Неврология и нейрохирургия / под ред. А.Н. Коновалова, А.В. Козлова ; Е.И. Гусев, А.Н. Коновалов, В.И. Скворцова : учебник : - т. 1. - 2009. - 624 с.
|
|
|
|
ГЛАВА 17. НЕРВНО-МЫШЕЧНЫЕ ЗАБОЛЕВАНИЯ
Наследственные нервно-мышечные заболевания - большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата (рис. 17.1, 17.2). Заболевание проявляется прогресирующими мышечными атрофиями, мышечной слабостью, парезами мускулатуры.
При диагностике учитывают возраст дебюта клинических проявлений, локализацию и прогрессирование миодистрофического процесса (наличие или отсутствие псевдогипертрофий, фасцикулярных подергиваний, эпизодов мышечной слабости, нарушений чувствительности), отягощенность семейного анамнеза, тип наследования заболевания.
К наследственным нервно-мышечным заболеваниям относят прогрессирующие мышечные дистрофии (первично-мышечные заболевания), наследственные мотосенсорные полинейропатии или невральные амиотрофии (заболевания с преимущественным первичным поражением двигательных волокон периферических нервов), а также спинальные амиотрофии (заболевания с первичным поражением мотонейронов спинного мозга).
17.1. Прогрессирующие мышечные дистрофии
В основе большинства миодистрофий лежат дефекты генов, кодирующих различные структурные белки мышечных волокон.
Прогрессирующая мышечная дистрофия Дюшенна
Заболевание связано с патологией гена, локализующегося на коротком плече Х-хромосомы в локусе Хр21 и ответственного за выработку дистрофина. До 60% всех случаев заболевания связано с делециями, в остальных случаях причинами заболевания являются дупликации или точечные мутации. Около 50% всех мутаций приходится на экзоны 5-20 или 45-53, что предположительно связано со структурой хроматина в этих областях. Продуктом гена является белок дистрофин, синтезирующийся в скелетных мышцах, миокарде и головном мозге. Дистро-
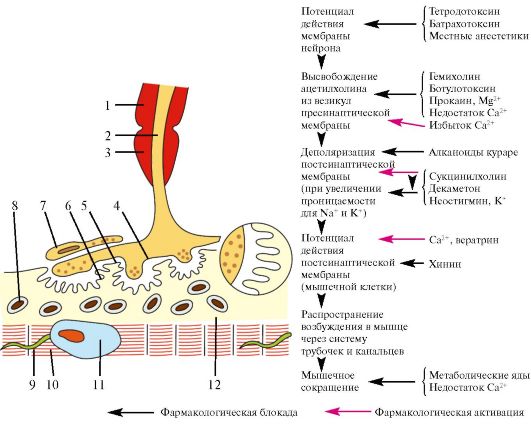 Рис. 17.1. Мионевральный
синапс. 1 - миелиновая оболочка; 2 - аксон; 3 - перехват Ранвье; 4 -
пресинаптическая мембрана; 5 - синаптическая щель; 6 - постсинаптическая
мембрана; 7- леммоцит (шванновская клетка); 8 - митохондрия; 9 -
поперечная система трубочек и канальцев; 10 - миофибриллы; 11 - ядро; 12
- саркоплазма
Рис. 17.1. Мионевральный
синапс. 1 - миелиновая оболочка; 2 - аксон; 3 - перехват Ранвье; 4 -
пресинаптическая мембрана; 5 - синаптическая щель; 6 - постсинаптическая
мембрана; 7- леммоцит (шванновская клетка); 8 - митохондрия; 9 -
поперечная система трубочек и канальцев; 10 - миофибриллы; 11 - ядро; 12
- саркоплазма
фин выполняет структурную функцию, а также различные модулирующие и сигнальные функции, связываясь с белками внеклеточного матрикса, плазматической мембраны, цитоскелета и других внутриклеточных структур. Отсутствие дистрофина в миофибриллах приводит к дезинтеграции дистрофингликанопротеинового комплекса, обеспечивающего структурно-функциональную организацию цитоскелета миофибрилл, утрате их устойчивости к циклическим актам сокращения и расслабления, разрывам. Его отсутствие повышает проницаемость мембраны для ионов кальция, что приводит к активации кальциевых протеаз, нарушению функционирования клетки и в конечном итоге к некрозу мышечных волокон.
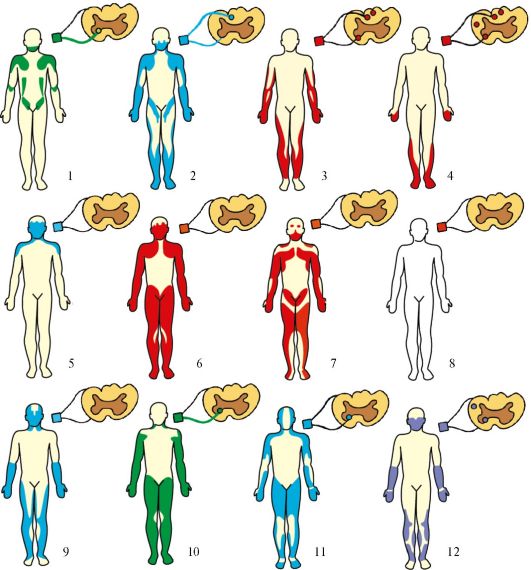 Рис. 17.2. Основные
типы мышечных атрофий. 1 - спинальная прогрессирующая детская
амиотрофия Верднига-Гоффманна; 2 - интерстициальная гипертрофическая
невропатия Дежерина-Сотта; 3 - невральная амиотрофия (прогрессирующая
невральная перонеальная мышечная атрофия Шарко-Мари-Тута); 4 - болезнь
Фридрейха (семейная спинно-мозжечковая атаксия); 5 - миастения; 6 -
пароксизмальная миоплегия (семейный периодический паралич); 7 -
прогрессирующая мышечная дистрофия; 8 - врожденная миотония Томсена
(неатрофическая); 9 - атрофическая миотония; 10 - врожденная амиотония
Оппенгейма; 11 - хроническая прогрессирующая семейная спинальная
амиотрофия взрослых Арана-Дюшенна; 12 - боковой амиотрофический склероз
Шарко. Зеленым цветом обозначен дебют заболевания в детстве, красным - в
подростковом возрасте, синим - в зрелом возрасте, фиолетовым - в
пожилом
Рис. 17.2. Основные
типы мышечных атрофий. 1 - спинальная прогрессирующая детская
амиотрофия Верднига-Гоффманна; 2 - интерстициальная гипертрофическая
невропатия Дежерина-Сотта; 3 - невральная амиотрофия (прогрессирующая
невральная перонеальная мышечная атрофия Шарко-Мари-Тута); 4 - болезнь
Фридрейха (семейная спинно-мозжечковая атаксия); 5 - миастения; 6 -
пароксизмальная миоплегия (семейный периодический паралич); 7 -
прогрессирующая мышечная дистрофия; 8 - врожденная миотония Томсена
(неатрофическая); 9 - атрофическая миотония; 10 - врожденная амиотония
Оппенгейма; 11 - хроническая прогрессирующая семейная спинальная
амиотрофия взрослых Арана-Дюшенна; 12 - боковой амиотрофический склероз
Шарко. Зеленым цветом обозначен дебют заболевания в детстве, красным - в
подростковом возрасте, синим - в зрелом возрасте, фиолетовым - в
пожилом
Мозговая изоформа экспрессируется в коре больших полушарий, гиппокампе и клетках Пуркинье. В головном мозге дистрофин участвует в процессах нейрональной пластичности, синаптической стабильности и интеграции сигнала на клеточном уровне. Кроме того, дистрофин принимает участие в нормальном функционировании глии. Снижение интеллекта, наблюдаемое примерно у 10-20% больных с формой Дюшенна, связано с нарушением синтеза этой изоформы дистрофина.
Частота заболевания составляет 3,3 на 100 000 населения, 14 на 100 000 родившихся. В большинстве случаев болеют мальчики. Случаи заболевания у девочек редки и возможны при кариотипе Х0, мозаицизме Х0/ХХ, Х0/ХХХ, Х0/ХХХ/ХХХ и при структурных аномалиях хромосом.
Патоморфология. Разрушение мышечных волокон, их замещение соединительной и жировой тканью.
Клинические проявления. Признаки заболевания проявляются на 1-3 годах жизни. Уже на 1-м году жизни обращает на себя внимание отставание детей в моторном развитии. Они с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, изменения походки по типу «утиной», что связано с поражением ягодичных мышц. Наблюдается своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, из положения на корточках или со стула. Вставание происходит поэтапно, с активным использованием рук - «взбирание лесенкой» или «взбирание по самому себе».
Атрофии мышц всегда симметричны. Сначала они локализуются в проксимальных группах мышц нижних конечностей и в мышцах тазового пояса, в 1-3 года распространяются на проксимальные группы мышц верхних конечностей - плечевой пояс, мышцы спины. Вследствие атрофий появляются лордоз, «крыловидные» лопатки, «осиная» талия (рис. 17.3). Типичным симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотны, безболезненны. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Сухожильные рефлексы изменяются с различной последовательностью: в ранних стадиях заболевания исчезают коленные рефлексы, затем рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы могут долго оставаться сохранными.
Одной из отличительных особенностей формы Дюшенна является патология костносуставной, сердечно-сосудистой и нейроэндокринной систем. Костно-суставные нарушения включают в себя деформации
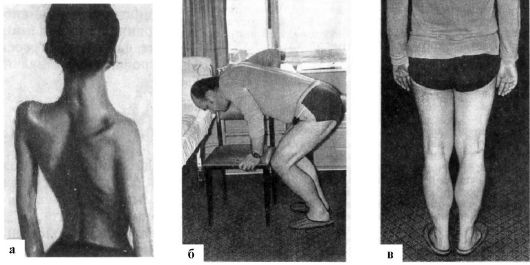 Рис. 17.3. Мышечная дистрофия. а - гипотрофия мышц плечевого и тазового пояса, проксимальных отделов конечностей; «крыловидные» лопатки, «осиная» талия; б - вставание с корточек «лесенкой» (прием миопата); в - псевдогипертрофия мышц икроножной группы
Рис. 17.3. Мышечная дистрофия. а - гипотрофия мышц плечевого и тазового пояса, проксимальных отделов конечностей; «крыловидные» лопатки, «осиная» талия; б - вставание с корточек «лесенкой» (прием миопата); в - псевдогипертрофия мышц икроножной группы
позвоночника, стоп, грудины. У многих больных в результате избирательного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и ретракции сухожилий. Сердечнососудистые расстройства проявляются дилатационной кардиомиопатией, которая может быть одной из причин неблагоприятного исхода заболевания. На ЭКГ регистрируются изменения миокарда (блокада пучка Гиса и др.). Среди нейроэндокринных расстройств чаще встречаются синдромы Иценко-Кушинга, Бабинского-Фрелиха.
Снижение интеллекта разной выраженности отмечается у большинства больных и, как правило, не соотносится с тяжестью поражения скелетной мускулатуры и тяжестью самого заболевания. Наиболее часто у детей выявляются относительно неспецифические изменения в виде затруднения концентрации внимания, сложности в воспроизведении недавно полученной информации, нарушения слуховой памяти, произношения, усвоения материала.
При позитронной эмиссионной томографии и при 31Р МР-спектроскопии наиболее выраженные изменения, отражающие нарушение утилизации глюкозы (по данным ПЭТ) и изменение соотношения неорганического фосфора, АТФ, фосфомоноэстеров и фосфокреатинина (МР-спектроскопия), отмечаются в коре лобных долей больших полушарий и в мозжечке, в меньшей степени - в гиппокампе.
Форма Беккера является аллельным вариантом миодистрофии Дюшенна и также связана со структурным дефектом гена дистрофина. Наследуется по рецессивному типу, сцепленному с Х-хромосомой. Первые клинические проявления отмечаются позднее, чем при форме Дюшенна, чаще в возрасте 10-15 лет, а само заболевание протекает значительно мягче. Мышечная слабость, повышенная мышечная утомляемость при физической нагрузке, псевдогипертрофии икроножных мышц не достигают такой выраженности, как при форме Дюшенна. Мышечный тонус снижен незначительно. Сухожильные рефлексы долго остаются сохранными. В поздних стадиях болезни могут наблюдаться изменения походки по типу «утиной», компенсаторные миопатические приемы при вставании. Заболевание прогрессирует медленно на протяжении многих лет. Сердечно-сосудистые расстройства выражены умеренно. Иногда наблюдается блокада ножек пучка Гиса. Эндокринные нарушения проявляются гинекомастией, снижением либидо, импотенцией. Выраженных изменений интеллекта не отмечается. Больные долго сохраняют работоспособность.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании клинических проявлений, данных биохимических исследований (повышение в крови активности КФК, ЛДГ), электромиографии (признаки первичного мышечного поражения) и молекулярно-генетического анализа. Для уточнения аллельной формы заболевания проводится биопсия мышц для определения дистрофина (при форме Дюшенна в скелетных мышцах дистрофин не выявляется; при форме Беккера дистрофин синтезируется, но в большинстве случаев его уровень снижен). При обследовании матерей - носителей патологического гена (биопсия ворсин хориона на 8-9 неделе) выявляют заболевание у мальчиков.
Дифференцировать форму Дюшенна следует от спинальной амиотрофии Верднига-Гофмана, а форму Беккера - от прогрессирующих мышечных дистрофий Дюшенна, конечностно-поясных форм прогрессирующих мышечных дистрофий, спинальной амиотрофии Кугельберга-Веландера, метаболических и эндокринных миопатических синдромов.
Прогрессирующая мышечная дистрофия Эмери-Дрейфуса
Наследуется по рецессивному типу, сцепленному с Х-хромосомой. Первичный молекулярно-генетический дефект находится на локусе Хq28, который отвечает за синтез белка эмерина. Эмерин относится к
белкам ядра клетки и предположительно стабилизирует ядерную мембрану при повторных актах сокращения - расслабления.
Клинические проявления. Первые признаки заболевания проявляются в 5-7 лет. Развиваются ранние контрактуры в локтевых суставах, ретракции ахилловых сухожилий. Как и при других формах прогрессирующих мышечных дистрофий, заболевание начинается с мышечной слабости, повышенной мышечной утомляемости при физической нагрузке. Атрофии возникают симметрично и сначала локализуются в проксимальных группах мышц нижних конечностей: тазового пояса, бедер. Проксимальные группы мышц верхних конечностей вовлекаются значительно позднее. Заболевание медленно прогрессирует. У многих больных имеются кардиальные нарушения, выраженность которых является важным признаком при определении прогноза заболевания.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании молекулярно-генетического исследования, а также особенностей клинической картины (ранние контрактуры локтевых суставов, ретракции ахилловых сухожилий, сердечно-сосудистые нарушения в виде аритмий, медленное прогрессирующее течение).
Дифференцировать болезнь следует от прогрессирующих мышечных дистрофий Беккера, Дюшенна, Эрба-Рота, спинальной амиотрофии Кугельберга-Веландер.
Конечностно-поясные формы прогрессирующих мышечных дистрофий
Конечностно-поясные формы (КПФ) прогрессирующих мышечных дистрофий - это генетически гетерогенная группа заболеваний, объединенная общим клиническим симптомокомплексом - нарастающей слабостью и атрофиями в проксимальных отделах конечностей. К настоящему времени выявлены варианты, наследуемые аутосомнодоминантно и аутосомно-рецессивно. Аутосомно-доминантный тип (тип I) передачи наблюдается в 5-10% случаев, аутосомно-рецессивный (тип II) - в 90-95% случаев. Аутосомно-доминантно передаются мышечные дистрофии, развивающиеся вследствие нарушения синтеза белков плазматической мембраны (кавеолин-3), белков саркомеры (миотилин) и ядерных белков (ламин А/С). Аутосомно-рецессивно наследуются варианты КПФ, при которых нарушается выработка саркогликанов α, β, γ, δ - белков, входящих в состав плазматической мембраны. Эта группа прогрессирующих мышечных дистрофий носит
название саркогликанопатий. Также аутосомно-рецессивно передаются мышечные дистрофии, связанные с нарушением синтеза дисферилина - белка плазматической мембраны, калпаина-3 и TRIM32 - белков цитозоля клетки, а также титина и телетонина - белков саркомеры. Форма прогрессирующей мышечной дистрофии, описанная Эрбом и Ротом, по современной классификации относится к типу IIА, и ген, ответственный за ее развитие, картирован на хромосоме 15 (15q15.1-21.1).
Клинические проявления. Первые признаки заболевания проявляются преимущественно в 14-16 лет, редко - в 5-10-летнем возрасте. При аутосомно-доминантной передаче дебют наблюдается позднее - в 20-25 лет. Необходимо отметить, что каждая из форм прогрессирующей мышечной дистрофии имеет определенные клинические особенности. Так, наиболее тяжело обычно протекают саркогликанопатии. Это связано с тем, что саркогликаны объединены в комплекс, участвующий вместе с дистрофином в поддержании целостности мембраны мышечной клетки при повторных актах сокращения-расслабления. Вовлечение в патологический процесс одного из саркогликанов нарушает структуру всего комплекса и приводит ко вторичному нарушению функционирования других саркогликанов. При всех вариантах заболевания начальными симптомами являются мышечная слабость, патологическая мышечная утомляемость при физической нагрузке, изменение походки по типу «утиной». В начале болезни атрофии локализуются в проксимальных группах мышц нижних конечностей (форма Эрба-Рота), в более поздних стадиях в процесс вовлекаются мышцы спины и живота. Иногда миодистрофический процесс одновременно поражает мышцы тазового и плечевого пояса. Вследствие атрофий возникают лордоз, «крыловидные» лопатки, «осиная» талия. При вставании больные применяют вспомогательные приемы (вставание «лесенкой»). Псевдогипертрофии мышц, контрактуры суставов, сухожильные ретракции, как правило, выражены умеренно. Характерно снижение сухожильных рефлексов (коленных, с двуглавой и трехглавой мышц плеча).
Диагностика и дифференциальная диагностика. Вследствие генетической гетерогенности этой группы прогрессирующих мышечных дистрофий основным при установлении диагноза является ДНК-анализ, что позволяет установить форму заболевания и определить прогноз. При необходимости проводят электромиографию и биопсию мышц. Дифференцировать эту группу заболеваний следует от прогрес-
сирующей мышечной дистрофии Беккера, спинальной амиотрофии Кугельберга-Веландера, миопатических синдромов.
Лицелопаточно-плечевая мышечная дистрофия Ландузи-Дежерина
Частота составляет 4-5 на 100 000 населения. Наследуется по аутосомно-доминантному типу с очень высокой пенетрантностью (до 95% к 20-25 годам жизни). До 20-30% заболевших не имеют положительного генеалогического анамнеза, и эти случаи рассматриваются как новые мутации. Заболевание развивается вследствие делеции на длинном плече хромосомы 4 (4q35), реже его развитие связано с локусом 10q26. Делеция локализуется в области, непосредственно прилегающей к теломере, и не захватывает смысловых последовательностей гена. Предполагается, что мутация в этой области приводит к изменению структуры хроматина, а это, в свою очередь, изменяет активность близлежащих генов (так называемый эффект положения), в частности генов, кодирующих транскрипционные факторы миогенеза.
Клинические проявления. Первые признаки появляются преимущественно в возрасте 10-20 лет. Мышечная слабость, атрофии затрагивают мимическую мускулатуру лица, лопаток, плеч. Вследствие атрофии лицо становится гипомимичным, типичны «полированный» лоб, лагофтальм, «поперечная» улыбка, толстые, иногда вывороченные губы («губы тапира»). Атрофии двуглавой и трехглавой мышц плеча, большой грудной, передней зубчатой, трапециевидной мышц обусловливают возникновение симптомов «свободных надплечий», «крыловидных» лопаток, появление широкого межлопаточного промежутка, уплощения грудной клетки, сколиоза. В ряде случаев атрофии распространяются на мышцы тазового пояса и ног. Псевдогипертрофии отмечаются в икроножных и дельтовидных мышцах. Мышечный тонус в ранних стадиях заболевания снижен в проксимальных группах мышц, затем - диффузно. Сухожильные рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча. Заболевание медленно прогрессирует, больные долго сохраняют работоспособность.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании клинической картины (преимущественно лицелопаточно-плечевая локализация миодистрофического процесса) и результатов молекулярно-генетического анализа. Дифференцировать заболевание следует от других прогрессирующих мышечных дистрофий.
17.2. Спинальные амиотрофии
Спинальные амиотрофии - одни из наиболее частых и тяжело протекающих заболеваний детского и подросткового возраста. Выделяют форму Верднига-Гофмана (тип I), промежуточную форму (тип II) и форму Ку- гельберга-Веландера (тип III), а также спинальные амиотрофии взрослых (IV тип). Все формы наследуются по аутосомно-рецессивному типу с локализацией дефекта на длинном плече хромосомы 5 (5q11.2-13.3). В 95-98% случаев обнаруживается делеция в 7-м экзоне гена, кодирующего синтез белка, поддерживающего жизнеспособность мотонейрона (motorneuron survival protein), предположительно участвующего в синтезе РНК.
Патоморфология. Обнаруживают дегенерацию клеток передних рогов спинного мозга, демиелинизацию передних корешков. Часто аналогичные изменения имеются в двигательных ядрах и корешках V, VI, VII, IX, X, XI и XII черепных нервов. В скелетных мышцах нейродегенеративные изменения проявляются «пучковой атрофией», чередованием атрофированных и сохранных пучков мышечных волокон, гиперплазией соединительной ткани.
Спинальная амиотрофия Верднига-Гофмана (тип I)
В Уз случаев клиническая картина разворачивается внутриутробно (вялое шевеление плода), у остальных пациентов симптомы появляются в течение первого полугодия жизни. У детей наблюдаются мышечная гипотония, гипотрофия мышц, преимущественно в проксимальных отделах конечностей, снижение либо отсутствие сухожильных рефлексов. Появляются бульбарные расстройства, проявляющиеся вялым сосанием, слабым криком, фибрилляциями языка, снижением глоточного рефлекса. Развитие статических и локомоторных функций резко замедлено. Лишь у ограниченного числа детей с большим опозданием формируется способность держать голову и самостоятельно садиться, но приобретенные навыки быстро регрессируют. Нарушения функций глазодвигательных и мимических мышц нет.
Заболевание сочетается с костно-суставными деформациями: сколиозом, воронкообразной или «куриной» грудной клеткой, контрактурами суставов. Могут быть врожденные пороки развития: врожденная гидроцефалия, крипторхизм, гемангиома, дисплазия тазобедренных суставов, косолапость и др. Болезнь быстро прогрессирует, летальный исход в большинстве случаев наступает до 2-летнего возраста. Одной из
основных причин смерти становится дыхательная недостаточность, обусловленная слабостью мускулатуры грудной клетки и диафрагмы.
Хроническая инфантильная спинальная амиотрофия (тип II)
Первые симптомы возникают в возрасте 6-24 мес. Моторное развитие в течение первых месяцев удовлетворительное. Дети своевременно начинают держать голову, сидеть, иногда стоять. Заболевание развивается подостро, нередко после инфекции, пищевой интоксикации. Вялые парезы первоначально локализуются в ногах, особенно часто в бедрах, затем распространяются на мышцы туловища и руки. Диффузные мышечные атрофии сочетаются с фасцикуляциями, фибрилляциями мышц языка, тремором пальцев, сухожильными контрактурами. Мышечный тонус, сухожильные и надкостничные рефлексы снижаются. В поздних стадиях возникают генерализованная мышечная гипотония, симптомы вовлечения двигательных ядер ствола головного мозга. Заболевание протекает злокачественно, хотя и мягче, чем врожденная форма. Летальный исход наступает к 14-15 годам.
Спинальная юношеская амиотрофия Кугельберга-Веландера (тип III)
Первые признаки болезни чаще всего возникают в интервале от 4 до 7 лет, когда появляются неловкость и неуверенность движений. Из-за нарастающей слабости дети спотыкаются, часто падают. Вялые парезы первоначально локализуются в проксимальных группах мышц нижних конечностей, в дальнейшем сравнительно медленно переходят на проксимальные группы мышц верхних конечностей, мышцы туловища; атрофии мышц обычно малозаметны вследствие хорошо развитого подкожного жирового слоя. Типичны фасцикуляции, мелкий тремор пальцев, бульбарные симптомы: фибрилляции и атрофия мышц языка, снижение глоточного и нёбного рефлексов, что позволяет клинически отличить эту форму от конечностно-поясных форм прогрессирующей мышечной дистрофии. Сухожильные и надкостничные рефлексы угасают уже в ранних стадиях болезни. Костно-суставные деформации развиваются параллельно основному заболеванию. Наиболее выражена деформация грудной клетки. Течение мягче, чем у первых двух форм. Способность к самостоятельной ходьбе нарушается через 10-12 лет после дебюта заболевания.
Диагностика и дифференциальная диагностика. Диагностика строится на данных молекулярно-генетического анализа, особенностях клинической картины (фасцикуляции и фибрилляции, бульбарные расстройства, отсутствие псевдогипертрофий), результатах электромиографии (признаки поражения переднего рога) и морфологического исследования скелетных мышц. Дифференцировать заболевания I и II типов следует от заболеваний, входящих в группу синдромов с врожденной мышечной гипотонией (синдром «вялого ребенка»): амиотонии Оппенгейма, врожденной доброкачественной формы мышечной дистрофии, атонической формы детского церебрального паралича, наследственных болезней обмена веществ, хромосомных синдромов. Заболевание типа III следует отграничивать от прогрессирующей мышечной дистрофии Дюшенна, конечностно-поясных форм прогрессирующей мышечной дистрофии.
Спинальные амиотрофии взрослых (тип IV)
Наследственная бульбоспинальная амиотрофия Кеннеди. Заболевание наследуется сцепленно с Х-хромосомой. Ген отвечает за функционирование рецепторов к андрогенам и картирован на коротком плече (Xq1.2-12), а мутация в нем включает увеличение числа (экспансию) ЦАГ(цитозин-аденин-гуанин) повторов (до 33-34). Следствием этого является увеличение полиглутаминовой последовательности в молекуле мутантного белка, патологическое взаимодействие его с рядом тканеспецифичных белков, формирование белковых агрегатов и включений в нейронах, запускающих процессы программированной клеточной гибели, нейродегенерация.
Клинические проявления. Первые признаки заболевания проявляются преимущественно после 25-30 лет жизни. Начальными симптомами болезни являются слабость и атрофии в проксимальных отделах ног. В последующем присоединяются симптомы вовлечения шейного утолщения и каудальных отделов ствола головного мозга. Отмечаются выраженные деформации стоп, ранняя утрата ахилловых рефлексов при сохранности коленных и глубоких рефлексов с рук, отсутствие чувствительных расстройств. Эндокринные нарушения имеют вид атрофии яичек, снижения потенции, гинекомастии, сахарного диабета. Болезнь медленно прогрессирует, больные долго сохраняют трудоспособность.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании генеалогического анализа (сцепленный с Х-хро-
мосомой тип наследования), особенностей клинической картины, результатов игольчатой электромиографии, позволяющей выявить вовлечение в процесс передних рогов спинного мозга. Дифференцировать заболевание следует от бокового амиотрофического склероза, дистальной миопатии Говерса-Веландера, невральной амиотрофии Шар- ко-Мари-Тута.
Наследственная дистальная спинальная амиотрофия. Болезнь дебютирует в раннем детском возрасте преимущественным поражением нижних конечностей (при аутосомно-рецессивном типе наследования) или в возрасте 20-25 лет преимущественным поражением верхних конечностей (при аутосомно-доминантном типе наследования). В первом случае генетический дефект связан с локусом 5q11, а во втором локус расположен на коротком плече хромосомы 7.
Клинические проявления. Начальными симптомами являются слабость и атрофия дистальной мускулатуры нижних конечностей. В 25% случаев наблюдаются слабость гипотрофии мышц рук. Отмечаются грубые деформации стоп, ранняя утрата ахилловых рефлексов при сохранных коленных рефлексах и рефлексах с рук. Чувствительные расстройства не характерны. Заболевание медленно прогрессирует.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании генеалогического анализа, клинических проявлений, результатов игольчатой электромиографии, позволяющей выявить поражение передних рогов спинного мозга. Дифференцировать заболевание следует от дистальной миопатии Говерса-Веландера, невральной амиотрофии Шарко-Мари-Тута.
17.3. Наследственные мотосенсорные невропатии типов I и II (невральные амиотрофии Шарко-Мари-Тута)
Невральные амиотрофии - электрофизиологически и генетически гетерогенная группа заболеваний, объединенная клинической картиной полиневропатии. В настоящее время установлено 7 типов наследственных мотосенсорных невропатий (НМСН). На основании электрофизиологических критериев выделяют две группы: демиелинизирующие НМСН (тип I) и аксональные (тип II), которые представляют собой варианты болезни Шарко-Мари-Тута.
Наследственные мотосенсорные невропатии типа I
Частота составляет 1 на 50 000 населения. Заболевания наследуются по аутосомно-доминантному, реже по аутосомно-рецессивному или сцепленному с полом типам. Известны также спорадические случаи. Выявляются мутации в генах синтеза белков миелина РМР 22 (локус 17q11.2-12) или МР 0 (локус 1q 22-23). Генетический дефект вызывает нарушение синтеза миелина с развитием демиелинизирующей полиневропатии.
 Патоморфология. Обнаруживается
сегментарная демиелинизиция в периферических нервах, наряду с этим
выявляются утолщения по типу «луковичных головок», отражающие процессы
ремиелинизации. В мышцах наблюдаются денервационные изменения с
явлениями «пучковой» атрофии мышечных волокон.
Патоморфология. Обнаруживается
сегментарная демиелинизиция в периферических нервах, наряду с этим
выявляются утолщения по типу «луковичных головок», отражающие процессы
ремиелинизации. В мышцах наблюдаются денервационные изменения с
явлениями «пучковой» атрофии мышечных волокон.
Клинические проявления. Первые признаки заболевания появляются в возрасте 15-30 лет, реже - в более молодом возрасте. В начале болезни отмечаются мышечная слабость, патологическая утомляемость, преимущественно в дистальных отделах конечностей. Больные быстро устают при стоянии на одном месте и нередко для уменьшения утомления прибегают к ходьбе на месте («симптом топтания»). Атрофии развиваются сначала в мышцах голеней и стоп и, как правило, симметричны. Поражаются преимущественно перонеальная группа мышц и передняя большеберцовая мышца, вследствие чего ноги приобретают форму «перевернутых бутылок» или «ног аиста». Стопы деформируются, становятся «выеденными», с высоким сводом («полая стопа», молоточкообразная деформация пальцев). Парез стоп изменяет походку больных. Они ходят, высоко поднимая ноги; ходьба на пятках невозможна. Атрофии в дистальных отделах рук - мышцах тенара, гипотенара, а также в мелких мышцах кистей присоединяются спустя несколько лет после развития изменений в ногах и всегда симметричны. С течением вре-
мени при выраженных атрофиях кисти приобретают форму «когтистых», «обезьяньих». Мышечный тонус диффузно снижен в дистальных отделах конечностей. Сухожильные рефлексы изменяются неравномерно: ахилловы рефлексы снижаются в ранних стадиях болезни, а коленный рефлекс, рефлексы с трех- и двуглавой мышц плеча долго остаются сохранными. Реже заболевание начинается с чувствительных расстройств: болей, парестезий, ощущения ползания мурашек в дистальных отделах нижних конечностей, хотя они присутствуют у большинства больных в развернутых стадиях заболевания. Часто имеются вегетативные нарушения в виде дистального гипергидроза, гиперемии кистей и стоп.
Течение. Заболевание медленно прогрессирует. Прогноз в большинстве случаев благоприятный.
Диагностика и дифференциальная диагностика. Диагноз устанвливается на основании результатов генеалогического анализа, клинической картины, результатов глобальной, стимуляционной и игольчатой электромиографии. При стимуляционной электронейрографии отмечается снижение скорости проведения по двигательным волокнам периферических нервов ниже 38 м/с на руках и 18 м/с на ногах. Одновременно наблюдаются снижение амплитуды потенциалов действия чувствительных волокон и снижение скорости проведения по ним. При игольчатой миографии выявляются спонтанная активность в виде потенциалов фибрилляций, фасцикуляций и положительных острых волн, снижение амплитуды и увеличение длительности и полифазии потенциалов двигательных единиц (изменения, подобные изменениям М-ответов). Для уточнения формы заболевания проводится анализ ДНК.
Дифференцировать следует от хронической демиелинизирующей воспалительной полиневропатии - курабельного заболевания, также дебютирующего в молодом возрасте. НМСН типа I также следует отличать от дистальной миодистрофии Говерса-Веландера, наследственной дистальной спинальной амиотрофии, мультифокальной моторной полиневропатии с блоками проведения.
Наследственные мотосенсорные невропатии типа II
При нейрональной (аксональной) форме болезни Шарко-Ма- ри-Тута первично поражаются аксоны двигательных волокон периферических нервов. Заболевание наследуется по аутосомно-доминантному или аутосомно-рецессивному типу. Генетический дефект связан с
большим количеством локусов (хромосомы 1, 2, 3, 5, 7, 8, 11). Один из установленных биохимических дефектов связан с геном, кодирующим легкие цепи нейрофиламентов (белки цитоскелета мотонеройнов).
Патоморфология. Обнаруживаются гибель аксонов периферических двигательных нервах и вторичная сегментарная демиелинизиция без формирования утолщений по типу «луковичных головок» (процессы ремиелинизации отсутствуют). Так же, как и при НМСН типа I, в мышцах развиваются денервационные изменения с явлениями «пучковой» атрофии мышечных волокон.
Клиническая картина сходна с НМСН типа I. Отличием является более поздний возраст дебюта заболевания, в патологический процесс реже вовлекаются мускулатура рук, менее выражены чувствительные нарушения. Заболевание прогрессирует медленно, прогноз благоприятный.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании результатов генеалогического анализа, молекулярногенетического анализа, клинической картины. При стимуляционной электромиографии в отличие от НМСН типа I выявляется снижение амплитуды и длительности М-ответа, скорости проведения по двигательным волокнам снижены минимально или нормальны. Значительно позже наблюдаются незначительное снижение амплитуд потенциалов чувствительных волокон и замедление проведения по ним.
Дифференциальная диагностика проводится с дистальной миодистрофией Говерса-Веландера, наследственной дистальной спинальной амиотрофией, другими наследственными и приобретенными полиневропатиями.
Лечение. Принципы лечения НМСН сходны. С целью улучшения трофики мышц и нервных волокон используют карнитин, церебролизин, креатин, кокарбоксилазу, фосфаден, рибоксин, аминокислотные и белковые препараты. Применяют витамины группы В, Е, А, липоевую кислоту, лекарственные средства, улучшающие микроциркуляцию (пентоксифиллин, ксантинола никотинат).
Наряду с курсовой лекарственной терапией широко используют немедикаментозные средства: ЛФК, массаж, физиотерапию (электромиостимуляция, аппликации озокерита, радоновые, хвойные, сульфидные ванны, гипербарическая оксигенация). При ортопедическом дефекте в виде контрактур, деформации позвоночника, асимметричном укорочении конечностей проводится соответствующая коррекция.
17.4. Пароксизмальные миоплегии
Наследственные пароксизмальные миоплегии - группа заболеваний, объединенных клиническим синдромом внезапных приступов мышечной слабости. Пароксизмальные миоплегии связаны с дисфункцией каналов клеточных мембран, регулирующих проникновение в клетку электролитов. Эта особенность патогенеза сближает пароксизмальные миоплегии с миотонией Томпсена, относящейся также к каналопатиям. При пароксизмальных миоплегиях нарушается проникновение хлора, натрия и кальция в клетку, что приводит к деполяризации мембраны с последующим снижением возбудимости сарколеммы и развитием пареза. Выделяют гипо- и гиперкалиемические формы пароксизмальных миоплегий.
Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля)
Заболевание наследуется по аутосомно-доминантному типу. Молекулярно-генетический дефект связан с точечными мутациями в гене, расположенном на хромосоме 1 (1q31-32) и регулирующим функционирование α1-субъединицы канала для вхождения кальция в клетку (дигидропиридиновый рецептор). Реже молекулярно-генетический дефект обнаруживается на хромосоме 17 (17q23) или хромосоме 11 (11q13-14) и связан с дисфункцией ионных каналов, отвечающих за проникновение в клетку натрия и калия. Дисфункция канала приводит к нарушению проницаемости мембраны и избирательному вхождению ионов калия из внеклеточного пространства внутрь клетки.
Клинические проявления. Первые симптомы чаще появляются в возрасте 6-15 лет. Пароксизмы заключаются во внезапном развитии мышечной слабости, обездвиженности, чаще в ночные или утренние часы. Отмечаются также снижение мышечного тонуса, сухожильных рефлексов, вегетативные расстройства: лабильность пульса, АД, гипергидроз. Приступы бывают парциальными, охватывающими небольшую группу мышц, и генерализованными. Во время приступа возникают нарушения сердечно-сосудистой деятельности: изменения на ЭКГ в виде уплощения зубцов Т, депрессии сегмента ST Сознание всегда сохранено. Продолжительность приступов несколько часов, их частота различна. Содержание калия в крови во время приступа менее 2 ммоль/л. Приступы провоцируются избыточным количеством углеводистой пищи, охлаждением, физическими нагрузками.
Лечение. Для купирования приступа назначают 10% раствор хлорида калия внутрь (по 1 столовой ложке каждые 30 мин) или 0,5% раствор на изотоническом растворе хлорида натрия внутривенно (2-2,5 г на 500 мл раствора вводят в течение часа) до купирования приступа. Возможно также внутривенное капельное введение панангина. Для профилактики назначают диакарб (ацетазоламид) по 0,125 мг через день; диету, богатую калием (чернослив, курага, картофель, изюм) и бедную углеводами и поваренной солью.
Гиперкалиемическая форма пароксизмальной миоплегии (болезнь Гамсторп)
Заболевание наследуется по аутосомно-доминантному типу. Молекулярно-генетический дефект локализуется на длинном плече хромосомы 17 (17q23) и связан с дисфункцией α-цепи белка, отвечающего за проникновение ионов натрия в клетку. При этом отмечается стойкая гиперполяризация сарколеммы.
Клинические проявления. Болезнь начинается в раннем детском возрасте, чаще до 5 лет. Внезапно развиваются мышечная слабость, снижение мышечного тонуса, сухожильных рефлексов, вегетативные расстройства. В отличие от гипокалиемического гиперкалиемический паралич развивается обычно днем, сопровождается выраженными парестезиями, сочетается со слабостью мышц лица, артикуляционного аппарата, имеет меньшую продолжительность (30-40 мин). Во время приступа содержание калия в крови повышается до 6-7 ммоль/л. Частота приступов различна: от ежедневных до нескольких раз в месяц. В межприступные периоды неврологическая симптоматика отсутствует. Приступы провоцируют голодание, физические нагрузки, вызывающие утомление. Кроме мышечной слабости отмечаются симптомы повышенной мышечной возбудимости с миотоническими феноменами.
Лечение. Во время приступа вводят 40 мл 40% раствора глюкозы внутривенно вместе с инсулином подкожно; 20 мл 10% раствора хлорида кальция внутривенно. Показана диета с повышенным содержанием углеводов, поваренной соли, ограниченным количеством калия.
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют. Прогноз при своевременно установленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
Диагностика и дифференциальная диагностика. Диагноз устанавливают на основании клинической картины, данных лабораторного биохимического исследования (снижение биоэлектрической активности мышц) и молекулярно-генетического анализа.
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний: тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Нормокалиемический (периодический) паралич
Заболевание наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь начинается в возрасте до 10 лет. Медленно (в течение нескольких суток) нарастает умеренная слабость в мышцах туловища, конечностей, жевательной мускулатуре, а затем также медленно (1-2 нед) происходит регресс симптоматики. Заболевание провоцируют продолжительный сон, длительное пребывание в фиксированной позе, переохлаждение.
Лечение. Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Диагностика и дифференциальная диагностика. Заболевание диагностируется на основании результатов генеалогического анализа, особенностей клинической картины (темп нарастания мышечной слабости, провоцирующие факторы), результатов лабораторного и биохимического обследования (нормальное содержание калия в сыворотке крови, электровозбудимость мышц).
Заболевание следует отличать от миоплегий, развивающихся на фоне эндокринных заболеваний: тиреотоксикоза, болезни Кона, болезни Аддисона и др.
17.5. Миотонии
Миотонии - гетерогенная группа нервно-мышечных заболеваний, объединенных затруднением расслабления мышц после активного сокращения. Патогенез миотоний разный: врожденная миотония относится в группе каналопатий, а в основе дистрофической миотонии лежит экспансия тринуклеотидных повторов.
Врожденная миотония (болезнь Лейдена-Томсена-Беккера)
Частота составляет 0,3-0,7 на 100 000 населения. Наследуется по аутосомно-доминантному (вариант Томсена), реже по аутосомно-рецессивному (вариант Беккера) типу с локализацией генетического дефекта на длинном плече хромосомы 7 (7q23-25).
Патогенез. Молекулярно-генетический дефект удается картировать примерно в 30% случаев, к настоящему времени установлено более 40 мутаций, распределяющихся приблизительно равномерно по всему гену. Мутации приводят к изменению проницаемости ионных каналов, отвечающих за поступление хлора в миоцит. Вследствие этого происходит накопление калия внутри клетки, что приводит к нарушению скорости де- и реполяризации мембраны миоцита и повышению возбудимости сарколеммы, что клинически проявляется повышением мышечного тонуса.
Патоморфология. При световой микроскопии обнаруживается гипертрофия отдельных мышечных волокон; гистохимически выявляется уменьшение размеров мышечных волокон II типа.
Клинические проявления. Первые симптомы заболевания проявляются в возрасте 8-15 лет. Ведущие признаки - миотонические спазмы: затруднения при расслаблении мышц после активного напряжения. Миотонические спазмы локализуются в различных группах мышц, чаще в мышцах кисти, ног, жевательных мышцах и круговых мышцах глаза. Сильное сжатие пальцев кисти, длительное статическое напряжение ног, смыкание челюстей, зажмуривание глаз вызывают тонические спазмы. Фаза расслабления мышц надолго задерживается, и больные не в состоянии быстро разжать кисти, изменить положение ног, открыть рот, глаза. Повторные движения уменьшают миотонические спазмы (симптом «врабатывания»). Повышение механической возбудимости мышц определяется с помощью специальных приемов: при ударе неврологическим молоточком по возвышению I пальца происходит его приведение к кисти (от нескольких секунд до минуты) - «симптом большого пальца», при ударе перкуссионным молоточком по языку на нем появляется ямка, перетяжка - «симптом языка». Внешний вид больных своеобразен: вследствие диффузных гипертрофий различных мышц они напоминают профессиональных атлетов. При пальпации мышцы плотные, твердые, но объективно мышечная сила снижена. Сухожильные рефлексы нормальны, в тяжелых случаях снижены. Болезнь медленно прогрессирует. Трудоспособность долго сохраняется.
Диагностика и дифференциальная диагностика. Диагноз устанавливается на основании особенностей клинической картины (миотонический синдром, атлетический тип телосложения, диффузные гипертрофии мышц), данных глобальной электромиографии (миотоническая реакция) и молекулярно-генетического анализа. Дифференцировать заболевание следует от других форм миотонии, иногда от псевдогипертрофических форм прогрессирующих мышечных дистрофий.
Лечение. При редких приступах могут быть показаны только ЛФК и физиотерапия. Больным с частыми приступами назначают дифенин (по 0,1-0,2 г 3 раза в день курсами по 2-3 нед), диакарб (по 0,125 г 2 раза в день в течение 2-3 нед), препараты кальция (внутривенно 10% раствор хлорида кальция по 10 мл или глюконат кальция внутримышечно), новокаинамид (по 200 мг 2 раза в день).
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Частота заболевания составляет 2,5-5,0 на 100 000 населения. Наследуется по аутосомно-доминантному типу и относится к группе мутаций с увеличением числа (экспансией) тринуклеотидных повторов. Кроме дистрофической миотонии, в эту группу заболеваний входят синдром нестабильной Х-хромосомы, бульбоспинальная атрофия Кеннеди, хорея Гентингтона, атаксия Фридрейха и спиномозжечковые атаксии, включая атаксию Пьера-Мари. При заболеваниях всей этой группы, и при миотонии в частности, увеличивается число повторов в последующих поколениях, что приводит к более раннему началу и тяжелому течению заболевания (феномен антиципации).
Развитие дистрофической миотонии обусловлено мутацией на длинном плече хромосомы 19 (19q13.2-13.3). При мутации отмечается экспансия тринуклеотидных повторов (цитозин-тимин-гуанин - CTG) в нетранслируемой области гена, отвечающего за выработку протеинкиназы, которая, как предполагается, участвует в фосфорилировании миотонина. В норме CTG повторов от 5 до 40, 50-70 повторов рассматривается как предмутация; это состояние крайне нестабильно и сопровождается высоким риском развития болезни в последующем поколении, особенно если патологический ген получен от матери. Более 80-90 повторов сопровождается развитием клинических проявлений заболевания.
Патоморфология. Методом световой микроскопии обнаруживают сочетание атрофированных и гипертрофированных мышечных воло-
кон, разрастание соединительной ткани, замещение мышечной ткани жировой и соединительной. При электронной микроскопии определяются изменение размеров митохондрий, деструкция миофибриллярного аппарата, саркоплазматической сети.
Клинические проявления. Первые признаки заболевания проявляются в 10-20 лет, это сочетание миотонических, миопатических, нейроэндокринных и сердечно-сосудистых нарушений. Миотонический симптомокомплекс, как и при врожденной миотонии Томсена, проявляется мышечными спазмами, повышенной механической возбудимостью. Выраженность миотонического феномена в поздних стадиях болезни может уменьшаться. Миопатический синдром проявляется мышечными атрофиями преимущественно в мышцах лица, шеи, дистальных отделов конечностей, повышенной мышечной утомляемостью, слабостью. Развиваются атрофии с вовлечением мимической мускулатуры и мышц конечностей. Отмечаются гипотрофии височных и жевательных мышц, частичный птоз, атрофии дистальных отделов конечностей. Выявляются гипотрофии дистальных отделов конечностей с развитием «выеденных» стоп, «обезьяньей» кисти. Мышечный тонус снижен, сухожильные рефлексы рано угасают. Нейроэндокринные расстройства многообразны: у мужчин наблюдаются крипторхизм, снижение либидо, импотенция, у женщин - нарушения менструального цикла. У многих больных отмечаются раннее облысение, истончение и сухость кожи, катаракта.
Диагностика и дифференциальная диагностика. Диагноз устанавливается на основании молекулярно-генетического анализа, клинической картины (сочетание миотонических, миопатических, нейроэндокринных, сердечно-сосудистых нарушений), результатов электромиографии (миотоническая реакция), биохимического исследования крови (инсулинорезистентность). Заболевание следует отличать от врожденной миотонии Томсена, других миотонических форм, прогрессирующих мышечных дистрофий: дистальной миопатии, невральной амиотрофии.
Лечение. Как и при врожденной миотонии, положительный эффект дают дифенин, диакарб. Показано применение анаболических стероидов (ретаболил, неробол, метиландростендиол). Диетотерапия включает ограничение количества калия в пище.