжелая цепь расположена снаружи клетки. β-Субъединица, или гликопротеин IIIa (молекулярная масса 92 кД), состоит из единственного полипептида (762 аминокислоты) с коротким цитоплазматическим хвостом, трансмембранной частью и большим внеклеточным доменом. Субъединицы нековалентно связаны друг с другом, для сохранения гетеродимерной структуры необходим кальций. IIb/IIIa-рецепторы наиболее распространены, на поверхности одного тромбоцита содержится примерно 50 тыс. рецепторов. IIb/IIIa-рецепторы тромбоцитов изучены в наибольшей степени благодаря проведению исследований по изучению тромбастении Глянцмана - врожденной патологии, связанной с отсутствием или резким снижением количества IIb/IIIa-рецепторов. Исследования крови больных с тромбастенией Глянцмана показали, что IIb/IIIa-рецепторы способны связываться не только с фибриногеном и тем самым осуществлять процесс агрегации тромбоцитов, но и с другими адгезивными гликопротеинами: фибронектином, фФВ,
витронектином, в большей степени отвечающими за адгезию тромбоцитов к субэндотелиальным структурам.
Механизм действия IIb/IIIa-рецептора заключается в его способности узнавать две характерные аминокислотные последовательности. Первая состоит из аминокислот Arg-Gly-Asp, она обнаружена в фибронектине, фФВ, витронектине, а также и в α-цепях молекул фибриногена, причем на каждую половину молекулы фибриногена приходится по две ключевые последовательности Arg-Gly-Asp. Следует подчеркнуть, что ключевая последовательность Arg-Gly-Asp узнаваема большинством представителей семейства интегринов. Тонкие механизмы взаимодействия IIb/IIIa-рецепторов с адгезивными молекулами до конца не изучены, но очевидно, что пептиды или мелкие молекулы, содержащие ключевую последовательность аминокислот Arg-Gly-Asp, могут быть потенциальными ингибиторами взаимодействия цепторов тромбоцитов с фибриногеном.
Вторая цепочка аминокислот, узнаваемая IIb/IIIa-рецепторами тромбоцитов, - Lys-Gln-Ala-Gly-Asp-Val. Она находится в карбоксильном конце γ-цепей фибриногена. В отличие от цепочки Arg-Gly-Asp цепочку Lys-Gln-Ala-Gly-Asp-Val обнаружили только в молекуле фибриногена и, вероятно, именно в этом месте фибриноген связывается с IIb/IIIa-рецепторами тромбоцитов. Взаимосвязь между двумя цепочками до конца не ясна. Показано, что пептиды из γ-цепей фибриногена ингибируют связывание фибронектина и фФВ с тромбоцитами. Это может быть связано с тем, что места на IIb/IIIa-рецепторах, узнающие как первую, так и вторую цепочку, частично перекрываются, или же с аллостерическим взаимодействием участков рецепторов, узнающих две упомянутые цепочки аминокислот. Предполагают, что обе цепочки аминокислот взаимодействуют с несколькими участками IIb/IIIa-рецептора.
РОЛЬ АРАХИДОНОВОЙ КИСЛОТЫ
Обсуждая роль тромбоцитов в патогенезе артериального тромбоза, необходимо упомянуть о двух прямо противоположных по своему действию на тромбоциты и гладкую мускулатуру веществах: тромбоксане А2 и простациклине. Оба соединения - конечные продукты метаболизма арахидоновой кислоты.
Арахидоновая кислота - предшественник всех классов простагландинов (PG). Синтез арахидоновой кислоты в организме осуществляется из фосфолипидов под действием фосфолипазы. Основной источник арахидоновой кислоты - ненасыщенные жирные кислоты,
поступающие в организм с пищей. Превращение арахидоновой кислоты в организме осуществляется под действием двух ферментов: липооксигеназы и циклооксигеназы. Под действием циклооксигеназы из арахидоновой кислоты образуются циклические эндоперекиси PGG2 и H2, которые в дальнейшем превращаются в тромбоксан А2 и простациклин, PGD2, E2, F2a. Тромбоксан А2, образующийся под действием фермента тромбоксансинтетазы, - нестабильное соединение (t1/2 составляет около 30 с), он быстро превращается в стабильный продукт тромбоксан B2. Тромбоксан А2 образуется в тромбоцитах и выделяется в кровоток в процессе реакции освобождения, однако было установлено, что небольшие количества тромбоксана А2 образуются в эндотелии внутренней оболочки аорты, а также в фибробластах легочной ткани, микросомах радужной оболочки глаза, в перфузируемой почке, пупочной артерии, плаценте; в малых количествах он образуется практически во всех сосудах человека. Тромбоксан А2 - мощный проагрегант и вазоконстриктор.
ПРОСТАЦИКЛИН
Простациклин образуется в эндотелиальных клетках сосудов под действием фермента простациклинсинтетазы. T1/2 составляет 2-3 мин. Простациклин имеет несколько стабильных метаболитов, основной из которых - 6-кетоPGF1a. Полагают, что его содержание отражает содержание простациклина в плазме крови. Простациклин - мощный системный вазодилататор и антиагрегант. Последнее обусловлено активацией в мембране тромбоцитов аденилатциклазного механизма, приводящего к увеличению в тромбоцитах содержания цАМФ, уменьшению свободного цитоплазматического кальция и снижению агрегационной способности тромбоцитов. Простациклин - вещество, образующееся in situ. Импульсом к образованию простациклина эндотелиальными клетками могут быть повреждение целостности эндотелия, а также появление в кровотоке тромбина. При адгезии тромбоцитов к месту поврежденного сосуда из них выделяется тромбоксан, одновременно с этим из эндотелиальных клеток выделяется простациклин, ограничивая или предотвращая процесс тромбообразования.
Известно, что серозные оболочки, в том числе и перикард, образуют простациклиноподобные субстанции, причем простациклин, содержащийся в перикардиальной жидкости, способен влиять на коронарный кровоток. С возрастом при развитии атеросклероза синтез простациклина сосудистой стенкой снижается.
ТРОМБОКСАН
С появлением исследований Монкада и Вейна, посвященных метаболитам арахидоновой кислоты, в начале 1970-х годов начался период активного изучения роли тромбоксана и простациклина в патогенезе коронарного спазма и тромбоза. В конце 70-х и 80-х годах была опубликована серия исследований, посвященных роли дисбаланса в соотношении тромбоксан/простациклин в патогенезе коронарного тромбоза и спазма. Результаты показали, что у больных стенокардией во время ишемии миокарда, вызванной стимуляцией предсердий, в крови, оттекающей непосредственно от миокарда, повышается содержание тромбоксана В2. Кроме того, было показано, что при отсутствии различий в покое у здоровых и больных стенокардией в содержании тромбоксана В2 и 6-кетоPGF1α по реакции на физическую нагрузку больные ИБС отличаются от здоровых преобладанием выброса тромбоксана и снижением выброса простациклина. Эти данные позволили группе исследователей во главе с Мехта выдвинуть гипотезу о роли дисбаланса в соотношении тромбоксан/простациклин в изменении сосудистого тонуса и происхождении ишемии миокарда. В дальнейшем появились данные о повышении содержания тромбоксана в крови, оттекающей непосредственно от миокарда (из коронарного синуса), у больных нестабильной стенокардией (Хирш и соавт., 1981) и во время индуцированной ишемии миокарда (Леви и соавт., 1980). Огромный интерес вызывали работы Мехта и соавт. (1984), Робертсона и соавт. (1981, 1983), продемонстрировавшие нарастание содержания тромбоксана В2 в крови коронарного синуса во время спонтанных приступов у больных с вазоспастической формой стенокардии. По мнению авторов, увеличение содержания тромбоксана определяло коронарный тонус и способствовало возникновению и усугублению ишемии. Высказывалась гипотеза, что происхождение приступов вазоспастической стенокардии может быть связано с активацией тромбоцитов, выбросом тромбоксана и быстрым образованием тромбоцитарного тромба в месте спазма коронарной артерии. Однако последующие исследования, проведенные у больных со спонтанной ишемией миокарда и взятием крови из коронарного синуса за несколько минут до возникновения приступа стенокардии, показали, что увеличение концентрации тромбоксана в момент приступа вторично по отношению к спазму и ишемии миокарда, а кроме того, угнетение синтеза тромбоксана с помощью аспирина, индометацина не уменьшало частоту эпизодов ишемии у больных вазоспастической стенокардией.
Итак, на сегодняшний день существуют экспериментальные и морфологические доказательства роли тромбоцитов в патогенезе атеротромбоза. Кроме того, результаты изучения маркеров активированных тромбоцитов (β-тромбоглобулин, тромбоцитарный фактор IV, тромбоксан В2) у больных с такими клиническими проявлениями атеросклероза, как инфаркт миокарда, нестабильная стенокардия, инсульт и перемежающаяся хромота, указывают на существенное повышение их содержания по сравнению со здоровыми людьми. Результаты исследований показывают, что подавляющее большинство признаков активации тромбоцитов при атеросклерозе вторично по отношению к патологии внутренней стенки сосудов. Вопрос о том, могут ли обнаруживаемые у отдельных больных гиперактивные тромбоциты per se участвовать в развитии атеросклероза и тромбоза, остается открытым. Имеются данные (Кристенсен, Мартин, 1992), что тромбоциты больших размеров обладают большей реактивностью, чем малые; осложнения инфаркта миокарда чаще отмечены у больных, имеющих тромбоциты больших размеров. Поскольку тромбоцит - безъядерная клетка, его размеры определяются размерами предшественника - мегакариоцита. Было показано, что тромбоциты больших размеров образуются из больших мегакариоцитов, а размеры последних контролируются цитокинами и ИЛ-6. Эти данные указывают на связь воспаления и тромбообразования.
И наконец, существенный вклад в доказательство роли тромбоцитов в патогенезе артериального тромбоза внесли многочисленные крупные проспективные исследования по изучению эффективности антитромбоцитарных препаратов у больных с различными клиническими проявлениями атеросклероза. Считается доказанным, что назначение аспирина (препарата, необратимо угнетающего циклооксигеназу тромбоцитов) в среднем на 25% снижает частоту развития инфаркта миокарда, инсульта и внезапной сердечно-сосудистой смерти. Антитромбоцитарный препарат клопидогрел, механизм действия которого связан с блокадой АДФ-индуцированной активации IIb/IIIa-рецепторов тромбоцитов, повышает этот процент до 33. Анализ трех исследований, посвященных применению абциксимаба (ингибитор IIb/IIIa-рецепторов антительной природы) перед процедурой коронарной баллонной ангиопластики, убедительно продемонстрировал его эффективность в отношении тромботических осложнений, которые в наибольшей степени выражены у больных нестабильной стенокардией.
2.5. МЕТОДЫ ИССЛЕДОВАНИЯ СИСТЕМЫ ГЕМОСТАЗА
Нормально функционирующая система гемостаза должна обеспечивать сохранность жидкого состояния крови в пределах сосудов и быстрое тромбирование поврежденных участков для предотвращения кровопотери и кровоизлияния в ткани. Нарушения системы гемостаза могут быть как врожденными, так и приобретенными и проявляться кровотечениями или тромбозами. В основе заболеваний могут лежать нарушения механизмов регуляции гемостаза: аномалии стенки сосудов, клеток крови (чаще всего тромбоцитов) и факторов плазмы. Применяемые в настоящее время методы лабораторного исследования системы гемостаза могут быть разделены на скрининговые (глобальные) и специальные.
2.5.1. ЗАБОР КРОВИ ДЛЯ КОАГУЛОЛОГИЧЕСКИХ АНАЛИЗОВ
Результат коагулологических исследований во многом зависит от того, как собиралась и обрабатывалась кровь. Активация свертывания во время получения или обработки крови может привести к непредсказуемому результату большинства тестов. Для предотвращения активации свертывания компрессия вены не должна превышать 1 мин, и первые капли крови, которые могут содержать тканевый фактор, лучше для этих анализов не использовать. Кровь необходимо собирать в вакутейнер или пластиковую пробирку, содержащие антикоагулянт.
Наиболее часто в качестве антикоагулянта используют 1/10 объема 0,11 М раствора цитрата натрия (3,2% раствор трехзамещенной двуводной соли - Na3C6H6O7 ? 2Н2О). Кровь необходимо немедленно и аккуратно перемешать с антикоагулянтом. Ионы Са2+ необходимы для активации, но образовавшийся тромбин может проявлять прокоагулянтное действие и в отсутствие Са2+. Поэтому задержка с перемешиванием крови увеличивает вероятность ошибочного результата при тестировании. Перед центрифугированием необходимо внимательно проверить пробирку на наличие сгустков. Лучший способ проверки - фильтрование через нейлоновую сетку. При обнаружении нитей фибрина образец необходимо выбраковывать.
Для получения бедной тромбоцитами плазмы кровь центрифугируют в течение 10 мин при 2000-3000 g (~3000 об/мин). Температурный режим зависит от того, какие анализы предполагается выполнять.
Для анализов на волчаночные антикоагулянты и маркеры активации тромбоцитов требуется дополнительная обработка плазмы, обеспечивающая более полное удаление клеток.
При значительном отклонении гематокрита от нормы может потребоваться изменение соотношения крови и антикоагулянта. При гематокрите < 0,25 1/10 объема цитрата может оказаться недостаточно для снижения концентрации Са2+ ниже пороговой величины, а при высоком гематокрите увеличится разведение плазмы цитратом, что приведет к кажущемуся снижению содержания факторов. Определить количество цитрата, которое необходимо внести в пробирку для получения 10 мл конечного объема крови, можно по формуле:
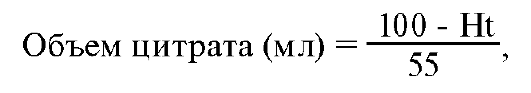 где Ht - гематокрит.
где Ht - гематокрит.
Цитрат не ингибирует активацию тромбоцитов и секрецию компонентов их α-гранул. Поэтому для точного определения содержания в плазме маркеров активации тромбоцитов (фактора IV и β-тромбоглобулина), Аг ИАП-1 или гепарина необходимо использовать специальный антикоагулянт, препятствующий активации тромбоцитов in vitro. Этот антикоагулянт получил название CTAD по первым буквам компонентов, входящих в его состав: 0,11 М - цитрат (С), 15 мМ - теофиллин (Т), 3,3 мМ - аденозин (А) и 0,2 мМ - дипиридамол (D), рН 5,0. Кровь, собранная с этим антикоагулянтом, может быть использована для большинства коагулологических анализов, за исключением, разумеется, исследования активации тромбоцитов.
2.5.2. СКРИНИНГОВЫЕ ТЕСТЫ
К скрининговым методам относят методы, в которых исследуют активацию ряда реакций, отражающих определенную последовательность превращений компонентов системы. Диагностическая значимость глобальных тестов определяется тем, что нормальные результаты позволяют исключить наличие значительных отклонений в содержании компонентов системы, в то время как аномальные результаты в большинстве случаев позволяют конкретизировать направление поиска дефектного звена. Кроме того, глобальные тесты используют также и для контроля антикоагулянтной терапии. Β настоящее время для начального исследования наиболее широко используют следующие тесты:
1) протромбиновый тест (ПТ);
2) активированное частичное тромбопластиновое время (АЧТВ);
3) тромбиновое время/рептилазное время (ТВ/РВ);
4) фибриноген;
5) лизис эуглобулинов;
6) время кровотечения.
К глобальным тестам могут быть отнесены также время свертывания, время рекальцификации, каолиновое и кефалиновое время, тромбоэластография и ряд других. Однако эти тесты, по сути, отражают те же реакции, что и перечисленные выше основные, и их использование в большинстве случаев не позволяет значительно расширить информативность основных методов.
Нормальные результаты первых двух глобальных тестов (ПТ и АЧТВ), отражающих активацию по внешнему и внутреннему пути свертывания крови, позволяют исключить наличие значительных дефектов большинства плазменных компонентов этой системы. Исключение составляет фактор XIII, дефицит которого в этих тестах не проявляется, но может быть причиной геморрагического синдрома у новорожденных, кровоточивости и медленного заживления ран. Направление дальнейших исследований зависит от клинических показаний.
При подозрении на гипокоагуляцию (кровоточивость) скрининговые тесты должны быть дополнены исследованием:
1) фФВ (активность - кофактор ристоцетина, Аг);
2) фактора XIII;
3) количества и агрегации тромбоцитов;
4) отдельных факторов и ингибиторов к ним;
5) ингибиторов фибринолиза.
При наличии признаков гиперкоагуляции (повышенный уровень маркеров активации) и тромбообразования целесообразно проведение исследования:
1) компонентов системы свертывания (фибриноген, фактор VII и его активированная форма, фактор VIII, резистентность фактора V к ПрСа);
2) наличия волчаночного антикоагулянта;
3) компонентов системы противосвертывания (антитромбин III, ПрС, ПрS, ГК-II);
4) активаторов фибринолиза, включая факторы XII и прекалликреин;
5) ингибиторов активаторов фибринолиза.
Диагностика причин тромбообразования - значительно более сложная задача, чем диагностика дефицита факторов системы свертывания. Врожденный дефицит компонентов системы противосвертывания обнаруживают довольно редко. Приобретенный дефицит отмечают значительно чаще, но в большинстве случаев его причина заключается в нарушении функций печени или регуляции системы гемостаза. Изменение функционального состояния эндотелия - повидимому, ведущая причина патологического тромбообразования при многих заболеваниях (атеросклероз, сахарный диабет, гомоцистеинурия, новообразования, сепсис и т.д.). Однако до настоящего времени не предложено приемлемого для клиники метода исследования антикоагулянтных свойств эндотелия. Возможность диагностики ограничивается измерением циркулирующих в крови компонентов и маркеров их активации.
2.5.3. ПРОТРОМБИНОВЫЙ ТЕСТ
ПТ - один из наиболее часто выполняемых коагулологических анализов. Он был предложен А. Квиком и соавт. в 1935 г., и его часто называют по имени первого из авторов методики тестом Квика. Принцип метода состоит в определении времени свертывания цитратной плазмы (в некоторых вариантах цельной крови) после добавления тромбопластина и Ca2+. Хотя в названии теста, которое было дано задолго до выяснения основных реакций свертывания, присутствует наименование только одного из факторов, в действительности результат теста зависит от:
1) содержания факторов VII, X, V, протромбина и фибриногена;
2) наличия патологических ингибиторов:
• полимеризации фибрина (продукты деградации фибрина, миеломные белки);
• фосфолипидзависимых реакций.
Техника выполнения ПТ очень проста. Достаточно иметь термостат на 37°С, секундомер, пробирки и пипетки. Однако при очевидной простоте выполнения самого теста оценка его результатов представляет серьезную проблему, окончательно не решенную до настоящего времени. Две основные причины обусловливают сложность проблемы.
• Во-первых, в тесте реализуется ряд последовательных и взаимовлияющих реакций и суммарная скорость зависит от множества параметров.
• Βо-вторых, тромбопластин - частично очищенный экстракт, получаемый из тканей мозга, легких или плаценты разных млекопитающих, включая человека. Βремя свертывания нормальной и, что исключительно важно, патологической плазмы значительно варьирует в зависимости от источника и метода получения тромбопластина.
Для выражения результатов ПТ используют:
1) время свертывания в секундах;
2) протромбиновое отношение (ПО), которое определяют как время свертывания плазмы больного, деленное на время свертывания нормальной плазмы:
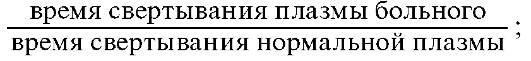 3)
процент протромбинового индекса, который определяют как кремя
свертывания нормальной плазмы, деленное на время свертывания плазмы
больного и умноженное на 100. Βеличине присваивают размерность %;
3)
процент протромбинового индекса, который определяют как кремя
свертывания нормальной плазмы, деленное на время свертывания плазмы
больного и умноженное на 100. Βеличине присваивают размерность %;
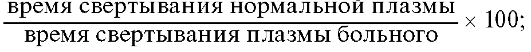 4) процент от нормы;
4) процент от нормы;
5) международное нормализованное отношение (МНО). Первые три системы выражения результатов ПТ имеют серьезные
недостатки, и от них следует отказаться. Βремя свертывания, ПО и процент протромбинового индекса свидетельствуют только о том, отличается ли плазма больного от нормальной, а информация о том, насколько они различаются в действительности, скрыта от исследователя.
По методу Квика для перевода времени свертывания в процент протромбина строится калибровочный график. Β основе его построения лежит простой логический принцип. Если содержание факторов протромбинового комплекса в нормальной плазме принять за 100%, то в разведенной в два раза оно будет 50%, в два раза - 25% и т. д. На рис. 2.10, А показаны примеры влияния снижения концентрации факторов на время свертывания при определении с использованием четырех разных тромбопластинов. Как видно из рисунка, тромбопластин может обладать высокой активностью* (~11 с) при
* Β отечественной литературе под активностью тромбопластина подразумевают время свертывания данным препаратом нормальной плазмы.
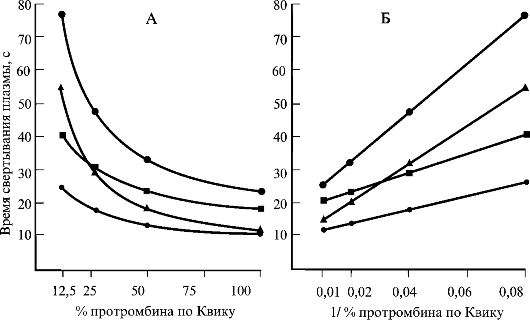 Рис. 2.10. Βлияние
разведения плазмы на время свертывания, измеряемое при использовании
разных тромбопластинов (А). Построение калибровочного графика по Квику в
координатах 1/% протромбина - секунды свертывания (Б)
Рис. 2.10. Βлияние
разведения плазмы на время свертывания, измеряемое при использовании
разных тромбопластинов (А). Построение калибровочного графика по Квику в
координатах 1/% протромбина - секунды свертывания (Б)
п лохой чувствительности к снижению концентрации факторов и низкой активностью (~23 с) при высокой чувствительности к дефициту факторов. Поэтому ни время свертывания, ни ПО и процент протромбинового индекса, представляющие собой время свертывания, умноженное или деленное на константу, не позволяют судить о том, насколько велика разница в содержании факторов. Другими словами, это некалиброванные величины, которые поэтому не должны использоваться. Проблема усложняется тем, что значения этих параметров варьируют не только между лабораториями, но и в пределах одной лаборатории при переходе от одной партии тромбопластина к другой, даже если они выпущены одним и тем же производителем.
Β ПТ графики зависимости времени свертывания от концентрации факторов протромбинового комплекса имеют форму обратной функции (y = 1/x). Использование нелинейных графиков для перевода секунд (с) в процент протромбина не очень удобно. Поэтому для вычислений применяют линеализацию графика в координатах 1/% протромбина (т.е. 100% - 0,01; 50% - 0,02; 25% - 0,04; 12,5% - 0,08) от времени свертывания, как показано на рис. 2.10, Б. Βо многих
современных коагулометрах используют именно этот подход и метод линейной регрессии для расчета калибровочного графика и обратного перевода секунд в процент протромбина по Квику. При такой оценке результатов нормальными принято считать значения протромбина ≥ 70%.
К недостаткам этого метода калибровки ПТ относят то, что разведение плазмы моделирует только снижение концентрации факторов, которое может наблюдаться, например, при нарушении синтеза белков в печени. Однако при другой патологии (дефицит витамина К или прием его антагонистов - пероральных антикоагулянтов) концентрация факторов может быть близкой к норме, но их функциональные свойства, определяющие взаимодействие и активацию факторов, существенно нарушены.
Международное нормализованное отношение (МНО). Другой метод стандартизации ПТ был принят ВОЗ в 1981 г. и модифицирован в 1983 г. В его основе лежит наличие линейной зависимости между логарифмами протромбинового времени, определенными с разными тромбопластинами, для плазмы больных, находящихся в стабильной фазе терапии пероральными антикоагулянтами. На практике это означает, что значения ПО, определенные с использованием разных тромбопластинов, могут быть приведены математически к одной величине, которая была бы получена при определении протромбина с неким стандартным тромбопластином. Эту величину предложили называть МНО - международным нормализованным отношением (от англ. INR - international normalized ratio).
Математическая операция, позволяющая перевести результаты ПТ в один масштаб, - возведение ПО в степень, характеризующую чувствительность используемого препарата тромбопластина. Чувствительность первичного стандарта тромбопластина, зарегистрированного ВОЗ, была принята за 1,0, а всех последующих, включая национальные, должна вычисляться по отношению к этому стандарту тромбопластина.
Для вычисления чувствительности тромбопластина определяют значения протромбинового времени для серии нормальных и патологических плазм с использованием двух тромбопластинов - стандартного (референтного) и анализируемого. Затем строится график, в котором по оси ординат откладывают значение логарифмов протромбинового времени, измеренного с референтным тромбопластином, а по оси абсцисс - с анализируемым. Тангенс угла наклона этого
графика - МИЧ (от англ. ISI - international sensitivity index - международный индекс чувствительности) анализируемого тромбопластина. Линейный характер зависимости между логарифмами протромбинового времени и ПО означает, что значение ПО, которое получили бы при определении данного образца плазмы с использованием референтного тромбопластина (МНО), может быть вычислено путем возведения ПО, определенного с любым тромбопластином, в степень, равную МИЧ этого тромбопластина:
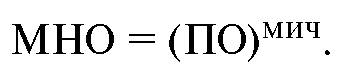 Определение
МИЧ - трудоемкая процедура. Первоначально для определения МИЧ
предлагалось измерение протромбинового времени с двумя тромбопластинами в
80 образцах, из которых 60 должны быть взяты от больных, которым
проводится терапия пероральными антикоагулянтами. Βпоследствии было
показано, что достаточная степень точности может быть достигнута при
использовании 20 патологических плазм.
Определение
МИЧ - трудоемкая процедура. Первоначально для определения МИЧ
предлагалось измерение протромбинового времени с двумя тромбопластинами в
80 образцах, из которых 60 должны быть взяты от больных, которым
проводится терапия пероральными антикоагулянтами. Βпоследствии было
показано, что достаточная степень точности может быть достигнута при
использовании 20 патологических плазм.
Βведение системы МНО ознаменовало значительный прогресс в стандартизации ПТ. Однако, несмотря на достижения, ряд проблем остались нерешенными. Одна из них - влияние типа коагулометра на результат.
Коагулометры - приборы, предназначенные для автоматического определения времени, которое проходит от момента внесения в измерительную кювету стартового реактива до начала свертывания (образования первых нитей фибрина). Полимеризация фибрина сопровождается увеличением вязкости и светорассеивания (мутности). Β зависимости от того, по какому из этих параметров определяют свертывание, коагулометры могут быть подразделены на механические и оптические. Более того, каждый из этих типов имеет значительное разнообразие конструкционных особенностей. Каждая из используемых систем обладает преимуществами и недостатками, чем, по-видимому, и объясняется наличие большого разнообразия конструкций. Различия в результатах измерений при использовании разных коагулометров обусловлены тем, что строго определить такое понятие, как начало свертывания, невозможно. Коагулометры запрограммированы на определение того отрезка времени, за который нарастание измеряемого параметра после внесения стартового реактива превысит определенный лимит. Способы программирования самые разнообразные: от соотношения между силой магнитного поля и размером/массой вращаемого шарика до определения порого-
вого значения изменения оптической плотности, которое принимают за начало свертывания.
Следствием этого разнообразия является то, что значения времени свертывания, определенные с использованием одних и тех же плазм и реактивов, но разных коагулометров, могут не совпадать. Хотя различий, обусловленных применением разных коагулометров, значительно меньше, чем при использовании разных тромбопластинов, тем не менее они есть и их необходимо учитывать. С этой целью разработан и уже применяется новый подход к стандартизации ПТ, заключающийся в использовании набора калибровочных плазм для локальной стандартизации.
Набор состоит из четырех лиофилизированных плазм: одной нормальной и трех, полученных от доноров, находящихся в стабильной фазе терапии пероральными антикоагулянтами низкой, средней и высокой интенсивности. С использованием этих плазм в лаборатории клиники строят калибровочный график в двойных логарифмических координатах для перевода секунд свертывания в МНО. Этот график уже учитывает характеристики как коагулометра, так и тромбопластина, используемых в лаборатории. По номограмме, предлагаемой производителем калибровочных плазм, может быть вычислен и МИЧ используемого тромбопластина. К важным преимуществам данного метода калибровки относят также и то, что он менее трудоемкий, чем принятый ВОЗ в 1983 г. Кроме того, построение калибровочного графика по четырем образцам плазмы уменьшает величину ошибки, обусловленной отклонением определенного в лаборатории значения времени свертывания нормальной плазмы от истинного. Вклад этой ошибки в конечный результат (вычисленное значение МНО) будет тем больше, чем ниже чувствительность (выше МИЧ) тромбопластина. Однако этот метод предложен относительно недавно, и только после проведения больших клинических исследований можно будет решить, какой из методов обеспечивает лучший контроль терапии.
МИЧ для различных тромбопластинов, получаемых из экстрактов тканей, варьирует от 0,9 до 2,8. Чем выше значение МИЧ, тем менее чувствителен тромбопластин к изменению содержания компонентов протромбинового комплекса и, следовательно, тем больше может быть ошибка результата ПТ. Тромбопластины с МИЧ более чем 2,0 лучше вообще не использовать, так как для достижения необходимой степени точности придется выполнять много параллельных определений одной пробы.
Основная цель, которую преследовало введение МНО, состояла в обеспечении оптимизации терапии пероральными антикоагулянтами. Использование единой системы контроля позволяет обобщать опыт, накопленный в разных клиниках, для подбора оптимального соотношения между риском развития тромбоза и кровотечения. В специальном докладе Американской кардиологической ассоциации, опубликованном в 1994 г., предложено при подборе дозы антикоагулянтов ориентироваться на терапевтические пределы МНО от 2,0 до 3,5.
Таким образом, целесообразно использовать два варианта представления результатов ПТ, что позволит объединить преимущества каждого из них. Калибровка по Квику моделирует дефицит факторов и переводит результат теста в линейную шкалу, по которой легче ориентироваться в динамике факторов свертывания, а использование системы МНО обеспечит более надежный контроль интенсивности антикоагулянтной терапии.
В завершение анализа ПТ остановимся на одной методической проблеме. Этот тест может выполняться с использованием как плазмы, полученной из венозной крови, так и капиллярной крови. Определение в капиллярной крови представляется предпочтительным, так как не требует пункции вены. Однако при кажущейся простоте в действительности оно сопряжено с рядом проблем.
Первая и главная проблема заключается в большей вероятности активации свертывания при заборе капиллярной крови. Кровь, проходя через поврежденную ткань, неизбежно контактирует с тромбогенной поверхностью и может активироваться. Это приводит вначале к кажущемуся увеличению содержания факторов (гиперкоагуляции), которое в действительности может быть лишь следствием их активации, а затем переходит в снижение содержания факторов (гипокоагуляцию), обусловленное низкой стабильностью активированных форм. Поэтому капиллярная кровь должна анализироваться как можно быстрее после забора.
Вторая проблема связана с объемом крови, необходимой для анализа. В стандартном варианте постановки ПТ используют 100 мкл плазмы и 200 мкл Са-тромбопластина. Быстро и аккуратно (без выдавливания из места прокола) отобрать даже 100 мкл крови удается не всегда. Поэтому для ПТ из капиллярной крови применяют реактив, содержащий, кроме тромбопластина, еще и плазму, из которой удалены факторы VII, X и протромбин. Это обеспечивает возможность выполнения теста с использованием всего 20-50 мкл капиллярной крови,
которая либо непосредственно вносится в реактив (при определении в присутствии больного), либо смешивается с буфером, содержащим цитрат, и доставляется в лабораторию для серийных исследований. Этот реактив может использоваться также и для определения протромбина в цитратной плазме. Преимущество постановки ПТ с использованием малого объема плазмы больного и адсорбированной плазмы как источника фибриногена и фактора V заключается в меньшей чувствительности этого метода к аномалиям фибриногена и ингибиторам полимеризации.
Наконец, третья проблема связана с влиянием гематокрита на результат теста. При внесении пипеткой в кювету стандартного объема цельной крови объем внесенной плазмы и, следовательно, концентрация факторов в реакционной смеси будут зависеть от величины гематокрита. При выражении результатов ПТ в виде процента по Квику влияние гематокрита может быть легко учтено путем умножения определенного по калибровке значения процента от нормы на корректировочный фактор
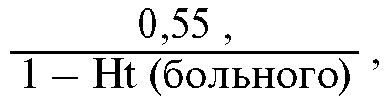 представляющий
собой соотношение удельных объемов плазмы в норме и у анализируемого
больного. Βлияние гематокрита на ПО и его производные (процент
протромбинового индекса и МНО) значительно сложнее и не может быть
скорригировано с помощью простого фактора.
представляющий
собой соотношение удельных объемов плазмы в норме и у анализируемого
больного. Βлияние гематокрита на ПО и его производные (процент
протромбинового индекса и МНО) значительно сложнее и не может быть
скорригировано с помощью простого фактора.
2.5.4. АКТИВИРОВАННОЕ ЧАСТИЧНОЕ ТРОМБОПЛАСТИНОВОЕ ВРЕМЯ
Β основе этого теста лежит определение времени свертывания плазмы в присутствии активатора контактной фазы и фосфолипидов (частичного тромбопластина) после добавления CaCl2. Β качестве активатора контактной фазы ранее широко использовалась суспензия каолина. Β последнее время предпочтение отдают эллаговой кислоте или микрочастицам окиси кремния. Эти реактивы не требуют постоянного перемешивания; они оптически прозрачные, что позволяет использовать любые типы коагулометров. Β качестве фосфолипидов, заменяющих собой фактор III тромбоцитов, чаще всего используют спиртовой экстракт ткани мозга, состоящий в основном из кефалина.
АЧТВ - тест на внутренний путь свертывания и зависит от всех факторов, за исключением VII и XIII. Его удлинение наблюдают при:
1) дефиците/аномалии факторов;
2) терапии гепарином и в меньшей степени антагонистами витамина К;
3) наличии патологических ингибиторов:
• полимеризации фибрина (продукты деградации фибрина, миеломные белки);
• инактивирующих факторов.
Следует отметить, что в тесте на АЧТВ реализуется целый набор реакций, включающих прямые (последовательные), шунтирующие и взаимоускоряющиеся, которые в конечном счете приводят к резкому нарастанию образования тромбина и полимеризации фибрина. Поэтому заметное превышение величины АЧТВ по сравнению с верхней границей нормального распределения наблюдают лишь при выраженном снижении уровня факторов (как правило, менее 30% от нормы).
Скорость процесса в таких сложных системах определяется в основном скоростью самой медленной реакции. В тесте определения АЧТВ такова реципрокная активация тромбином фактора VIII, протекающая в растворе и эффективно ингибируемая комплексом антитромбин III-гепарин. Это делает АЧТВ удобным показателем лабораторного контроля интенсивности гепарино-терапии. Недостаток определения АЧТВ (как метода контроля гепаринотерапии) - зависимость этого теста от многих факторов, что при некоторых патологических состояниях может приводить к несоответствию между истинной концентрацией гепарина в крови и ожидаемой, исходя из данных АЧТВ. Например, в послеоперационном периоде и при активации воспалительных реакций повышение уровня фактора VIII и фибриногена, фибронектина, факторов системы комплемента и др. может уменьшать влияние гепарина на АЧТВ, а дефицит факторов и наличие аномальных антикоагулянтов - усиливать его эффект. Очевидно, что влияние гепарина на эту систему зависит также и от уровня антитромбина III.
Удлинение АЧТВ при нормальном значении протромбинового времени может быть обусловлено дефицитом факторов XII, XI, IX, VIII, а также прекалликреина и ВМК или наличием ингибиторов к этим факторам. Простейший способ дифференцировки дефицита от присутствия ингибиторов - исследование коррекции АЧТВ после
добавления к исследуемой плазме небольшого количества нормальной. В случае дефицита факторов добавление уже 10-25% объема нормальной плазмы к исследуемой приводит к значительному сокращению (нормализации) АЧТВ, что не наблюдается при наличии ингибитора(ов) к факторам. Правда, если ингибитор медленно действующий (так называемый прогрессивный), то для его выявления смесь плазм необходимо выдержать в течение 1-2 ч до начала измерения. Длительная инкубация исследуемой плазмы с нормальной повышает вероятность выявления патологических ингибиторов свертывания. Наиболее часто ингибиторы к факторам свертывания выявляют у больных гемофилией А (антифактор VIII:C), находящихся на заместительной терапии, и при аутоиммунных заболеваниях. В последнем случае это иммуноглобулины, влияющие на взаимодействие факторов систем свертывания и противосвертывания с фосфолипидами.
На основе АЧТВ разработаны методы количественного определения факторов XII, XI, IX и VIII. В так называемых одностадийных тестах определение этих факторов проводят по калибровочным графикам, которые строятся с использованием смеси дефицитной по исследуемому фактору плазмы и разведений в геометрической прогрессии стандартной (с известным содержанием фактора) плазмы. Из-за каскадной природы реакций теста зависимость времени свертывания от содержания фактора имеет экспоненциальный характер и для линеализации графиков используют логарифмирование. Обычно линеализацию удается получить при содержании/активности фактора в смеси от 1 до 10% от нормы. Для наиболее точного определения уровня фактора у больного желательно подобрать такое разведение исследуемой плазмы, которое обеспечивает время свертывания вблизи средней области калибровочного графика. В области высокой концентрации фактора снижение точности определения обусловлено увеличением относительного вклада других реакций в суммарную скорость теста, а в области низкой концентрации фактора - увеличением вклада остаточной активности фактора в дефицитной плазме. Остаточная активность обусловлена не только примесью фактора, которая может быть очень низкой в плазме, очищенной на колонках с АТ, но и наличием шунтирующих (обходных) путей активации.
Серьезная и пока нерешенная проблема использования АЧТВ в диагностике - стандартизация теста. Очевидно, что стандартизировать тест, который определяет набор реакций и зависит от многих факторов, крайне сложно или, что вполне вероятно, невозможно.
Реактивы, выпускаемые разными производителями, отличаются не только по разбросу значений, определяемых в норме, но и по чувствительности к ингибиторам и дефициту факторов. Лаборатории целесообразно подобрать в соответствии со специализацией наиболее подходящий реактив, используемый в качестве основного, и иметь некоторое количество специализированного реактива, обладающего, например, повышенной чувствительностью к волчаночному антикоагулянту.
2.5.5. ТРОМБИНОВОЕ ВРЕМЯ
Этот тест относят к числу скрининговых, и его необходимо выполнять, если результаты ПТ или АЧТВ превышают нормальные значения. В этом случае определение времени свертывания плазмы после добавления стандартного количества тромбина может сузить диапазон поиска дефектного звена. Удлинение ТВ наблюдают при:
1) аномалии фибриногена - выраженная (< 0,3 г/л) гипофибриногенемия, дисфибриногенемия;
2) антикоагулянтной терапии гепарином или прямыми ингибиторами тромбина (гирудин, гирулог, синтетические ингибиторы), но не антагонистами витамина К;
3) наличии патологических ингибиторов полимеризации фибрина:
• продуктов деградации фибриногена/фибрина;
• аномальных антикоагулянтов (миеломных белков). Нормальные значения ТВ зависят от активности тромбина. Для
наиболее часто используемых растворов, содержащих три или шесть МЕ тромбина/мл, нормальные значения находятся соответственно в пределах 10-13 и 17-22 с. Чем ниже концентрация тромбина, тем выше чувствительность теста к аномалиям. В связи с этим выбор варианта постановки теста на определение ТВ определяется задачей исследования. Для диагностики продуктов расщепления фибриногена, гипофибриногенемии или дисфибриногенемии целесообразнее использовать реактив меньшей активности. В то же время этот тест обладает очень высокой чувствительностью к гепарину и при использовании реактива с активностью 17-22 с ТВ выходит за пределы определения даже при терапии низкими дозами гепарина. В связи с этим для контроля гепаринотерапии более подходят реактивы высокой активности. На чувствительность теста к гепарину влияют также рН и состав буфера, используемого для стабилизации тромбина.
В контроле гепаринотерапии АЧТВ и ТВ в большей степени взаимодополняют друг друга, чем служат альтернативными тестами. К преимуществам ТВ относят то, что на результат этого теста влияет меньшее число факторов, чем на АЧТВ. Он более специфично, чем АЧТВ, отражает активность комплекса антитромбин III-гепарин в плазме. В то же время с помощью АЧТВ контролируют влияние гепарина на активацию свертывания, что представляется более важным и могло бы быть аргументом в пользу бесспорного преимущества этого теста перед ТВ. Однако активация свертывания и антикоагулянтный эффект гепарина в условиях проведения АЧТВ, так же как и антикоагулянтный эффект гепарина в ТВ, существенно отличаются от таковых при тромбообразовании in vivo. Другими словами, корреляция между антикоагулянтными эффектами гепарина in vitro и антитромботической активностью in vivo пока оставляет желать лучшего. Диагностическая значимость этих тестов в контроле гепаринотерапии ограничивается информацией о приблизительном уровне циркулирующих в крови комплексов гепарина с антитромбином III, что может служить основанием для изменения в соответствии с клиническими показаниями режима гепаринотерапии. Снижение чувствительности к гепарину в ходе терапии может свидетельствовать о продолжающейся активации свертывания (тромбоцитов) и потреблении антитромбина III.
Более точное определение гепарина возможно по ингибированию тромбина или фактора Xa (последний тест используют также и для определения гепарина низкой молекулярной массы) при измерении их остаточной активности с помощью хромогенных субстратов. Однако стоимость этих тестов выше, чем ТВ или АЧТВ, а данных о более высокой диагностической значимости точного определения гепарина в клинических исследованиях не получено. Бесспорное преимущество этих тестов - возможность определения очень высоких концентраций гепарина, используемых при искусственном кровообращении. Альтернативный экспресс-метод мониторирования гепаринотерапии высокой интенсивности - определение активированного времени свертывания (АВС). В этом тесте определяют время свертывания цельной крови после внесения в пробирку, содержащую стандартное количество активатора - измельченной диатомовой земли. Этот активатор вызывает лизис клеток крови и выброс фактора четырех тромбоцитов, что значительно снижает антикоагулянтный эффект гепарина и обеспечивает свертывание даже в присутствии исходно высокой концентрации гепарина. На основании АВС разработаны номограммы для
расчета количества протаминсульфата, необходимого для нейтрализации циркулирующего гепарина после завершения операции.
2.5.6. РЕПТИЛАЗНОЕ ВРЕМЯ
Рептилаза (батроксобин) - тромбиноподобный фермент, выделяемый из яда змеи Bothrops atrox. В отличие от тромбина этот фермент не активирует тромбоциты, не инактивируется плазменными ингибиторами тромбина и гирудином. Поэтому он нечувствителен к гепарину. От фибриногена рептилаза отщепляет только фибринопептид А. Удлинение РВ наблюдают при:
1) аномалии фибриногена - выраженная (< 0,3 г/л) гипофибриногенемия, дисфибриногенемия;
2) наличии патологических ингибиторов полимеризации фибрина:
• продуктов разрушения фибриногена/фибрина;
• аномальных антикоагулянтов (миеломных белков). Известны редкие случаи, например фибриноген «Мадебург I» и
специфически инактивирующие тромбин иммуноглобулины, при которых может быть удлинено ТВ, но не РВ. Отсутствие зависимости от гепарина позволяет мониторировать с помощью РВ динамику фибриногена и продуктов его разрушения при тромболизисе и лечении ДВС антикоагулянтами. Рептилаза может использоваться также для получения сыворотки из гепаринизированной крови для количественного определения продуктов разрушения фибриногена/фибрина.
2.5.7. ОПРЕДЕЛЕНИЕ ФИБРИНОГЕНА
В число скрининговых тестов должно входить определение фибриногена. Важность этого теста определяется двумя причинами. Во-первых, повышение концентрации фибриногена - фактор риска заболеваний ССС, по значимости сравнимый с гиперхолестеринемией. Во-вторых, фибриноген - белок острой фазы, поэтому в ряде случаев может оказаться необходимым наблюдение за его динамикой. Предложено несколько неравноценных подходов к определению фибриногена, и вполне реальной может оказаться ситуация, когда для диагностики потребуется применение двух различных методов. Применяемые методы могут быть разделены на три группы: функциональные (наиболее универсальные и должны занимать ведущее место в диагностике); паракоагуляционные; иммунологические. Каждый из подходов имеет свои преимущества и недостатки.
Метод Клаусса. Из функциональных тестов наиболее часто используют метод Клаусса. Метод основан на том, что при добавлении избытка тромбина к разбавленной плазме (обычно в разведении 1 : 10) скорость образования сгустка пропорциональна концентрации фибриногена. Как правило, в реактив добавляют инактиватор гепарина. Метод Клаусса можно использовать для определения фибриногена и у больных, находящихся на стандартной гепаринотерапии*, но при очень высоком уровне гепаринизации, применяемой при экстракорпоральном кровообращении, концентрации инактиватора может оказаться недостаточно. К достоинствам метода относят то, что он позволяет быстро получить результат, и поэтому с равным успехом его можно использовать как для серийных, так и для одиночных экспресс-анализов.
Основной недостаток метода Клаусса - высокая чувствительность к ингибиторам полимеризации фибриногена (продуктам разрушения фибрина и др.), способным занижать результат. К другим недостаткам метода относят сравнительно узкий рабочий диапазон, зависящий от типа коагулометра (для большинства приборов максимальная концентрация отличается от минимальной лишь в три раза). Поскольку вариабельность уровня фибриногена у больных значительно больше, чем рабочий диапазон метода Клаусса, то часто приходится повторять определение с использованием другого разведения плазмы.
Метод, основанный на фотометрическом определении скорости преципитации дезАА-фибрина. Относительно недавно был предложен новый метод определения фибриногена, основанный на фотометрическом (турбодиметрическом) определении скорости преципитации дезАА-фибрина, образующегося под действием рептилазы. Основные достоинства этого метода - отсутствие чувствительности к применяемым в клинике ингибиторам тромбина, меньшая, чем у метода Клаусса, чувствительность к ингибиторам полимеризации, более широкий рабочий диапазон (от 0,8 до 8 г/л) и возможность применения на любом фотометре, включая автоматические биохимические анализаторы. Важное достоинство этого метода (реализуемое лишь при определении на регистрирующем фотометре) - возможность получения в тесте данных не только о концентрации фибриногена,
* Опытом применения метода Клаусса для определения фибриногена у больных, получающих прямые ингибиторы тромбина (гирудин, аргатробан и др.) мы не располагаем. Вполне вероятно, что благодаря высокой концентрации тромбина в реактиве эти препараты в терапевтических концентрациях не окажут заметного влияния на результат теста
но и об активации фибриногенолиза. Накопление в плазме ранних продуктов разрушения фибриногена (в основном Х-фрагментов) приводит к увеличению продолжительности лагфазы реакции.
Методы на основе определения свертываемого белка.
Некоторые тесты на фибриноген могут быть объединены в группу методов, позволяющих определить содержание свертываемого белка. Фибриноген в стандартном объеме плазмы свертывается (чаще всего тромбином, но могут использоваться рептилаза, Са-тромбопластин и другие активаторы), а количество свернувшегося белка определяют фотометрически или взвешиванием после высушивания. Ранее для фотометрического определения сгусток отмывали в 0,9% растворе NaCl от других белков плазмы, затем растворяли в NaOH и измеряли А280 или белок по биуретовой реакции. Эти методы трудоемкие, требуют большого количества плазмы и из-за большого количества ручных операций увеличивают вероятность инфицирования персонала.
Усовершенствованный вариант этого подхода к определению фибриногена - измерение оптической плотности или светорассеяния до и после свертывания плазмы; разница нарастает при увеличении концентрации фибриногена в плазме. В некоторых современных анализаторах этот принцип измерения лежит в основе одновременного определения фибриногена с ПТ, АЧТВ или ТВ.
Недостаток этих методов - отсутствие учета скорости полимеризации, поэтому аномалии фибриногена, заключающиеся в снижении скорости полимеризации, могут быть не обнаружены. Очевидно также, что при значительном (по сравнению с нормой) увеличении времени свертывания в скрининговом тесте выданный результат определения фибриногена подчас занижен, так как нарастание светорассеяния может не достичь плато за заданное анализатору время определения.
Преципитационные методы, основанные на осаждении фибриногена с последующим определением объема осадка или величины светорассеяния, наименее специфичны, их используют в крайнем случае.
Иммунологические методы. Большое разнообразие возможностей определения фибриногена (и что очень важно, его производных) представляют иммунологические методы. Эти методы можно использовать для качественного, полуколичественного (при анализе разведений образца) и количественного определения. Трудоемкость и время, необходимые для выполнения анализа, определяются применяемой методикой. Для экспресс-анализов могут применяться качественные методы и методы, основанные на нефелометрическом
определении комплексов Аг-АТ. Прост метод иммунодиффузии, однако он требует значительного времени для проведения. Наиболее высокую чувствительность обеспечивает ИФА. Применение комбинации двух АТ, выработанных к разным антигенным детерминантам, и использование в анализе системы усиления сигнала открывают практически неограниченные возможности для этого метода. Однако из-за длительности анализа и необходимости построения калибровочного графика (обычно нелинейного и поэтому требующего набора стандартов) в каждой серии определений этот метод малопригоден для одиночных анализов. Правда, методики быстро совершенствуются и уже появляются варианты ИФА, применимые и для одиночных определений.
2.5.8. ОПРЕДЕЛЕНИЕ ВОЛЧАНОЧНЫХ АНТИКОАГУЛЯНТОВ
Подкомитетом по волчаночным антикоагулянтам/АТ к фосфолипидам Международного общества по тромбозам и гемостазу в 1995 г. были внесены поправки в предложенные ранее четыре основных критерия для определения волчаночных антикоагулянтов (ВА). Согласно новым рекомендациям, об определении ВА можно судить, если наблюдают каждый из следующих признаков:
1) удлинение как минимум одного из фосфолипидзависимых тестов свертывания;
2) наличие ингибирования в тестах по смешиванию плазмы больного с пулом нормальных плазм;
3) доказана зависимость ингибирования от фосфолипидов. Для этого может использоваться изменение концентрации или состава фосфолипидов;
4) тщательно отдифференцированы ВА от других лабораторных показателей при других коагулопатиях. Для этого могут оказаться необходимыми специфические исследования факторов свертывания.
AT к фосфолипидам обнаруживают при многих заболеваниях, поэтому определение ВА может быть актуальной задачей в клиниках широкого профиля. Сложность проблемы заключается в том, что не существует ни одного метода, который обеспечивал бы близкую к 100% чувствительность тестов. Это обусловлено как разнообразием свойств образующихся АТ, которые теоретически могут ингибировать любую из фосфолипидзависимых реакций, так и сложной природой реакций системы гемостаза.
СКРИHИHΓОΒЫЕ ТЕСТЫ ДЛЯ ОПРЕДЕЛЕНИЯ ΒА
1. Тесты свертывания ядом змеи Рассела (Vipera russelli) или тайпана (Oxyuranus scutellatus), содержащих ферменты, активирующие на поверхности фосфолипидов фактор X и протромбин соответственно;
2. АЧТΒ с увеличенным временем инкубации и/или разбавленным реактивом;
3. Тест с разбавленным тромбопластином (ингибирование тромбопластина);
4. Каолиновое (или силикатное) время.
Если хотя бы один из двух скрининговых тестов (ПТ и АЧТΒ) позволяет выявить удлинение времени свертывания, а ТΒ нормальное, то это можно рассматривать как важный аргумент в пользу подозрений на ΒА. Учитывая невысокую чувствительность к ΒА скрининговых тестов в стандартной постановке, при всяком подозрении на наличие таких АТ необходимо выполнение четырех скрининговых тестов, обладающих более высокой чувствительностью к ΒА. Порядок выполнения может соответствовать порядку перечисления (тесты расположены в порядке снижения чувствительности или специфичности), но не обязательно. При получении первого аномального результата необходимо перейти от скрининговых к подтверждающим тестам, которые должны показать или исключить связь с зависимостью от фосфолипидов. Для дифференцировки между этой патологией и дефицитом факторов и/или наличием ингибиторов к ним необходимо выполнение исследования на смеси плазмы больного с донорской с применением метода, выявившего аномалию. Βозможная схема исследования представлена на рис. 2.11.
Чаще всего плазму больного и нормального донора смешивают в равной пропорции. Увеличение доли плазмы больного в смеси повышает чувствительность, но снижает специфичность. За отсутствие коррекции принимают результат, превышающий средние значения теста на два-три стандартных отклонения. Предложена следующая формула для вычисления индекса коррекции тестов каолинового времени и ингибирования тромбопластина:
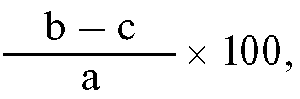 где a - время свертывания плазмы больного; b - время свертывания смеси плазм, c -
время свертывания нормальной плазмы. Значения индекса коррекции,
превышающие 15, рассматривают как свидетельство в пользу ΒА. Примерно в
1/3 выявляемых случаев ΒА проявляются
где a - время свертывания плазмы больного; b - время свертывания смеси плазм, c -
время свертывания нормальной плазмы. Значения индекса коррекции,
превышающие 15, рассматривают как свидетельство в пользу ΒА. Примерно в
1/3 выявляемых случаев ΒА проявляются
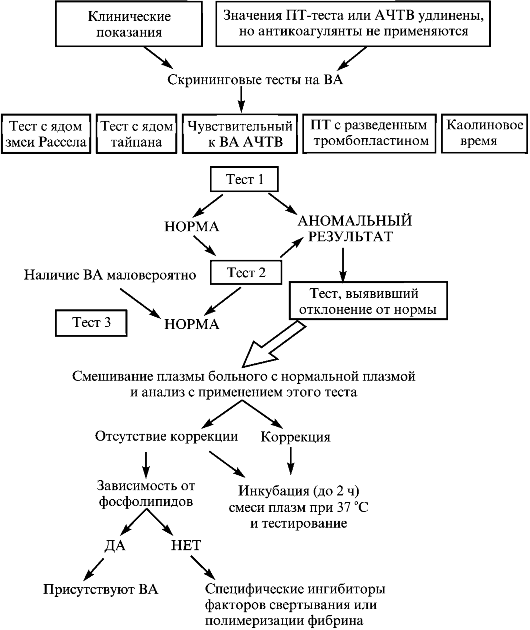 Рис. 2.11. Примерная
схема анализов при подозрении на наличие волчаночного антикоагулянта.
Для исключения необходимо выполнение не менее двух скрининговых тестов
Рис. 2.11. Примерная
схема анализов при подозрении на наличие волчаночного антикоагулянта.
Для исключения необходимо выполнение не менее двух скрининговых тестов
как прогрессивные ингибиторы, и для их выявления необходима длительная (до 2 ч) инкубация смеси плазм при 37 °С. В этом случае в качестве контрольной плазмы используют смесь, приготовленную из этих плазм, инкубировавшихся раздельно в этих же условиях.
Если в экспериментах по смешиванию получены данные в пользу ВА, то следующим этапом исследования должно быть доказательство связи патологии с зависимостью от фосфолипидов. Для этого может выполняться исследование нейтрализации ингибитора синтетическими фосфолипидами или лизатами тромбоцитов, получаемых замораживанием и оттаиванием суспензии клеток. Плазму больного смешивают с равным объемом лизатов, а в качестве контроля используют плазму больного, разведенную в два раза раствором 0,9% NaCl.
2.5.9. СПЕЦИАЛЬНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ КОМПОНЕНТОВ СИСТЕМЫ ГЕМОСТАЗА
В зависимости от результатов скрининговых тестов или клинических показаний может оказаться необходимым исследование отдельных компонентов системы гемостаза. Применяемые методы могут быть разделены на функциональные, основанные на определении специфической активности компонента, и иммунологические, в которых измеряют концентрацию Аг. Иммунологические методы незаменимы в тех случаях, когда исследуемый компонент не обладает легко измеряемой функциональной активностью. Кроме того, возможность применения в анализе комбинации двух АТ, выработанных к разным антигенным детерминантам, и использование системы усиления сигнала открывают практически неограниченные возможности этих методов. Именно благодаря иммунологическим методам существенно расширились возможности исследования активации системы гемостаза in vivo.
Большинство факторов системы свертывания может быть определено с использованием дефицитной по исследуемому фактору плазмы и одного из двух глобальных тестов. АЧТВ обычно применяют для определения факторов XII, XI, IX и VIII, а Са-тромбопластиновый тест - для определения факторов VII, X, V и протромбина. Особые подходы применяют для исследования факторов (фФВ и XIII), не определяемых в глобальных тестах.
Нелинейный характер зависимости времени свертывания от концентрации исследуемого фактора в этих тестах требует построения калибровочных графиков с использованием геометрического разведения стандарта, обычно от 1:10 до 1:100 и более. Для повышения точности определения рекомендуют использование разных разведений исследуемой плазмы. Это обусловлено тем, что в области высоких концентраций фактора отмечают низкую чувствительность, а в области
низких концентраций возможно завышение из-за вклада остаточной активности или шунтирующих реакций. Особое внимание этому следует уделить при анализе с использованием Са-тромбопластина. Одна из причин высокой активности тромбопластина - наличие в препарате, получаемом из тканей, факторов свертывания. Пологий характер калибровочного графика свидетельствует о необходимости замены тромбопластина.
Определение фФВ возможно по его кофакторной активности в индуцируемой ристоцетином агрегации тромбоцитов. Для количественного определения используют иммунологические методы или сравниваются скорости агрегации отмытых от плазмы тромбоцитов, суспендированных в разведениях исследуемой и нормальной плазмы. Можно использовать не только свежевыделенные тромбоциты, но и клетки, фиксированные 2% раствором формальдегида.
Для полуколичественного определения фактора XIII (более точного определения обычно не требуется) к разведениям нормальной плазмы и плазмы больного добавляют очищенный (не содержащий фактора XIII) фибриноген, тромбин и CaCl2. После инкубации в течение 30 мин при 37 °С сгусток аккуратно отделяют от стенок, в пробирку вносят 8-кратный объем 5% монойодуксусной кислоты и через 5 мин инкубации при 37 °С проверяют наличие сгустков. Содержание фактора XIII вычисляют как соотношение максимального разведения плазмы больного к максимальному разведению контрольной плазмы, при которых в пробирке обнаруживают сгусток.
Значительный прогресс в исследованиях компонентов системы гемостаза был обеспечен разработкой хромогенных субстратов, представляющих собой р-нитроанилиды, как правило, трипептидов. Протеазы отщепляют от этих субстратов р-нитроанилин, обладающий достаточно высоким поглощением при 405 нм, что обеспечивает возможность прямого и простого определения большинства протеаз системы гемостаза, их кофакторов и ингибиторов.
К несомненным преимуществам хромогенных субстратов относят также возможность исследования отдельных стадий последовательных превращений при минимальном влиянии других реакций, чего трудно избежать в глобальных тестах, где измеряют конечные стадии - образование или растворение фибрина.
В то же время взаимодействие хромогенных субстратов с протеазами в отличие от белковых определяется в основном структурой активного центра. Поэтому амидолитическая и протеолитическая
активность протеаз может существенно различаться. Например, β- и γ-тромбины, а также комплекс тромбин-тромбомодулин обладают сходной с α-тромбином амидолитической активностью, но их свертывающая активность в отношении фибриногена на два порядка ниже, чем у α-тромбина. Комплексы протеаз с α2-макроглобулином не обладают протеолитической активностью, но расщепляют хромогенные субстраты. Плазмин и комплекс стрептокиназа-Пг обладают сходной специфичностью в отношении хромогенных субстратов, но значительно отличаются по протеолитической активности. Эти примеры свидетельствуют о том, что протеазы, как правило, менее требовательны к хромогенным субстратам, чем к белковым. Можно привести и пример другого рода. Одноцепочечная урокиназа расщепляет Пг, хотя и с низкой скоростью, но не обладает амидолитической активностью.
На основе хромогенных субстратов разработаны методы определения большинства компонентов системы гемостаза, а также и варианты двух глобальных коагуляционных тестов - протромбинового и АЧТВ. Однако преимущества хромогенных субстратов наиболее заметно выявляются при определении компонентов систем противосвертывания и фибринолиза.
2.5.10. АНТИТРОМБИН III
Антитромбин III определяют по ингибированию тромбина или фактора Xa. Принцип метода состоит в определении остаточной активности фермента после инкубации в присутствии гепарина избытка фермента с разведенной плазмой. Определение остаточной активности тромбина возможно с помощью хромогенных субстратов (фотометрические методы) или по скорости свертывания фибриногена (коагулометрические методы). К преимуществам определения по ингибированию фактора Xa относят отсутствие влияния на результат ГК-II. Его недостаток - необходимость добавления в состав реактивов гирудина или синтетических ингибиторов тромбина.
При определении антитромбина III по ингибированию тромбина желательно использовать фермент из плазмы быка, так как он более стабилен и менее чувствителен к ГК-II. Расчеты проводят по калибровочным графикам, для построения которых используют разведения стандартной плазмы. При фотометрических определениях обычно достаточно трех разведений плазмы, соответствующих, например, 100, 50 и 25% от нормы. Из-за нелинейного характера зависимости времени свертывания фибриногена от остаточной
активности тромбина для калибровки коагулометрического метода необходимо большее количество разведений плазмы.
При фотометрических определениях альтернативным вариантом расчета служит определение исходной активности фермента, из которой вычитают остаточную активность, а разницу умножают на коэффициент, представляющий собой дробь (числитель - активность ингибитора в референтной плазме, знаменатель - разница между исходной активностью фермента, измеренной после инкубации фермента с 0,9% NaCl вместо плазмы, и активностью, измеренной после инкубации фермента с референтной плазмой).
К недостаткам этого метода относят то, что из-за высокой стоимости хромогенных субстратов производители реактивов обычно дают минимально возможную концентрацию субстрата. Вследствие этого измеренная величина исходной (максимальной) активности фермента может быть ниже истинной, что приведет к занижению всех результатов определения. В то же время при построении калибровочного графика максимальное разведение плазмы можно ограничить точкой, соответствующей 25% содержания антитромбина III. Остаточная активность тромбина при этом будет ниже исходной и ближе к истинному значению (субстрат меньше израсходуется за время реакции), что повысит точность определения.
2.5.11. ПРОТЕИН С
Наиболее удобный метод определения ПрС - измерение его функциональной активности после активации ферментом из яда Agkistrodon contortrix. Этот фермент, получивший название «протак», оказался очень удобным инструментом для определения ПрС. Высокоочищенные препараты этого фермента активируют ПрС в отсутствие Са2+ с высокой скоростью, но не расщепляют ни такие белки плазмы, как фибриноген и факторы V и VIII, ни хромогенные субстраты ПрС. Это позволяет использовать протак как для коагулометрического, так и для фотометрического определения ПрС.
Коагулометрическое измерение ПрС основано на определении остаточной активности фактора VIII в субстратной плазме после инкубации с исследуемой плазмой в присутствии активатора, состоящего из АЧТВ-реактива и протака. Смесь достаточно сложная, и точность определения во многом зависит от строгого соблюдения условий реакции. Кроме того, высокое содержание фактора VIII в исследуемой плазме может приводить к занижению результата определения.
Более просты метод активации ПрС протаком и прямое определение ПрСа по величине амидолитической активности. Однако протак активирует и неполноценный ПрС, образующийся в условиях дефицита витамина К, что может приводить к несоответствию между активностью ПрСа, определяемой этим методом, и его истинной антикоагулянтной активностью. Нельзя исключить также и возможность несоответствия амидолитической и антикоагулянтной активности ПрСа при некоторых генетических дефектах ПрС. В этом плане фотометрический метод определения ПрС, активированного протаком, следует рассматривать как полуфункциональный и по значимости сопоставимый с методами ИФА.
РЕЗИСТЕНТНОСТЬ К АКТИВИРОВАННОМУ ПРОТЕИНУ С
Для определения резистентности к ПрСа исследуют влияние добавления стандартного количества ПрСа на АЧТВ плазмы больного. При необходимости определения резистентности к ПрСа у больных, находящихся на лечении гепарином или антагонистами витамина К, плазму больного разводят дефицитной по фактору V плазмой.
В подавляющем большинстве врожденных случаев резистентность к ПрСа обусловлена мутацией по 1691 нуклеотиду (G→A) гена фактора V. Для обнаружения этой мутации из лейкоцитов экстрагируют ДНК и фрагмент, содержащий участок мутации, амплифицируют в полимеразной цепной реакции. Синтезированная ДНК расщепляется рестриктазой и анализируется с помощью ЭФ. Наличие замены G- A приводит к изменению числа и размера образующихся фрагментов.
2.5.12. ПРОТЕИН S, ПЛАЗМИНОГЕН, а2-АНТИПЛАЗМИН И ТКАНЕВЫЙ АКТИВАТОР ПЛАЗМИНОГЕНА
Определение ПрS возможно с помощью ИФА и коагулометрического теста, основанного на определении его кофакторной активности в инактивации фактора Va под действием ПрСа. Активированный фактор Va характеризуется низкой стабильностью, а в условиях in vitro ПрS ускоряет его инактивацию лишь в 2-3 раза. Поэтому точное определение ПрS в коагулометрическом тесте требует очень строгого соблюдения условий реакций.
В зависимости от специфичности применяемых АТ с помощью ИФА может определяться как свободный, так и суммарный (свободный + связанный с C4b-связывaющим белком комплемента) ПрS.
Наиболее удобный метод определения Пг - измерение амидолитической активности комплекса Пг-стрептокиназа. Стрептокиназа связывает Пг с высоким сродством и открывает в нем активный центр, расщепляющий хромогенные субстраты. К уникальным свойствам человеческого Пг относят высокую устойчивость его комплексов со стрептокиназой к белковым ингибиторам плазмы, что в большинстве случаев позволяет прямо определять Пг по скорости расщепления хромогенного субстрата плазмой после добавления избытка стрептокиназы. Для расчета используют калибровочный график или произведение значения ΔА405/мин и коэффициента. Способ расчета принципиального значения не имеет. Однако в некоторых случаях амидолитический метод определения Пг может давать ошибочные результаты.
Основная проблема - завышение результата определения в образцах плазмы с высоким уровнем фибриногена или продуктов его разрушения. Эту ошибку можно устранить введением в среду реакции очищенного (не содержащего Пг) фибриногена.
Более сложная проблема - точное определение Пг при тромболизисе. В первые часы тромболитической терапии содержание Пг (профермента) резко снижается, а вклад его производных (комплексов со стрептокиназой и/или плазмин-α2-макроглобулин) в суммарную амидолитическую активность плазмы увеличивается. При тромболизисе урокиназой или тАП основным считают вклад комплексов с α2-макроглобулином, который может быть легко учтен путем вычитания из суммарной активности той активности, которая измеряется после замены стрептокиназы соответствующим буферным раствором.
Для определения α2-АП к плазме добавляют избыток плазмина, а остаточную активность фермента измеряют с помощью хромогенных субстратов. Инактивация плазмина α2-АП протекает очень быстро. Для уменьшения относительного вклада других ингибиторов желательно сокращать до минимума время инкубации плазмы с плазмином и определять начальную скорость хромогенной реакции. Для расчетов лучше использовать калибровочный график, построенный с использованием трех разведений плазмы. Стабильность плазмина в буферах с нейтральными или щелочными значениями рН невысокая. Для повышения стабильности плазмина в буферы часто добавляют глицерин. Работа с такими растворами, обладающими высокой вязкостью и плотностью, требует особой аккуратности при пипетировании и тщательного перемешивания после внесения в кювету.
Определение активности тАП в плазме больных - сложная задача. Сложность обусловлена двумя причинами. Во-первых, в норме концентрация тАП в плазме ниже, чем его быстродействующего ингибитора, и активатор, секретируемый эндотелием, быстро нейтрализуется. Во-вторых, кроме тАП, Пг могут активировать урокиназа, компоненты контактной фазы системы свертывания и фактор XIa. Хотя скорость активации Пг этими компонентами намного ниже, чем тАП или урокиназой, их концентрация в плазме значительно выше, чем активаторов, поэтому они могут вносить существенный вклад в определяемую in vitro активацию Пг.
Для определения тАП кровь немедленно после забора смешивают с 1/2 объема 0,5 М Na-ацетатного буфера рН 3,9 и центрифугируют. Закисление предотвращает инактивацию тАП. Прямое определение тАП в плазме с использованием хромогенных субстратов невозможно из-за низкого содержания активатора и невысокой селективности субстратов. Для повышения чувствительности и специфичности закисленную плазму вносят в буфер, содержащий Пг, кофактор (фибринмономеры или BrCN-фрагменты фибриногена) и хромогенный субстрат. В смеси одновременно протекают две реакции: образование плазмина и расщепление образующимся плазмином хромогенного субстрата. Графики нарастания А405 имеют вид параболы. Их можно линеализовать в координатах А405 от t2 и по наклону определить скорость образования плазмина, но обычно определяется увеличение поглощения за фиксированный промежуток времени (~1 ч). Активность тАП выражают в международных единицах (МЕ/мл), вычисляемых как соотношение ΔА405 в исследуемых образцах/ ΔА405 стандарта тАП, умноженное на активность стандарта.
2.5.13. ИНГИБИТОР АКТИВАТОРОВ ПЛАЗМИНОГЕНА
Для определения активности ИАП исследуемую плазму инкубируют в течение 10 мин с избытком активатора. Затем смесь закисляют добавлением равного объема 0,5 М Na-ацетатного буфера рН 3,9, а остаточную активность активатора определяют так, как описано выше. Активность ИАП выражают в условных единицах (УЕ/мл). За 1 УЕ принимают такое количество ингибитора, которое нейтрализует 1 МЕ активатора. Для определения активности ИАП может использоваться как тАП (определяют только ИАП-1), так и урокиназа (определяют сумму ИАП-1 и ИАП-2). Для расчета активности ингибитора используют либо калибровочный график, либо формулу:
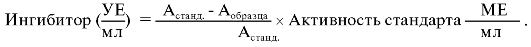 При
построении калибровочного графика ингибирование активатора моделируют
разведением стандарта дефицитной по ингибитору плазмой.
При
построении калибровочного графика ингибирование активатора моделируют
разведением стандарта дефицитной по ингибитору плазмой.
2.5.14. ИММУНОЛОГИЧЕСКИЕ МЕТОДЫ ИССЛЕДОВАНИЯ СИСТЕМЫ ГЕМОСТАЗА
На основе АТ разработаны как качественные и полуколичественные экспресс-методы, так и высокочувствительные системы ИФА. АТ фактически незаменимы для определения компонентов, не обладающих легко измеряемой активностью. Таковы, например, маркеры фибринообразования, расширяющие возможности неинвазивной диагностики венозных тромбозов и тромбоэмболий, и активации свертывания, позволяющие определить состояние гиперкоагуляции и контролировать эффективность терапии. Использовать ИФА очень удобно при серийных определениях большого количества образцов. Β то же время вряд ли стоит рекомендовать эти методы для определения тех компонентов, которые легко могут быть измерены с помощью функциональных тестов. Более того, уровень Аг фактора может не совпадать с уровнем его функциональной активности, что важно для исследования некоторых заболеваний. Примером могут быть тип II дефицита антитромбина III или ПрС, а также соотношение между Аг тАП и его активностью.
Ранний маркер активации фибринообразования - фибринмономер. Паракоагуляционные протамин-сульфатный и этаноловый тесты обладают низкой чувствительностью и специфичностью. Для более точного определения были предложены качественный метод, основанный на агглютинации эритроцитов, покрытых фибрин-мономерами, и количественный по кофакторной активности фибринмономера в активации Пг тАП метод.
Маркер сформированных тромбов в венозной системе, создающих угрозу тромбоэмболий, - D-димер. К важной характеристике этого маркера относят высокую достоверность положительного результата. D-димер в отличие от других маркеров активации не может образоваться вследствие ошибки при заборе крови (разумеется, если анализ не проводят спустя несколько часов после образования сгустка). Для полуколичественного определения D-димера используют тест по агглютинации частиц латекса, покрытых моноклональными
АТ, реагирующими только с областью сшивки D-доменов. Для этого теста используют плазму больного. Более быстрым и чувствительным является тест, основанный на применении биспецифических АТ, взаимодействующих с областью сшивки и поверхностным белком мембран эритроцитов. В присутствии D-димеров эти АТ вызывают агглютинацию собственных эритроцитов больного. Тест выполняют на цельной капиллярной крови, что позволяет получить результат через несколько минут после взятия крови.
Использование двух АТ, взаимодействующих с разными антигенными детерминантами, позволяет определять с помощью ИФА не только исходные белки, но и их фрагменты и нео-Аг, образующиеся в результате сшивания, расщепления или взаимодействия с ингибиторами.
Диапазон возможностей метода может быть продемонстрирован на примере фибриногена и его производных. С помощью ИФА можно определять: только интактный фибриноген (первые АТ взаимодействуют с С-концевым доменом α-цепей, вторые - с фибринопептидом А); активацию фибриногена тромбином - фибрин-мономер, или фибринопептид А; ранние продукты разрушения фибриногена плазмином (Х-фрагменты или пептид (β1-42); поздние продукты разрушения фибриногена (D- и E-фрагменты) и фибрина (D-димер).
Благодаря ИФА стало возможным определение: активированных форм факторов XII и XI; пептидов активации факторов IX, X, протромбина и ПрС; протеаз, инактивированных ингибиторами (тромбин-антитромбин III, ПрСа-ИАП-3/α1-антитрипсин, плазмин-α2-АП, тАП-ИАП-1). Естественно, что для селективного определения следов модифицированных белков при многократно превосходящей концентрации исходных форм необходимы АТ высокой специфичности.