Дерматовенерология : учебник для студентов высших учебных заведений / В. В. Чеботарёв, О. Б. Тамразова, Н. В. Чеботарёва, А. В. Одинец. -2013. - 584 с. : ил.
|
|
|
|
Глава 16. НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ КОЖИ
16.1. ИХТИОЗ
Ихтиозы (ichthyosis) - группа наследственных заболеваний, характеризующаяся генерализованным нарушением процессов ороговения. Общие клинические особенности данной группы - раннее начало (не позднее первого года жизни), сухость кожи, чрезмерное шелушение, напоминающее чешую рыбы, сезонность обострений в зимние месяцы.
Этиология и патогенез
Этиология заболевания неизвестна, патогенетическая основа ихтиоза - мутации и особенности экспрессии генов, ответственных за образование кератина. Тип наследования ихтиозов различен.
Клиническая картина
Наиболее часто встречаются следующие формы ихтиозов: вульгарный ихтиоз, Х-сцепленный ихтиоз, ламеллярный ихтиоз, ихтиозиформная эритродермия, врожденный ихтиоз (синдром Арлекина).
Вульгарный ихтиоз
Вульгарный ихтиоз - наиболее часто встречаемая форма ихтиоза. Наследуется аутосомно-доминантно.
Заболевание проявляется с 3-12-месячного возраста, когда возникают выраженная сухость кожи и шелушение. Максимальные клинические проявления развиваются к периоду полового созревания и ослабевают к зрелому возрасту. Заболевание протекает волнообразно: летом наступает некоторое улучшение, зимой - усиление сухости и шелушения.
Для клинической картины вульгарного ихтиоза характерна триада.
• Мелкопластинчатое диффузное шелушение кожи (нетипично поражение крупных складок и лица), воспалительные явления отсутствуют (рис. 16-1).
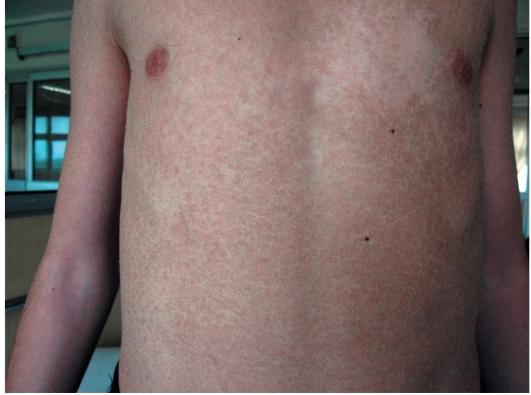
Рис. 16-1. Вульгарный ихтиоз
• Поражение ладоней и подошв в виде усиления кожного рисунка - появление «старческих ладоней» (рис. 16-2).
• Фолликулярный гиперкератоз. На разгибательных поверхностях бедер и плеч образуются роговые пробки в устьях фолликулов - на ощупь кожа напоминает терку (рис. 16-3).
В зависимости от степени выраженности шелушения и цвета чешуек выделяют несколько клинических вариантов ихтиоза: простой, блестящий, змеевидный и др. Наиболее часто встречается абортивная форма вульгарного ихтиоза - ксеродермия, выявляемая у 3-5% населения и характеризующаяся сухостью и легким шелушением на разгибательных поверхностях конечностей (наиболее заметно на голенях в зимнее время).
В 50% случаях вульгарный ихтиоз сочетается с атопическим дерматитом.
Х-сцепленный ихтиоз
Х-сцепленный ихтиоз - редкая разновидность ихтиоза. Болеют мужчины, матери которых являются гетерозиготными носительницами патологического гена, локализованного в одной из половых хромосом. Основной наследуемый биохимический дефект - нарушение синтеза стероидной сульфатазы.
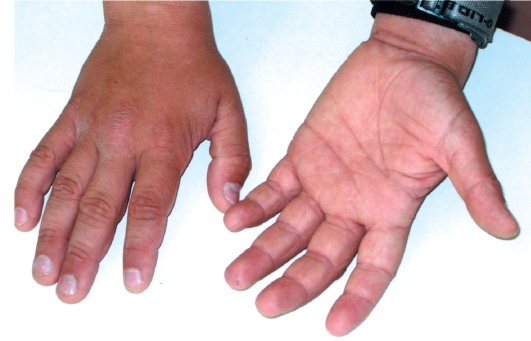
Рис. 16-2. Вульгарный ихтиоз. Гиперлинеарность ладоней
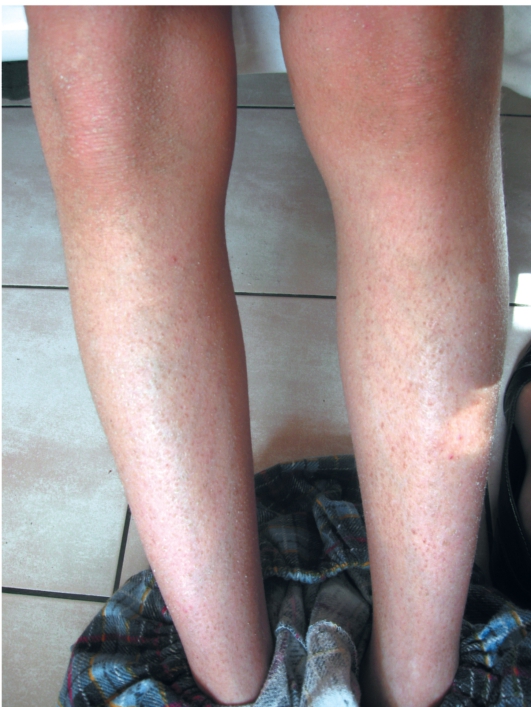
Рис. 16-3. Вульгарный ихтиоз. Фолликулярный кератоз
Х-сцепленный ихтиоз развивается у мальчиков сразу после рождения и проявляется темно-коричневыми чешуйками. Для данного ихтиоза характерна своеобразная локализация высыпаний: наиболее заметны высыпания в крупных складках; ладони, подошвы и лицо не поражаются. У маленьких детей черные чешуйки-щитки всегда возникают на волосистой части головы и шее. Часто возникают помутнение роговицы, гипогонадизм, крипторхизм, умственная отсталость. С возрастом положительная динамика заболевания не возникает.
Коллодийный плод
Коллодийный плод - ихтиоз новорожденных. Данное состояние не относится к самостоятельным заболеваниям, оно является начальным проявлением целой группы ихтиозов. При рождении плод заключен в похожую на коллодий прозрачную желто-коричневую пленку, которая с течением времени подсыхает и трескается. У данных детей нарушается терморегуляция, а также защитные свойства кожи. В 70% случаев у них впоследствии развивается та или иная форма ихтиоза (ламеллярный ихтиоз, сухая ихтиозиформная эритродермия и др.).
Ламеллярный ихтиоз
После высыхания пленка на эритематозной коже новорожденного превращается в крупные темные чешуйки, между которыми появляются трещины, что приводит к избыточной потере жидкости. В связи с избыточным натяжением кожи лица происходи выворот век (эктропион) и губ (эклабиум). Распространенное поражение кожи сохраняется на всю жизнь. На ладонях и подошвах - диффузный кератоз и болезненные трещины (рис. 16-4).
Сухая ихтиозиформная эритродермия
Сухая ихтиозиформная эритродермия также является вариантом развития коллодийного плода. После отторжения пленки развивается выраженная эритродермия. С возрастом гиперемия кожных покровов стихает, сменяясь выраженным шелушением. В отличие от ламеллярного ихтиоза при данном заболевании на коже возникают мелкие серебристые чешуйки. Так же как и при ламеллярном ихтиозе, для данных больных характерны выворот век и стянутость кожи лица. У некоторых больных формируется деформация ушных раковин (рис. 16-5, 16-6).
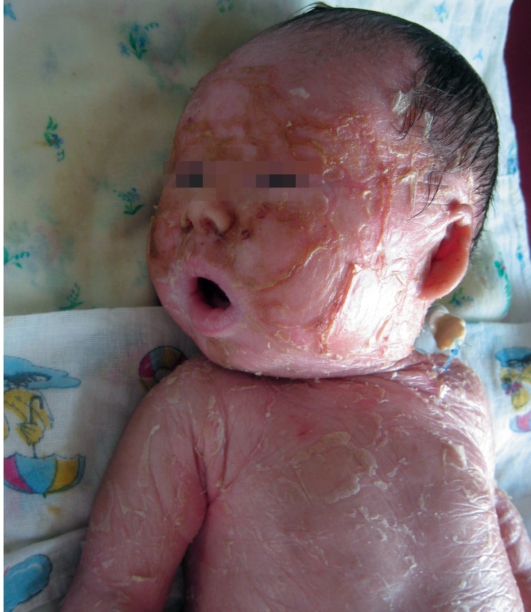
Рис. 16-4. Ламеллярный ихтиоз
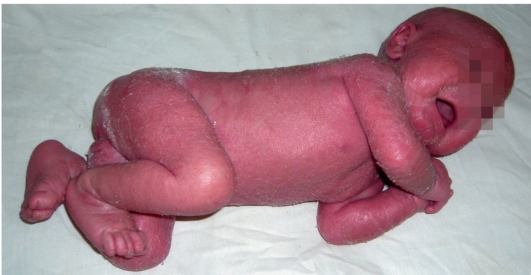
Рис. 16-5. Сухая ихтиозиформная эритродермия у ребенка
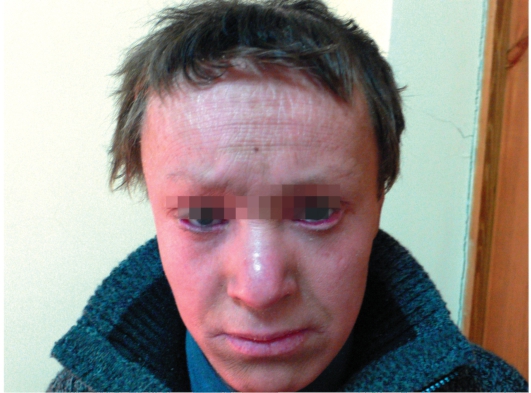
Рис. 16-6. Сухая ихтиозиформная эритродермия у взрослого
Буллезная форма ихтиозиформной эритродермии
Буллезная форма ихтиозиформной эритродермии сразу после рождения проявляется выраженной эритродермией, на фоне которой формируются множественные пузыри и эрозии. С возрастом в области крупных складок возникает гиперкератоз с крупнопластинчатыми роговыми крошкоподобными образованиями серо-бурого цвета, напоминающими вельветовую ткань. При отторжении роговых наслоений остаются эрозированные очаги с заметными сосочковыми разрастаниями.
Врожденный ихтиоз
Врожденный ихтиоз (синдром Арлекина) - редкая и самая тяжелая форма ихтиоза. Характеризуется резким утолщением рогового слоя кожи. Клинически проявляется выворотом век, рта, глубокими трещинами кожи, микроцефалией. Течение - крайне неблагоприятное, прогноз плохой (рис. 16-7).
Дифференциальная диагностика
Дифференциальную диагностику проводят с остаточным поствоспалительным шелушением (эритродермия, распространенный контактный дерматит и др.), постдегидрационным состоянием. Буллезную ихтиозиформную эритродермию дифференцируют от буллезного эпидермолиза, буллезной пиодермии, эксфолиативного дерматита Риттера.
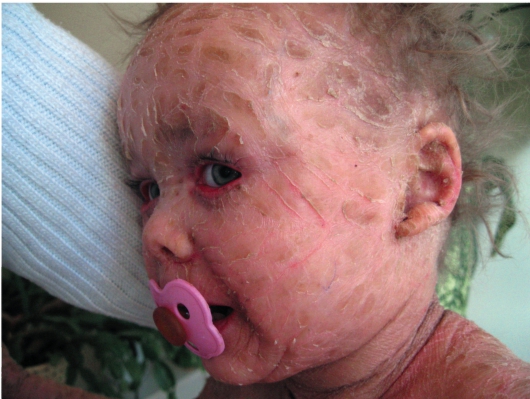
Рис. 16-7. Врожденный ихтиоз
Лечение
Общее лечение
При рождении коллодийного плода и синдроме Арлекина показаны госпитализация и инкубация с режимом высокой влажности. Сразу после рождения новорожденным назначают глюкокортикоидные препараты - преднизолон в дозе 1,5-3 мг/кг в сутки длительно (1-1,5 мес). Затем дозу постепенно снижают. Для профилактики вторичной инфекции больным проводят антибиотикотерапию, для коррекции водноэлектролитного и белкового баланса показаны инфузионная терапия, вливание белковых препаратов (10% раствор альбумина человека, нативная плазма).
В терапии тяжелых форм ихтиозов рекомендован прием витамина А (ретинол 10 тыс. МЕ на 1 кг/сут, ацитретин 0,5-1 мг на 1 кг массы тела). Длительно ретиноиды назначать нельзя из-за выраженных побочных эффектов.
Наружная терапия
Наружная терапия при ихтиозах носит симптоматический характер, но имеет исключительное значение. Применяют увлажняющие и кератолитические средства.
К увлажняющим и влагосохраняющим средствам относят средства с 2% мочевиной, вазелин, ланолин, минеральное масло, эмоленты (атодерм*, трикзера*, топикрем*, крем фореталь*, дардиа*, эмолеум* и др.).
Кератолитические средства - мази с 2-10% салициловой, молочной, лимонной кислотой*, а также 5-10% мази с мочевиной. Детям до 3 лет не рекомендуют применять средства с салициловой кислотой. Эффективное наружное средство в терапии ихтиозов - мази, содержащие витамин А.
Физиолечение
Показаны ванны: кислородные, соляные (хлоридно-натриевые, с морской солью), сероводородные, а также масляно-скипидарные, масляно-молочные, крахмальные и другие, УФО, курортотерапия (влажный и теплый воздух, морские купания).
Консультирование
При тяжелых формах ихтиозов рекомендовано медико-генетическое консультирование. Больным ихтиозами необходим уход за кожей - постоянное использование увлажняющих, влагосберегающих и кератолитических средств. Необходимо избегать гипертермии.
16.2. БУЛЛЕЗНЫЙ ЭПИДЕРМОЛИЗ
Буллезный эпидермолиз (epidermolysis bullosa congenital), или наследственная пузырчатка, - большая группа невоспалительных заболеваний кожи, характеризующихся склонностью кожи и слизистых оболочек к развитию пузырей преимущественно на местах травмы. Раньше буллезный эпидермолиз называли «механобуллезная болезнь», подчеркивая выраженную связь с травматическим фактором.
Этиология и патогенез
Развитие всех форм буллезного эпидермолиза определяется многочисленными генными мутациями в различных хромосомах, которые влекут за собой структурные дефекты тонофиламентов и полудесмосом. При этом нарушается соединение кератиноцитов, базальной мембраны и дермы, вызывая формирование пузырей.
Тип наследования многочисленных форм буллезного дерматита различен: аутосомно-доминантный или аутосомно-рецессивный.
Клиническая картина
Общие клинические признаки, характерные для всех форм:
• ранний дебют заболевания (или сразу с рождения, или в раннем детском возрасте);
• пузыри с серозным или серозно-геморрагическим содержимым, эрозии;
• появление пузырей после незначительной травмы (трение, давление);
• ухудшение течения заболевания летом, при УФО;
• возможно поражение ногтей, зубов, развитие рубцовой алопеции, отставание в физическом развитии;
• анемия.
Наличие или отсутствие рубцов после заживления эрозий дает основание для разделения всех форм буллезного эпидермолиза на простые и дистрофические.
Простой (нерубцующийся) буллезный эпидермолиз
При данной форме пузыри располагаются на уровне эпителиоцитов базального слоя (над базальной мембраной).
Клинические признаки заболевания проявляются на первом году жизни ребенка. Для данного заболевания характерно появление на неизмененной коже различных размеров пузырей. Наиболее часто у маленьких детей пузыри появляются после касательных травм на кистях, стопах, локтях и коленях, в области подмышечных впадин (формируются при подъеме ребенка), на животе (место давления резинки памперсов) (рис. 16-8). После вскрытия эрозии быстро эпителизируются без рубца. Типично появление новых пузырей на участках бывших элемен-
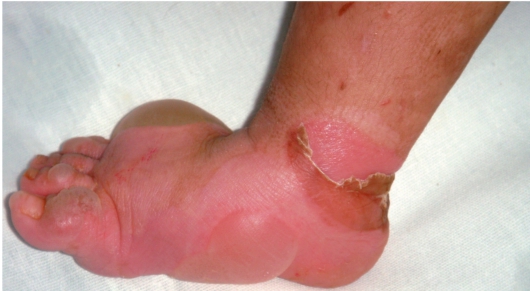
Рис. 16-8. Буллезный эпидермолиз. Простая нерубцующаяся форма
тов. При частом рецидивировании пузырей и эрозий на одних и тех же местах формируется рубцовая атрофия кожи. Ногти поражаются редко. С возрастом состояние больных улучшается. Прогноз благоприятный.
Дистрофический (рубцующийся) буллезный эпидермолиз
Заболевание начинается в раннем детском возрасте (до 3 лет).
При дистрофическом буллезном эпидермолизе пузыри формируются глубоко, под базальной мембраной, вследствие нарушения связи между мембраной и коллагеновыми волокнами. При данной форме определяется повышенная активность фермента коллагеназы.
Дистрофический буллезный эпидермолиз - тяжелое заболевание, при котором на коже формируются глубокие пузыри, длительно заживающие с образованием рубца, имеются участки гипо- и гиперпигментации (рис. 16-9). На коже выявляются милиарные кисты. При локализации пузырей на кистях и стопах происходит сращение пальцев, деформирующие кисти по типу варежек, возникают контрактуры (рис. 16-10). Появляющиеся пузыри под ногтевыми пластинками приводят к их дальнейшему отслаиванию и замещению рубцовой тканью.
Появление пузырей на слизистых оболочках полости рта, глотки, гортани сопровождается болью, затруднением глотания, голос становится хриплым. Образование рубцов приводит к развитию микросто-
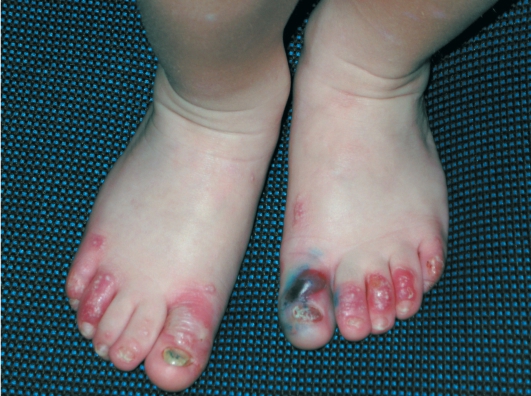
Рис. 16-9. Буллезный эпидермолиз. Дистрофическая форма. Ступни
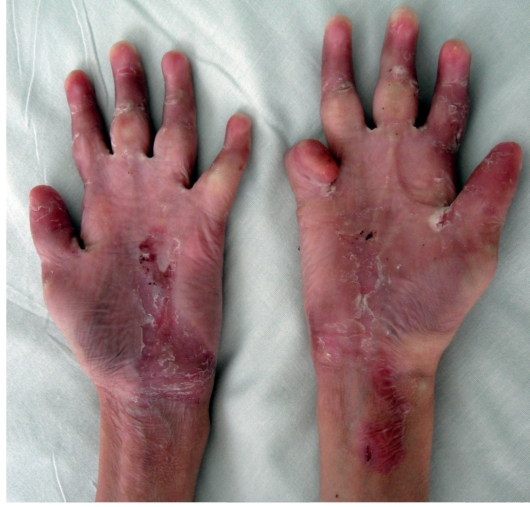
Рис. 16-10. Буллезный эпидермолиз. Дистрофическая форма. Ладони
мы, сужению пищевода, анального отверстия. Локализация пузырей на конъюнктиве, роговице может привести к потере зрения.
Характерны дистрофии зубов, рубцовая алопеция, анемия, отставание в росте и физическом развитии. Умственное отставание не характерно!
При дистрофическом буллезном эпидермолизе прогноз серьезный, характерна высокая летальность в детском возрасте (асфиксия, сепсис, почечная и сердечно-сосудистая недостаточность, плоскоклеточный рак).
Дифференциальная диагностика
Дифференциальную диагностику проводят с буллезной пиодермией, пузырчаткой новорожденных, простым герпесом, аплазиями кожи.
Лечение
Лечение при буллезном эпидермолизе только облегчает состояние больного, не влияя на течение и прогноз заболевания.
Прежде всего необходимо обеспечить правильный уход за кожей. Резистентность кожи к механическим воздействиям обеспечивают ожири-
ванием и гидратацией. Увлажняющие и смягчающие средства помогают уменьшить трение в области заживающей поверхности.
При появлении пузырей рекомендуют удалять пузырную жидкость пункционной иглой, стараясь сохранить покрышку пузыря. Эрозии обрабатывают водными растворами антисептиков, затем возможно наложение раневых пленок (парапран*, воскопран* и др.), эпителизирующих средств (кремы солкосерил*, актовегин*, гель с гиалуронатом цинка и др.).
При присоединении вторичной инфекции рекомендовано местное или системное использование антибактериальных средств.
Общая терапия включает назначение витаминов А и Е (витамин Е 500-700 мг/сут, витамин Е + ретинол 1-2 капсулы в день, ретинол и др.), фенитоин по 8 мг/кг в сутки, анаболических гормонов, оксида цинка.
При развитии контрактур и деформаций кистей рекомендуют проводить лечебную гимнастику, при необходимости - реконструктивное хирургическое вмешательство.
Консультирование
При всех формах буллезного эпидермолиза необходимо провести медико-генетическое консультирование семьи.
С целью уменьшить возможность травматизации кожи больным буллезным эпидермолизом требуется бережный уход. Рекомендовано:
• минимизировать ятрогенные воздействия (инъекции, применение зондов, пластырей и т.д.);
• исключить касательное травмирование кожи (избегать резких движений), а также тепловое воздействие на кожу;
• использование свободной одежды из мягких тканей без завязок и шнурков, мягкой, нетесной обуви;
• кормить мягкой, протертой едой; избегать жесткой и сухой пищи; в бутылочках с питанием должна быть широкая дырочка;
• профилактика инфекций, при необходимости назначение обезболивающих препаратов;
• контроль за анемией: прием железосодержащих препаратов, богатая белком пища;
• социальная адаптация больных.
16.3. НЕЙРОФИБРОМАТОЗ
Нейрофиброматоз (neurofibromatosis) - группа наследственных заболеваний с преимущественным поражением кожи и нервной системы, с повышенным риском развития злокачественных опухолей.
Этиология и патогенез
Нейрофиброматоз относят к нейрокожным заболеваниям, связанным с наследственными патологическими изменениями кожи и ЦНС. В связи с тем что в эмбрионезе кожа и нервная система развиваются из одного и того же неврального гребня, многие врожденные неврологические заболевания сочетаются с заболеваниями кожи. Нейрофиброматоз наследуется аутосомно-доминантно, чаще передается по мужской линии.
В патогенезе заболевания большую роль играет нарушение синтеза белков нейрофибромина и мерлина, тормозящих рост опухолей.
Заболевание встречается с частотой 1:3000 новорожденных. Болеют чаще мальчики.
Клиническая картина
Основные клинические проявления нейрофиброматоза - пятна цвета «кофе с молоком», нейрофибромы, поражение нервной системы, нарушение роста костей, умственная отсталость. Выделяют 7 типов заболевания. Наиболее часто встречаются 1 и 2-й типы.
Нейрофиброматоз 1-го типа, или болезнь Реклингаузена
Нейрофиброматоз 1-го типа, или болезнь Реклингаузена, - наиболее часто встречающаяся форма. Клинические проявления зависят от возраста ребенка, с годами становятся все более заметными.
Заболевание развивается не позднее первого года жизни ребенка с появления нескольких крупных пигментных пятен цвета «кофе с молоком». С возрастом пятна увеличиваются в размерах и появляются новые. Диагностически значимы 5 пятен и более: для маленьких детей диаметром более 5 мм, для подростков - более 15 мм (рис. 16-11).
Важный клинический признак - появление мелких пигментных пятен типа веснушек в подмышечной и паховой областях (у детей после 6 лет) (рис. 16-12). В течение ряда лет пигментные пятна могут оставаться единственными проявлениями заболевания.
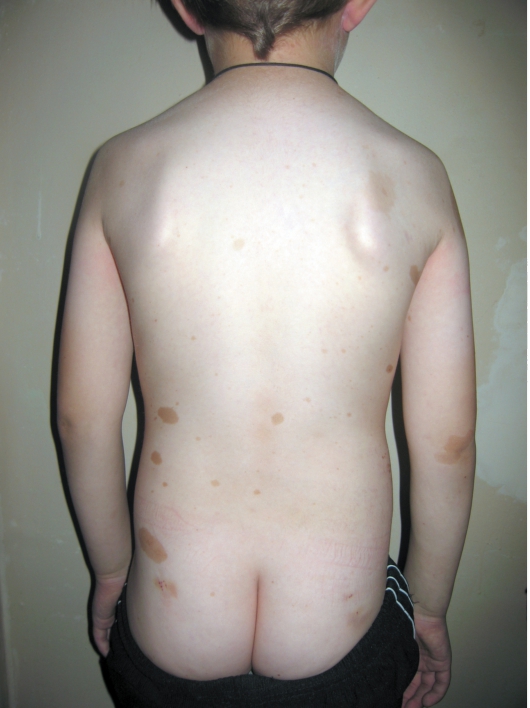
Рис. 16-11. Нейрофиброматоз. Многочисленные пятна цвета «кофе с молоком»
К подростковому возрасту появляются первые нейрофибромы - доброкачественные опухоли нервных стволов. Множественные опухоли различных размеров (от 1 мм до 15 см) розового или бледнокоричневого цвета расположены в коже или в подкожной жировой клетчатке (рис. 16-13). При надавливании на нейрофибромы пальцем характерно ощущение его проваливания в ткани - симптом «кнопки звонка».
Опасные проявления нейрофиброматоза - плексиформные нейрофибромы - диффузные опухолевидные разрастания по ходу нервных
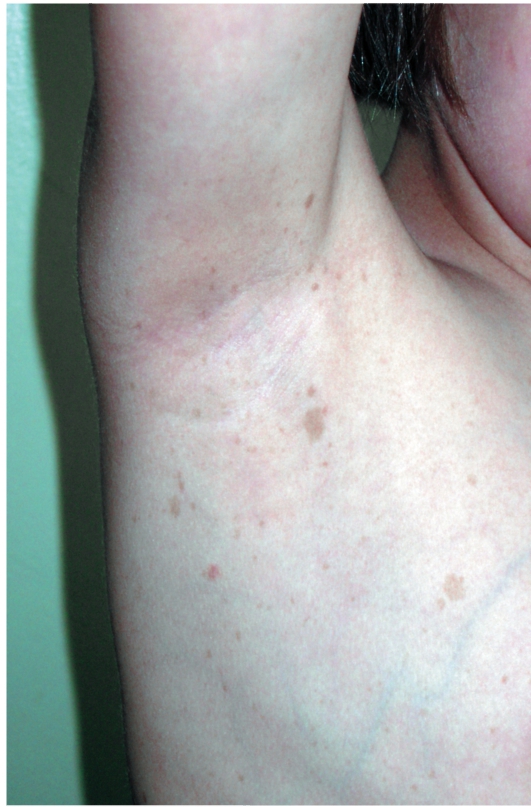
Рис. 16-12. Нейрофиброматоз. Веснушчатые высыпания в подмышечных впадинах
стволов, которые часто подвергаются озлокачествлению с развитием нейрофибросаркомы. Имеют вид мешковидно свисающих, массивных дольчатых опухолей, нередко гиперпигментированных (рис. 1614). В глубине пальпируются утолщенные извилистые нервные стволы.
Характерно поражение глаз: пигментные гамартомы радужной оболочки (узелки Лиша), глиома зрительного нерва и др.
Для больных нейрофиброматозом характерны низкий рост, прогрессирующий сколиоз, а также снижение интеллекта.
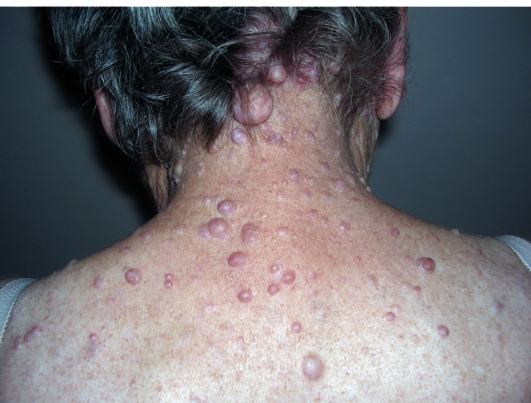
Рис. 16-13. Нейрофиброматоз. Нейрофибромы
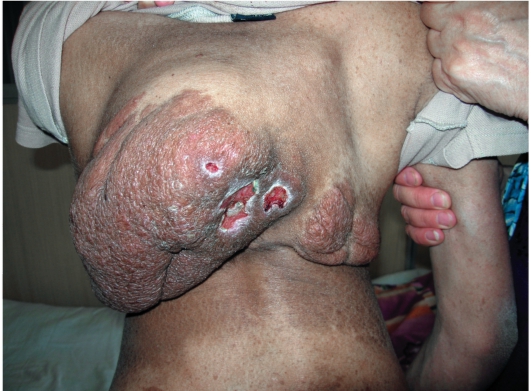
Рис. 16-14. Нейрофиброматоз. Плексиформные нейрофибромы
Нейрофиброматоз 2-го типа
Клинические проявления сходны с 1-м типом, но кожные проявления минимальны. Более характерны болезненные, плотные подвижные подкожные опухоли.
Отличительная особенность данного типа - развитие двусторонней невриномы слухового прохода, приводящей к потере слуха, обычно в
возрасте 20-30 лет (подтверждают рентгенологическим и аудиологическим исследованием, КТ).
Характерны эпилептиформные припадки, судороги, умственная отсталость, менингеальные симптомы, обусловленные развитием опухолей в головном мозге.
Прогноз для выздоровления неблагоприятный, тяжесть течения определяется возможным озлокачествлением опухолей и степенью вовлечения в процесс внутренних органов и систем.
Дифференциальная диагностика
Дифференциальную диагностику проводят с пигментными врожденными невусами, поствоспалительной гиперпигментацией, дерматофибромой и другими опухолями.
Лечение
Специфического лечения нейрофиброматоза не существует. Проводят лечение осложнений.
Консультирование
Показано медико-генетическое консультирование.
16.4. ЭНТЕРОПАТИЧЕСКИЙ АКРОДЕРМАТИТ
Энтеропатический акродерматит (синдром Данболта-Клосса, acrodermatitis enteropathica) - обменное заболевание, сопровождаемое недостатком цинка в организме. Заболевание развивается у детей в период с 3-месячного возраста до 1,5 лет.
Этиология и патогенез
В организме цинк играет важную регулирующую роль, будучи кофактором ряда ферментов: щелочной фосфатазы, карбоангидразы, тиаминкиназы и др. Дефицит цинка вызывает уменьшение ферментативной активности и синтеза нуклеиновых кислот, нарушает гуморальный и клеточный иммунитет, что создает предпосылки для развития оппортунистических инфекций.
В основе заболевания лежит наследственный или приобретенный синдром, приводящий к дефициту цинка. Врожденное
нарушение всасывания цинка наследуется аутосомно-рецессивно, а выраженность клинических проявлений напрямую связана со сниженной секрецией поджелудочной железой низкомолекулярного белка - цинксвязывающего фактора. Данный фермент также поступает в организм ребенка с материнским молоком (но отсутствует в коровьем), поэтому первые проявления заболевания возникают при переводе ребенка с естественного вскармливания на искусственное.
Среди приобретенных причин дефицита цинка наиболее часто бывает недостаточное поступление данного элемента с пищей (вегетарианство, диета, алкоголизм), поражение кишечника при болезни Крона, ВИЧ-инфекции. Возраст пациентов при приобретенной форме энтеропатического дерматита различный.
Клиническая картина
Начало заболевания у маленьких детей совпадает с отменой грудного вскармливания.
Клиническая картина данного заболевания представлена триадой основных проявлений: кожными высыпаниями, алопецией и желудочно-кишечными расстройствами.
• Поражения кожи симметричны и отличаются типичной локализацией: вокруг естественных отверстий (вокруг рта, глаз, половых органов), а также на разгибательных поверхностях конечностей (локти, колени) и пальцах. Высыпания представлены эритематозносквамозными очагами и везикулобуллезными элементами ярко красного цвета с четкими границами, эрозиями, корками (рис. 16-15).
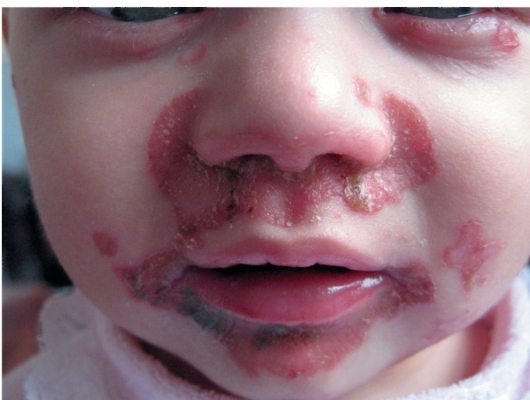
Рис. 16-15. Акродерматит энтеропатический
• Алопеция проявляется выпадением волос на голове, нарушением роста ресниц и бровей.
• Поражения ЖКТ характеризуются длительно протекающей диареей, глосситами, стоматитами.
Для клинической картины энтеропатического дерматита характерны также потеря массы тела, задержка роста, раздражительность, беспокойство, возможны психические расстройства.
Лабораторная диагностика
Определение низкой концентрации цинка в сыворотке крови (нормальные показатели - 0,78-1,2 мг/л), а также снижение активности щелочной фосфатазы, секреция которой напрямую зависит от концентрации цинка, подтверждают диагноз.
Дифференциальная диагностика
Дифференциальную диагностику проводят с атопическим и контактно-аллергическим дерматитом, псориазом, экземой, кандидозом.
Лечение
Основная терапия заключается в назначении пациентам внутрь препаратов цинка: оксида цинка или сульфата цинка 7-10 мг/кг в сутки в течение 2-4 нед с дальнейшем постепенным снижением дозы до 2-5 мг/кг в сутки, которую пациенты получают в течение 4-6 мес.
Наружная терапия включает применение комбинированных глюкокортикоидных препаратов с антимикотиками и антибиотиками (крем гидрокортизон + натамицин + неомицин, гидрокортизон + окситетрациклин, гидрокортизон + окситетрациклин и т.д.), назначение индифферентных цинковых паст.
Прогноз
При своевременной диагностике и правильном лечении заболевание быстро регрессирует.
Без лечения пациенты погибают от оппортунистических инфекций.
Консультирование
Рекомендуют грудное вскармливание, полноценное питание.