Внутренние болезни: учебник: в 2 т. / под ред. В.С. Моисеева, А.И. Мартынова, Н.А. Мухина. - 3-е изд., испр. и доп. - 2013. - Т.2. - 896 с.: ил.
|
|
|
|
ЧАСТЬ VI. ЗАБОЛЕВАНИЯ КРОВИ
Глава 54. НОРМАЛЬНЫЙ ГЕМОПОЭЗ
Кроветворение (гемопоэз) - тонко регулируемый процесс последовательных дифференцировок родоначальных клеток, проводящий к образованию зрелых клеток крови всех восьми линий:
• миелоидных:
- эритроциты;
- базофильные, эозинофильные и нейтрофильные гранулоциты;
- мегакариоциты;
- моноциты - макрофаги;
• лимфоидных:
- Т- лимфоциты;
- В-лимфоциты.
КРОВЕТВОРНЫЕ ОРГАНЫ
Кроветворные клетки образуются в органах кроветворения, которые подразделяют:
• на эмбриональные (желточный мешок, эмбриональная печень, селезенка и костный мозг);
• взрослые (костный мозг, селезенка, тимус, лимфатические узлы и пейеровы бляшки).
Кроветворение в костном мозге происходит в полости всех трубчатых и плоских костей в пространстве между синусами - в так называемом кроветворном (стромальном) микроокружении. К клеткам микроокружения относят:
• эндотелиальные клетки;
• адвентициальные клетки;
• ретикулярные клетки (фибробласты костного мозга);
• макрофаги;
• жировые клетки;
• остеокласты;
• остеоциты.
Внеклеточный матрикс представлен набором нерастворимых белков (глюкозаминогликанов, протеогликанов, фибронектина, гликопротеинов), коллагеновыми и эластиновыми волокнами, в сети которых расположены тяжи кроветворных клеток и основное вещество кости. Способность кроветворных клеток узнавать клетки стромы и распределяться там (хоуминг) обусловлена молекулами клеточной адгезии, интегринами и непосредственными клеточными контактами. Это свойство клеток проявляется при трансплантации костного мозга: 85% введенных внутривенно клеток попадает в костный мозг, масса которого составляет 6% массы тела. Оставшиеся 15% распределяются между печенью, легкими, селезенкой и другими органами.
Родоначальные кроветворные клетки локализуются в костном мозге. Предшественники Т- и В-лимфоцитов также образуются в костном мозге, однако их окончательная дифференцировка происходит в тимусе (Т-лимфоциты) и селезенке, лимфатических узлах и пейеровых бляшках (В-лимфоциты).
Селезенка морфологически состоит из двух отделов - красной и белой пульпы. В красной пульпе происходит депонирование и разрушение эритроцитов. Большинство макрофагов красной пульпы фагоцитируют разрушенные эритроциты и пигменты железа. Белая пульпа, образованная артериями и окружающими их лимфоидными футлярами, в основном заселена Т-лимфоцитами. Кластеры В-лимфоцитов расположены по периферии периартериальных зон. После антигенной стимуляции первичные фолликулы развиваются во вторичные с зародышевыми центрами. В них развиваются В-лимфоциты и плазматические клетки.
Тимус - центральный и высокоспециализированный орган лимфопоэза, в котором происходят созревание и клональная селекция Т-лимфоцитов, а также удаление аутореактивных клонов. Предшественники Т-лимфоцитов попадают в корковое вещество тимуса из костного мозга. Для тимоцитов коркового вещества характерна высокая скорость пролиферации, однако большая часть из них гибнет, а часть популяции приобретает специфические маркеры Т-хелперов и Т-супрессоров и мигрирует через мозговое вещество тимуса во вторичные лимфоидные органы (селезенку, лимфатические узлы).
С возрастом происходит инволюция тимуса, однако он никогда не замещается жировой тканью полностью и в нем продолжается выработка гуморальных факторов. Лимфопоэтическую функцию принимают на себя клетки Лангерганса в коже и брыжеечные лимфоидные клеточные скопления.
Лимфатические узлы - основа формирования иммунного ответа. В синусах лимфатических узлов макрофаги, захватив антиген, презентируют его В-лимфоцитам, непосредственно осуществляющим иммунный ответ. Субкапсулярная зона лимфатических узлов заполнена преимущественно Т-лимфоцитами и дендритными клетками, несущими большое количество молекул гистосовместимости II класса, необходимых для активации Т-лимфоцитов. Медуллярная зона заполнена более зрелыми клетками, секретирующими антитела.
Строение и функция пейеровых бляшек, расположенных по ходу тонкой кишки, аналогичны лимфоидным фолликулам селезенки и лимфатических узлов.
Схема кроветворения
Основные положения схемы кроветворения (отсутствие бессмертных, «самоподдерживающихся» стволовых клеток, возможность сокращения числа митозов в процессе созревания, клональный характер кроветворения со сменой клонов, наличие еще не выявленных, более ранних, чем стволовая клетка, предшественников и др.) были многократно подтверждены в течение многих лет. После утраты основного свойства стволовых и только стволовых клеток - способности к самоподдержанию и невозможности определения самого понятия «стволовость», единственным критерием принадлежности клеток к стволовому отделу остается пролиферативный потенциал, достаточный для мультилинейного восстановления кроветворения после депрессии собственного кроветворения.
Время определения молекулярных основ биологии стволовых клеток еще не пришло, и составить более или менее ясную картину оркестровки генов в ходе кроветворных дифференцировок пока не удается.
В нижних этажах кроветворного дерева изменений очень немного. Помимо восьми ранее известных линий кроветворных дифференцировок, выделены еще новые:
• естественные киллеры (клетки, участвующие в природном иммунитете, в том числе и против злокачественных клеток);
• профессиональные антигенпрезентирующие дендритные клетки.
В настоящее время различают 11-12 линий дифференцировки кроветворных клеток.
Схема кроветворения (рис. 54-1) начинается с единственного члена отдела тотипотентных предшественников - эмбриональной стволовой клетки. Эта клетка способна к образованию клеток всех тканей организма. Эмбриональные стволовые клетки выделяют из внутренней массы бластоциста на стадии примерно 100-120 клеток. В эмбриогенезе эти клетки быстро переходят на следующие стадии дифференцировки. Эмбриональные стволовые клетки образуются только в условиях остановки созревания клеток бластоциста вне организма, например, при культивировании в определенных условиях. Дифференцировка клеток бластоциста блокируется, и они способны пролиферировать практически бесконечно (больше 120 удвоений) без дифференцировки, малигнизации, изменений кариотипа и др. Снятие блока приводит к беспорядочной дифференцировке эмбриональной стволовой клетки. И хотя до разумного клинического применения эмбриональных стволовых клеток еще далеко, уже сейчас целесообразно поместить этот предшественник на вершину кроветворной иерархии.
Собственно кроветворение начинается с клеток стволового отдела. Популяция стволовых кроветворных клеток (СКК) немногочисленна и крайне гетерогенна. Клетки этого отдела находятся в состоянии дифференцировки и созревания, продвигаясь вниз по кроветворной иерархии. Никакого набора одинаковых клеток даже в суботделах стволовых кроветворных клеток не существует, нет дискретных отделов, разделенных четкими границами.
Отдел стволовых клеток включает предшественников, способных к мультипотентным дифференцировкам по всем линиям кроветворных клеток и обладающих высоким пролиферативным потенциалом. Этот отдел пока включает 3 члена. Первый и наиболее ранний из них - проСКК. Эта клетка, видимо, близка к промежуточным элементам, расположенным между тотипотентными эмбриональными клетками и ранними кроветворными предшественниками. Про-СКК находятся в состоянии глубокого покоя, они не пролиферируют в культуре в ответ на цитокины, не образуют колоний в селезенке in vivo или в полутвердых средах in vitro, при трансплантации начинают образовывать миелоидные клетки только через 8 мес, а лимфоидные - только после 10 мес. Неясно, участвуют ли вообще про-СКК в нормальном кроветворении или существуют в качестве резерва для особых ситуаций.
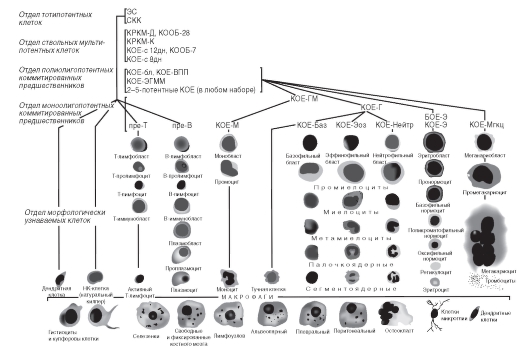
Рис. S4-1. Схема кроветворения: ЭС - эмбриональная стволовая клетка; СКК - стволовая кроветворная клетка; КОЕ - колониеобразующая единица
Второй член стволового отдела - клетка, способная длительно репопулировать облученное животное (ДР-СКК). Главная ее характеристика - высокий пролиферативный потенциал. Одна такая клетка может обеспечить поддержание мультилинейного кроветворения в течение всей жизни животного. Она даже способна восстановить кроветворение при пассаже к вторично-облученному реципиенту. ДР-СКК активно функционирует при трансплантации кроветворных тканей. Именно она обеспечивает эффекты, определяемые термином «трансплантация костного мозга». Стволовые клетки кроветворения человека имеют фенотип Lin-c-Kit+CD34+CD38-.
Последний член отдела - клетка, способная кратковременно репопулировать облученное животное (КР-СКК). Ее отличие от ДР-СКК только количественное: она способна полностью мультилинейно репопулировать облученный организм, однако эффект кратковременный, и через 4-6 нед ее кроветворные способности истощаются.
Очень близок к клеткам стволового отдела мультипотентный предшественник (МКП): эти клетки не способны длительно поддерживать кроветворение, хотя они мультипотентны и сохраняют весь набор кроветворных дифференцировок.
Клетки всей кроветворной иерархии представляют собой континуум клеток, пролиферативный потенциал которых постепенно снижается, а степень дифференцировки нарастает. В отделе стволовых клеток возможно движение не строго в одном направлении: КР-СКК и даже МКП могут вернуться к свойствам ДР-СКК или остановиться в этом движении, возвращаясь к состоянию глубокого покоя.
Следующий отдел - полипотентные коммитированные предшественники. Выделены 2 популяции предшественников:
• общий лимфоидный предшественник (ОЛП) - способен только к лимфоидным дифференцировкам без промежуточных стадий;
• общий миелоидный предшественник (ОМП) - дифференцируется только по миелоидным направлениям.
Они неспособны к сколько-нибудь длительному поддержанию кроветворения без подсева из стволового отдела.
Дифференцировка общего миелоидного предшественника включает ряд промежуточных стадий:
• общий предшественник гранулоцитов и макрофагов;
• гранулоцитарно-моноцитарную колониеобразующую единицу, из которой дифференцируются монопотентные предшественники:
- моноцитарная колониеобразующая единица;
- гранулоцитарная колониеобразующая единица;
- общий предшественник эритроцитов и мегакариоцитов, который дает начало эритроидному и тромбоцитарному ростку в костном мозге.
В схему кроветворения всегда включали только паренхиму костного мозга, т.е. кроветворные клетки, производные стволовой клетки кроветворения. Между тем в костном мозге существует еще одна стволовая клетка - мезенхимальная стволовая клетка (МСК), которая строит «дом» для кроветворных клеток, строму костного мозга. Целесообразно сопроводить схему иерархии кроветворных клеток иерархическим деревом мезенхимальных стволовых клеток, обеспечивающих не только поддержание кроветворения, но и в значительной степени его регуляцию (рис. 54-2).
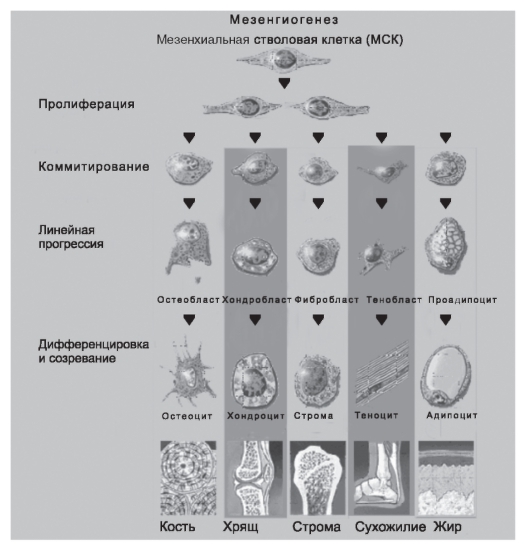
Рис. S4-2. Схема дифференцировки мезенхимальной стволовой клетки
Мезенхимальные стволовые клетки охарактеризованы значительно хуже, чем стволовые кроветворные клетки. В костном мозге обнаружены клетки, способные при культивировании давать колонии прилипающих клеток разной морфологии, главным образом фибробластоподобных. По мере пассажей клетки приобретают более однообразную морфологию и могут расти бесконечно. Такие клетки названы мультипотентными взрослыми предшественниками (МАРС - Multipotent Adult Progenitor Cells).
Основные данные о существовании мезенхимальной стволовой клетки и ее физиологическом значении были получены in vivo при имплантации фрагментов костного мозга, например, под капсулу почки. В этих условиях мезенхимальные стволовые клетки строят кроветворное микроокружение заново - образуются кость, строма костного мозга, внеклеточный матрикс и все компоненты нормальной костномозговой стромы. На такую строму из крови мигрируют стволовые кроветворные клетки, начинающие кроветворную дифференцировку. Остеокласты ремодулируют кость, образуется костный футляр с губчатой костью внутри, возникает эктопический очаг кроветворения.
Одной из главных характеристик стволовой кроветворной клетки считают ее способность к миграции с повторным заселением кроветворных территорий (инстинкт «дома»). Именно эта ее способность позволила использовать внутривенную трансплантацию стволовых кроветворных клеток. И в этом отношении мезенхимальная стволовая клетка существенно отличается от стволовой кроветворной клетки: она неспособна к миграции, не попадает в пригодные для заселения участки при внутривенном введении и даже у полных радиационных химер не участвует в создании кроветворного микроокружения. Различные миграционные характеристики - еще одно веское доказательство независимости стволовой кроветворной и мезенхимальной стволовой клеток.
Мезенхимальная стволовая клетка обладает способностью к дифференцировке во все клетки кроветворного микроокружения и характеризуется высоким «самоподдержанием». Открыты, по меньшей мере, 2 стромальных предшественника:
• мезенхимальная стволовая клетка - исходная мультипотентная клетка, способная к повторному переносу микроокружения, т.е. обладающая «самоподдержанием»;
• индуцибильный предшественник стромы - более зрелый мультипотентный элемент, который отвечает на индукционные влияния и при стимуляции (переносе в облученный организм) строит очаг
кроветворения, размер которого гораздо больше обычного (в нормальном реципиенте).
Существует внешнее сходство кроветворной и стромальной иерархий костного мозга. Однако по существу эти ткани принципиально отличаются. Основная задача кроветворной ткани - производство огромного количества клеток крови, имеющих относительно короткий жизненный цикл и потому нуждающихся в постоянном пополнении. Строма же кроветворной ткани представляет собой основу, «дом» для кроветворных клеток. Кроветворное микроокружение и обновление стромальных клеток происходят очень медленно, так как они имеют длительный жизненный цикл. Хотя строма постоянно перестраивается, интенсивность перестройки не идет ни в какое сравнение с темпом кроветворения. Например, для замены скелета у человека требуется около 10 лет. Представленная картина только намечает линии стромальных дифференцировок. Неизвестно, существуют ли олигомонопотентные стромальные предшественники и сколько клеточных элементов располагается на пути от мезенхимальной стволовой клетки до терминально дифференцированных клеток ряда. Пока доказана дифференцировка мезенхимальной стволовой клетки в костную ткань, хрящ, строму костного мозга, сухожилия, жировую ткань. Весьма вероятно, что мезенхимальная стволовая клетка способна дифференцироваться в гладкие мышцы сосудов. Менее ясна дифференцировка в эндотелий с последующим васкуло- и ангиогенезом.
В очень примитивном виде стромы можно ограничить двумя функциями:
• механической (образование скелета);
• кроветворной (создание кроветворного микроокружения). Основная его роль заключается в осуществлении регулирующих
влияний. В этом «доме» есть привилегированные помещения, ниши, в которых стволовые кроветворные клетки защищены как от внешних индуцирующих воздействий, так и от внутренних сигналов, что блокирует их дифференцировку и обеспечивает сохранение резерва стволовых кроветворных клеток. Другие участки стромы, включая клеточный матрикс, принимают участие в регуляции более зрелых клеток, разграничивая строму на участки преимущественно эритроидного или миелоидного кроветворения. Основы такой регуляции, видимо, связаны с градиентом концентрации цитокинов и ростовых факторов в зависимости либо от близости клеток, продуцирующих цитокины, либо от взаимодействия «рецептор-лиганд» на кроветворных и стромальных клетках.
В отличие от стволовой кроветворной клетки, использование в клинической практике мезенхимальной стволовой клетки только начато. Применение ее обычно требует создания искусственных трехмерных структур из биодеградированных материалов или двухмерных пленок. Существующие данные показывают перспективность этих исследований для ускорения заживления переломов, создания синовиальных поверхностей и даже ремоделирования суставов.
В предлагаемой схеме кроветворения впервые объединены обе категории стволовых клеток костного мозга, демонстрируется простой и очевидный факт: в физиологических условиях дифференцировки строго специфичны. Даже при общем происхождении (в данном случае - мезенхимальном) и расположении в одной и той же ткани (костном мозге) линии дифференцировок стволовой кроветворной клетки и мезенхимальной стволовой клетки никогда не перекрываются.
Глава 55. АНЕМИИ
55.1. ЖЕЛЕЗОДЕФИЦИТНЫЕ АНЕМИИ
Железодефицитная анемия (ЖДА) - гипохромная микроцитарная анемия, развивающаяся вследствие снижения количества железа в организме. Дефицит железа приводит к нарушению синтеза гемоглобина и уменьшению его содержания в эритроцитах.
ЭПИДЕМИОЛОГИЯ
Железодефицитная анемия - повсеместно распространенное заболевание, наиболее часто наблюдаемое у женщин репродуктивного возраста.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Дефицит железа может быть обусловлен следующими факторами:
• уменьшением поступления железа в организм на фоне длительного соблюдения несбалансированных диет, при вегетарианстве;
• нарушением всасывания железа в ЖКТ при ахлоргидрии, заболеваниях тонкой кишки или ее резекции;
• потерями железа при обильных менструациях, кровотечениях из ЖКТ (например, на фоне приема НПВС, при язвенном колите, геморрое), геморрагических синдромах, глистной инвазии, изолированном легочном гемосидерозе, телеангиэктазиях, гемоглобинурии и пр. Кроме того, дефицитом железа часто сопровождаются некоторые физиологические (беременность, лактация, период активного роста) и патологические (ХПН) состояния.
Патогенез железодефицитной анемии определяется 3 основными звеньями:
• нарушением синтеза гемоглобина в результате уменьшения запасов железа;
• генерализованными нарушениями пролиферации клеток;
• укорочением продолжительности жизни эритроцитов (последнее наблюдают преимущественно при тяжелом дефиците железа).
Железо входит в состав миоглобина, цитохромов, каталаз, пероксидаз, поэтому его дефицит, помимо гематологических проявлений, сопровождается возникновением целого ряда нарушений, связанных с патологией клеточных мембран и развитием трофических нарушений.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Железодефицитная анемия в большинстве случаев развивается постепенно, после длительного периода латентного дефицита железа, поэтому больные обычно адаптируются к низкому уровню гемоглобина, и симптоматика появляется только при достаточно выраженной анемии (концентрация гемоглобина - 80-100 г/л).
• Вне зависимости от причин дефицита железа, его клинические проявления стереотипны. Основные жалобы больных железодефицитной анемией - повышенная утомляемость, раздражительность, трудности в концентрации внимания, головокружение, головная боль, ощущение сердцебиения. При снижении концентрации гемоглобина до 70-80 г/л развиваются выраженные метаболические нарушения - боли в мышцах, вызванные накоплением лактата в тканях при физической нагрузке, парестезии, вазомоторные расстройства, снижение температуры тела.
• Дефицит железа сопровождается нарушением структуры и функций эпителиальных тканей, что проявляется уплощением, исчерченностью и ломкостью ногтей, сухостью, ломкостью и усиленным выпадением волос, болезненностью языка и атрофией его сосочков, возникновением ангулярного стоматита, дисфагии, ахлоргидрии и гастрита.
• Для железодефицитной анемии характерны извращение вкуса (желание есть мел, землю, испорченные мясные продукты, лед) и пристрастие к резким, иногда неприятным запахам (бензина, гуталина и пр.).
• Даже при латентном дефиците железа отмечают склонность к частым инфекциям, что связано с уменьшением количества Т-лимфоцитов и нарушением синтеза ИЛ-1 и ИЛ-2.
• В анализах крови выявляют снижение концентрации гемоглобина, уменьшение содержания гемоглобина в эритроцитах, гипохромию, микроцитоз и анизоцитоз эритроцитов, снижение гематокрита. Содержание лейкоцитов обычно в норме, СОЭ повышена, возможен незначительный ретикулоцитоз. Основные диагностические критерии железодефицитной анемии: снижение концентрации
железа в сыворотке крови, повышение общей и латентной железосвязывающей способности сыворотки крови, уменьшение степени насыщения трансферрина железом. • В красном костном мозге выявляют умеренную гиперплазию эритроидного ростка, не связанную с тяжестью клинических симптомов, обнаруживают эритрокариоциты с фрагментированным ядром и многоядерные клетки красного ряда.
Дифференциальная диагностика
Основные критерии дифференциальной диагностики железодефицитных состояний приведены в табл. 55-1.
Таблица 55-1. Дифференциальная диагностика железодефицитных состояний
ЛЕЧЕНИЕ
Попытки компенсировать дефицит железа диетой несостоятельны. Для успешной терапии железодефицитной анемии необходимо назначение ЛС с высоким содержанием железа и хорошей всасываемостью. Терапия железодефицитной анемии должна быть длительной, поскольку восстановление запасов железа в депо происходит не ранее чем через 3 мес от начала лечения (хотя концентрация гемоглобина может нормализоваться к 8-й неделе).
Обычно применяют препараты железа сульфата (в таблетках или в виде сиропа). При индивидуальной непереносимости железа сульфата применяют препараты железа глюконата и фумарата. Доза для взрослых составляет приблизительно 200 мг железа, для детей - 1,5-2 мг/кг. В среднем в течение первых 20 дней терапии усваивается 605 мг железа (13,5% принятого количества). Дополнительное назначение аскорбиновой кислоты (по 200 мг на каждые 30 мг железа) повышает абсорбцию на 30%, а янтарной кислоты (185 мг на 37 мг железа) - с 13,5 до
21%.
Основные побочные эффекты пероральных препаратов железа - тошнота, боли в животе, запор. При выраженных побочных эффектах назначают препарат, содержащий другую соль железа, либо уменьшают дозу в 2 раза. Отсутствие эффекта от терапии пероральными препаратами железа может быть связано с наличием сопутствующей патологии или сохранением причины дефицита железа, неправильным подбором дозы, мальабсорбцией железа.
Парентеральное введение препаратов железа более эффективно, однако чаще сопровождается выраженными побочными эффектами. Назначение парентеральных препаратов показано в следующих случаях:
• при осутствии эффекта адекватной терапии пероральными препаратами железа или наличие противопоказаний к их применению (например, они могут ухудшить течение язвенного колита);
• невозможности соблюдения пациентом режима приема и дозирования препарата;
• невозможности корригировать потери железа с помощью пероральных препаратов (потери железа превышают возможный эффект терапии, например, при врожденных телеангиэктазиях);
• нарушении всасывания железа в кишечнике;
• невозможности определения обмена железа (у пациентов на гемодиализе).
Для парентерального введения используют комплекс железа с декстранами. Непосредственно во время введения возможны развитие анафилактических реакций, снижение АД, тошнота, головная боль, появление уртикарной сыпи. Отсроченные реакции проявляются артралгиями, миалгиями, лимфаденопатией, лихорадкой. Парентеральное введение железа значительно повышает риск развития гемосидероза. Трансфузии эритроцитарной массы проводят только при выраженной анемии, угрожающей жизни пациента.
55.2. АНЕМИИ, СВЯЗАННЫЕ С НАРУШЕНИЕМ СИНТЕЗА ДНК (МЕГАЛОБЛАСТНЫЕ АНЕМИИ)
Мегалобластные анемии - группа заболеваний, характеризуемых появлением в красном костном мозге мегалобластов - клеток красного ряда больших размеров с измененной структурой ядра, которые прослеживаются на всех стадиях дифференцировки эритроидных предшественников. Появление мегалобластов связано с нарушением синтеза ДНК и замедлением созревания клеток. Образование РНК не нарушается, поэтому за увеличившийся промежуток между делениями клеток происходят избыточный синтез, накопление гемоглобина и увеличение размеров цитоплазмы.
Нарушение синтеза ДНК с формированием характерной мегалобластной картины в красном костном мозге чаще всего связано с дефицитом витамина B12 или фолиевой кислоты.
Витамин B12-дефицитная анемия
Витамин В12-дефицитная анемия - группа заболеваний, связанных с дефицитом цианокобаламина или нарушением его метаболизма. Витамин В12-дефицитную анемию в основном наблюдают в пожилом возрасте, несколько чаще у женщин.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Идиопатическая форма витамин В12-дефицитной анемии (пернициозная анемия) развивается в результате недостаточного поступления в организм цианокобаламина вследствие нарушения выработки внутреннего фактора (Касла) - гликопротеина, синтезируемого париетальными клетками слизистой оболочки желудка. Пернициозная анемия - аутоиммунное заболевание, при котором происходит образование АТ к париетальным клеткам желудка или к внутреннему фактору, в большинстве случаев сочетается с фундальным гастритом и ахлоргидрией. Появление АТ к париетальным клеткам или фактору Касла также возможно при других аутоиммунных заболеваниях - сахарном диабете, тиреоидите Хасимото, болезни Аддисона, микседеме и др. У перенесших тотальную гастрэктомию пациентов витамин В12-дефицитная анемия развивается через 5-8 лет и более после операции (до этого гемопоэз происходит за счет витамина В12, депонированного в печени).
Нарушение синтеза внутреннего фактора возможно при алкоголизме, вследствие токсического поражения слизистой оболочки желудка.
Дефицит витамина B12 может быть связан с нарушением его всасывания при заболеваниях тонкой кишки (тяжелом хроническом энтерите, терминальном илеите, дивертикулезе тонкой кишки), а также с инвазией широким лентецом и избыточным ростом кишечной микрофлоры при синдроме слепой кишки (слепая петля тонкой кишки после операции), поглощающими большое количество цианокобаламина.
Кроме того, существуют редкие наследственные формы пернициозной анемии:
• пернициозная анемия у подростков с полигландулярным аутоиммунным синдромом (*240300, 21q22.3, р);
• пернициозная анемия ювенильная с относительной недостаточностью всасывания витамина B12 и протеинурией (*261100, синдром Иммурслунда-Грасбека, 10p12.1, MGA1, р);
• врожденная пернициозная анемия (*261000, хромосома 11, мутация гена GIF, р).
Коферментные формы витамина B12 (метилкобаламин и дезоксиаденозинкобаламин) участвуют в переносе метильных групп (трансметилировании) и водорода, в частности, при биосинтезе метионина из гомоцистеина. Нарушение этого процесса при дефиците витамина B12 приводит к недостаточному образованию активных метаболитов фолиевой кислоты, что проявляется мегалобластным кроветворением (см. ниже «Фолиеводефицитная анемия»). Вследствие недостаточного синтеза метионина также нарушается образование компонентов миелина, что обусловливает демиелинизацию, приводящую к неврологическим расстройствам.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Клинически дефицит витамина B12 характеризуется поражением кроветворной ткани, пищеварительной и нервной систем. В связи с медленным развитием анемии больные обычно поздно обращаются к врачу, и между возникновением симптомов заболевания (слабости, повышенной утомляемости, сердцебиени при физической нагрузке) и постановкой диагноза в среднем проходит не менее 15 мес.
При тяжелой анемии кожные покровы приобретают лимонножелтый оттенок, на них появляются участки гипер- и гипопигментации, напоминающие витилиго. Иктеричность склер возникает редко.
У 50% больных дефицитом витамина B12 в дебюте заболевания наблюдают болезненность языка, появление на нем участков воспаления и атрофии сосочков. В 65% случаев пациенты жалуются на снижение
аппетита, иногда - на чувство дискомфорта в эпигастральной области. Желудочная секреция, как правило, снижена, возможна стойкая ахлоргидрия. Иногда отмечают незначительное увеличение селезенки и печени.
Тяжесть неврологической симптоматики при пернициозной анемии не коррелирует с выраженностью гематологических нарушений. Наиболее типичное неврологическое проявление дефицита витамина B12 - фуникулярный миелоз (дегенерация задних и боковых столбов спинного мозга), характеризуемый расстройством глубокой чувствительности, заднестолбовой атаксией и спастическими парезами конечностей. Также возможны полиневропатия, деменция и психические расстройства (бред, галлюцинации и пр.).
В анализах крови выявляют умеренную, как правило, гиперхромную анемию, наличие мелких фрагментов эритроцитов (шизоцитов) наряду с очень крупными (диаметром >12 мкм) мегалоцитами, выраженный пойкилоцитоз, лейкопению, гиперсегментацию ядер нейтрофилов, тромбоцитопению. В сыворотке крови повышена концентрация непрямого билирубина (за счет разрушения мегалобластов в красном костном мозге), в отличие от гемолитических анемий, одновременно определяют значительное увеличение активности ЛДГ. При подозрении на дефицит витамина B12 обязательно нужно провести определение его концентрации в сыворотке крови (в норме - 160-950 пг/мл).
В красном костном мозге выявляют большое количество мегалобластов, однако если пациент за несколько дней до исследования принимал даже минимальное количество цианокобаламина (например, в составе поливитаминных препаратов), мегалобластоз красного костного мозга может быть выражен слабо либо вообще отсутствовать.
Дифференциальная диагностика
Витамин В12-дефицитные анемии, вне зависимости от их этиологии, имеют схожие клинические и морфологические проявления, поэтому во всех случаях необходимо всестороннее обследование пациента для выявления причины дефицита цианокобаламина (инвазии широким лентецом, хронического энтерита и т.п.). Следует помнить, что у больных пернициозной анемией повышен риск развития рака желудка.
Клетки красного ряда, очень напоминающие мегалобласты, могут появляться при остром эритробластном лейкозе, причем, как и при витамин В12-дефицитной анемии, также возможны легкая желтушность кожных покровов, лейко- и тромбоцитопения. Однако при остром эритробластном лейкозе отсутствуют выраженные анизо- и пойкилоцитоз,
в красном костном мозге наряду с мегалобластоподобными клетками в большом количестве обнаруживают бластные клетки, кроме того, лечение витамином B12 не оказывает влияния ни на картину крови, ни на состояние больного.
ЛЕЧЕНИЕ
Для лечения применяют цианокобаламин по 200-500 мкг 1 раз в день подкожно в течение 4-6 нед. Через 8-10 дней после начала лечения происходит резкое увеличение количества ретикулоцитов в крови (ретикулоцитарный криз), повышается концентрация гемоглобина, в крови исчезает выраженный анизоцитоз, а в красном костном мозге - мегалобластоз. После нормализации состава крови (обычно через 1,5-2 мес) цианокобаламин вводят 1 раз в неделю в течение 2-3 мес, затем в течение полугода 2 раза в месяц (в тех же дозах, что и в начале курса). В дальнейшем с профилактической целью проводят 1-2 курса лечения в год (по 5-6 инъекций на курс).
Фолиеводефицитная анемия
Фолиеводефицитная анемия - мегалобластная анемия, развивающаяся вследствие дефицита фолиевой кислоты или нарушения ее утилизации в процессе эритропоэза.
ЭПИДЕМИОЛОГИЯ
Фолиеводефицитную анемию наблюдают преимущественно у беременных, страдающих гемолитической анемией, недоношенных детей, при заболеваниях тонкой кишки, алкоголизме, а также при длительном приеме противосудорожных препаратов (фенобарбитала, фенитоина).
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Дефицит фолиевой кислоты может быть связан:
• с ее недостаточным поступлением [несбалансированная диета с малым количеством свежих овощей и фруктов, вскармливание грудных детей козьим молоком (содержит малое количество фолиевой кислоты - 6 нг/г, в коровьем и женском молоке - 50 нг/г)];
• нарушением всасывания при синдроме мальабсорбции любой этиологии (после резекции тонкой кишки, при тропической спру, целиакии и пр.);
• повышенной утилизацией [при гемолизе, гемобластозах и других онкологических заболеваниях (в последнем случае причиной так-
же может быть метотрексат - антагонист фолиевой кислоты, входящий в состав многих схем химиотерапии)]. Мегалобластную анемию, связанную с дефицитом фолиевой кислоты, выявляют у 20-40% лиц, страдающих алкоголизмом. Цирроз печени практически всегда сопровождается дефицитом фолиевой кислоты, хотя корреляция между тяжестью поражения печени и выраженностью анемии отсутствует. Механизм развития фолиеводефицитной анемии в этих случаях связан с нарушением накопления фолиевой кислоты в печени.
Активные метаболиты фолиевой кислоты осуществляют перенос одноуглеродных групп (формильной, метильной, оксиметильной и метиленовой), в том числе при биосинтезе пуринов и пиримидинов. Именно поэтому дефицит фолиевой кислоты сопровождается нарушением синтеза ДНК, что замедляет процесс нормального созревания гемопоэтических клеток и расстраивает синхронность созревания и гемоглобинизации эритроцитов, приводя к мегалобластному кроветворению.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Клиническая картина, изменения в анализах крови и красном костном мозге аналогичны таковым при дефиците витамина В12. От последнего фолиеводефицитная анемия отличается отсутствием неврологических проявлений и глоссита. Подтвердить диагноз фолиеводефицитной анемии можно по снижению концентрации фолиевой кислоты в эритроцитах и сыворотке крови (однако на практике эти исследования малодоступны).
ЛЕЧЕНИЕ
Обнаружение мегалобластной анемии при состояниях, которые могут сопровождаться дефицитом фолиевой кислоты, считают достаточным основанием для ее назначения по 5-15 мг/сут внутрь (указанная доза обеспечивает лечебный эффект даже после резекции тонкой кишки, при энтеритах и т.п.). Ретикулоцитарный криз через 1,5-2 нед после начала лечения свидетельствует об эффективности терапии.
55.3. ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
Гемолитические анемии связаны с усиленным разрушением эритроцитов. При любых гемолитических анемиях в крови повышается концентрация продуктов распада эритроцитов - билирубина или сво-
бодного гемоглобина. Другой важный признак - значительное увеличение в крови количества ретикулоцитов за счет повышения продукции клеток красной крови. В красном костном мозге при гемолитических анемиях значительно увеличивается количество клеток красного ряда.
Аутоиммунные гемолитические анемии
Аутоиммунные гемолитические анемии - группа заболеваний, характеризуемых усиленным разрушением эритроцитов под воздействием аутоантител.
ЭПИДЕМИОЛОГИЯ
Аутоиммунные гемолитические анемии - наиболее частая причина гемолиза. Заболеваемость составляет 1 на 75 000 населения.
КЛАССИФИКАЦИЯ, ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
В основу классификации аутоиммунных гемолитических анемий положены особенности АТ, вызывающих гемолиз. Чаще всего АТ классифицируют по диапазону температур, при которых возможно возникновение гемолиза. Тепловые АТ разрушают эритроциты при температуре не менее 37 °С, в основном они представлены IgG, реже - IgM и IgA. Холодовые AT разрушают эритроциты при температуре менее 37 °C (их действие достигает максимума при 0 °C); они представлены преимущественно и значительно реже - IgG. При аутоиммунной гемолитической анемии с тепловыми АТ разрушение эритроцитов происходит в селезенке, а при аутоиммунной гемолитической анемии с холодовыми АТ гемолиз преимущественно внутрисосудистый, опосредованный системой комплемента. Кроме того, существуют двухфазные гемолизины (фиксация АТ на эритроцитах происходит при низкой температуре, а гемолиз - при последующем повышении температуры тела до 37 °C), обусловливающие развитие пароксизмальной холодовой гемоглобинурии. • Тепловые антиэритроцитарные АТ могут образовываться при многих заболеваниях - вирусных инфекциях (гепатитах, инфекциях цитомегаловирусом, инфекционном мононуклеозе, краснухе), хроническом лимфолейкозе, лимфомах, злокачественных опухолях, аутоиммунных болезнях (например, СКВ), иммунодефицитных состояниях, а также при приеме некоторых ЛС - метилдопы, пенициллинов, сульфаниламидов. Риск развития аутоиммунных гемолитических анемий повышен у пациентов с HLA-B7 (при приеме метилдопы гемолиз развивается у 60% носителей этого фенотипа, а в остальной популяции - не более чем у 20%).
• Развитие аутоиммунной гемолитической анемии с холодовыми АТ возможно при микоплазменной инфекции, инфекционном мононуклеозе, инфекции цитомегаловирусом, паротите, сифилисе, малярии, инфекционном эндокардите и иммунокомплексной патологии.
• Пароксизмальная холодовая гемоглобинурия - относительно редкое заболевание, на нее приходится 1,6-5,1% случаев гемолиза у взрослых (у детей до 5 лет - до 40%). Большинство случаев пароксизмальной холодовой гемоглобинурии описаны при третичном и врожденном сифилисе, однако в редких случаях она может развиться при кори, паротите, инфекционном мононуклеозе.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Течение аутоиммунного гемолиза, связанного с наличием тепловых АТ, очень вариабельно - от незначительного, выявляемого только по уменьшению продолжительности жизни эритроцитов, до фульминантного, угрожающего жизни пациента. Основные симптомы аутоиммунных гемолитических анемий:
• слабость;
• головокружение;
• лихорадка;
• желтуха;
• бледность кожных покровов;
• снижение массы тела;
• одышка.
Потемнение мочи наблюдают у 30% больных. Спленомегалию выявляют практически у всех пациентов, увеличение размеров печени - у 45%, лимфаденопатию - у 34%, повышение концентрации билирубина в сыворотке крови - у 21%, отеки - у 6%. Возможен цианоз губ, крыльев носа, ушей, связанный с внутрисосудистой агглютинацией эритроцитов и нарушением микроциркуляции.
Идиопатическая аутоиммунная гемолитическая анемия с холодовыми AT обычно возникает в возрасте 70-80 лет. Основные клинические признаки этого заболевания - внутрисосудистый гемолиз и цианоз пальцев рук, носа, ушей при воздействии низких температур (по типу синдрома Рейно), иногда с развитием необратимого поражения тканей и некрозов. Концентрация гемоглобина редко бывает ниже 70 г/л.
Наиболее типичный симптом пароксизмальной холодовой гемоглобинурии - появление через несколько часов после локального или
общего переохлаждения на фоне озноба и лихорадки темно-коричневой или черной мочи, тянущих болей в спине, ногах, животе, сопровождающихся рвотой, диареей. Концентрация гемоглобина снижается до
50 г/л.
Лабораторные исследования
При аутоиммунной гемолитической анемии, обусловленной тепловыми АТ, в анализах крови определяют значительное увеличение содержания ретикулоцитов (до 1,0х1012/л). Концентрация билирубина повышается до 45 ммоль/л в основном за счет непрямой фракции, в моче увеличивается содержание уробилина, в кале - стеркобилина.
У 65% пациентов положительна прямая проба Кумбса, выявляющая АТ, фиксированные на поверхности эритроцитов. Более достоверны результаты агрегат-гемагглютинационной пробы (используют тестэритроциты, к которым ковалентно присоединены белки иммунной сыворотки). С ее помощью можно обнаружить незначительные количества фиксированных на эритроцитах Ig.
ЛЕЧЕНИЕ
При аутоиммунной гемолитической анемии с тепловыми АТ препараты выбора - ГК. Преднизолон назначают в начальной дозе 40 мг/ м2 в сутки до достижения концентрации гемоглобина 100 г/л с последующим уменьшением дозы до 20 мг в течение 4-6 нед и медленной отменой в течение 3-4 мес. Введение препаратов IgG в больших дозах (до 1000 мг/кг) в течение 5 дней приводит к прекращению гемолиза у 50-90% больных.
Спленэктомию проводят только при неэффективности терапии ГК (у 15-20% пациентов), при возникновении тяжелых осложнений терапии ГК и после получения доказательств усиленной секвестрации эритроцитов в селезенке (сцинтиграфия с 51Cr).
Цитостатики назначают при непереносимости больших доз преднизолона и невозможности спленэктомии. Чаще всего применяют азатиоприн или циклофосфамид в сочетании с преднизолоном.
При аутоиммунной гемолитической анемии с холодовыми АТ наиболее важным считают соблюдение соответствующего образа жизни (исключение переохлаждений) и улучшение реологических свойств крови для предотвращения развития периферических некрозов. Эффективность спленэктомии и ГК сомнительна. Патогенетически наиболее оправдано применение циклофосфамида (для подавления синтеза АТ). Плазмаферез применяют в сочетании с химиотерапией.
Пароксизмальная холодовая гемоглобинурия самостоятельно проходит в течение нескольких дней. Необходима адекватная терапия заболевания, ставшего причиной гемолиза (например, сифилиса).
Пароксизмальная ночная гемоглобинурия
Пароксизмальная ночная гемоглобинурия (синдром МаркиафавыМикели) - заболевание, характеризуемое преходящими эпизодами внутрисосудистого гемолиза, возникающего преимущественно в ночное время. В ряде случаев заболевание протекает в виде вялотекущего гемолиза, сопровождаемого панцитопенией, дефицитом железа, эпизодами тромботических осложнений.
ЭПИДЕМИОЛОГИЯ
Пароксизмальная ночная гемоглобинурия (ПНГ) - редкая форма приобретенных гемолитических анемий. Заболеваемость не превышает 1 случая на 500 000 населения.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Гемолиз при пароксизмальной ночной гемоглобинурии обусловлен появлением клона эритроцитов с выраженными дефектами белковых и липидных компонентов клеточной мембраны. Нарушение структуры мембраны обусловливает повышенную чувствительность эритроцитов к комплементассоциированному лизису. Гемолиз происходит при изменении pH крови и при активации системы комплемента.
Среди лейкоцитов (за исключением Т-лимфоцитов) также обнаруживают патологические популяции с повышенной чувствительностью к лизису, нарушенной способностью к миграции, уменьшением фагоцитарной активности.
Усиление тромботической активности при пароксизмальной ночной гемоглобинурии связано как с внутрисосудистым разрушением эритроцитов и стимуляцией ДВС, так и с изменениями мембраны тромбоцитов (на тромбоцитах патологического клона фиксируется С3-компонент комплемента, стимулирующий выброс активаторов свертывания крови).
Патологический клон при пароксизмальной ночной гемоглобинурии присутствует и на уровне кроветворных предшественников: с помощью цитогенетических методов в красном костном мозге обнаруживают 2 или 3 патологических клона, не несущих хромосомных аберраций, специфичных для какой-либо нозологии. С другой стороны, существует этио-
логическая связь пароксизмальной ночной гемоглобинурии с острым миелобластным лейкозом, апластической и сидеробластной анемиями.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Наиболее частые симптомы - жалобы, связанные с анемией вследствие гемолиза и нарастающего дефицита железа (общая слабость, повышенная утомляемость и пр.), желтушность кожных покровов и слизистых оболочек. Характерный симптом гемоглобинурии - изменение цвета мочи - наблюдают только у 25% пациентов, причем во многих случаях он не связан с эпизодом гемолиза. Усиление гемолиза провоцируют инфекции, переохлаждение, гемотрансфузии, вакцинации, оперативные вмешательства. Гемолиз клинически проявляется:
• болями за грудиной, в животе, поясничной области;
• сонливостью;
• головной болью;
• лихорадкой.
У некоторых больных манифестация заболевания происходит по типу аплазии кроветворения (лейкопения, тромбоцитопения, анемия, геморрагический синдром, присоединение тяжелых инфекционных осложнений). При гистологическом исследовании красного костного мозга обнаруживают преобладание жирового компонента над активным кроветворным. Панцитопению в таких случаях рассматривают как признак прогрессирования пароксизмальной ночной гемоглобинурии.
Наиболее тяжелым из тромботических осложнений, характерных для пароксизмальной ночной гемоглобинурии, считают тромбоз печеночных вен (синдром Бадда-Киари), сопровождаемый резким увеличением размеров печени, нарастающим асцитом, расширением вен пищевода. Поражение почек с развитием ОПН наиболее вероятно в момент гемолитического криза, однако постоянная гемосидеринурия может сопровождаться развитием канальцевого нефрита с гематурией, протеинурией, снижением клиренса креатинина.
В анализах крови выявляют анемию (концентрация гемоглобина снижается до 60 г/л), возможны гипохромия и микроцитоз, лейкопения с относительным лимфоцитозом. Тромбоцитопению не считают обязательным признаком пароксизмальной ночной гемоглобинурии.
В моче выявляют уробилиноген, при интенсивном гемолизе - гемоглобин, повышенное содержание железа. Как следствие канальцевого нефрита, возможны гипостенурия, гематурия, снижение клиренса креатинина.
Специфичны для пароксизмальной ночной гемоглобинурии пробы Хема (кислотный тест) и Хартманна (сахарозный тест), основанные на выявлении характерной для эритроцитов патологического клона чувствительности к комплементу.
Дифференциальная диагностика
Пароксизмальную ночную гемоглобинурию необходимо исключать:
• во всех случаях внутрисосудистого гемолиза, особенно сопровождаемых гемоглобинурией;
• при сочетании панцитопении с гемолизом;
• наличии множественных тромбозов, особенно локализуемых в брюшной полости.
Наибольшую трудность представляет дифференциальная диагностика пароксизмальной ночной гемоглобинурии с аутоиммунной гемолитической анемией (АИГА), связанной с холодовыми АТ (табл. 55-2), и наследственной дизэритропоэтической анемией (в последнем случае основным диагностическим критерием являются положительные результаты сахарозного теста).
Таблица 55-2. Дифференциальная диагностика пароксизмальной ночной гемоглобинурии с аутоиммунной гемолитической анемией
ЛЕЧЕНИЕ
Учитывая наличие патологического клона, определяемого на уровне ранних кроветворных предшественников, наиболее эффективным методом лечения пароксизмальной ночной гемоглобинурии считают трансплантацию аллогенного красного костного мозга.
ГК (преднизолон по 15-40 мг/сут) и андрогены показаны в случаях гипоплазии красного костного мозга. Подобная терапия приводит к уменьшению потребности в трансфузиях, однако часто сопровождается многочисленными осложнениями.
Трансфузии отмытых эритроцитов проводят для купирования анемии. После трансфузий возможно длительное улучшение состояния больных в связи с уменьшением продукции собственных патологических эритроцитов.
Для профилактики тромботических осложнений назначают непрямые антикоагулянты. При наличии доказанных тромбозов любой локализации необходимо введение гепарина натрия.
Эффективность спленэктомии и применения антиоксидантов (витамина E) не доказана.
Наследственные гемолитические анемии
Причиной наследственных гемолитических анемий могут быть следующие нарушения.
• Патологические изменения мембраны эритроцитов, определяющие изменение их формы и уменьшение устойчивости к механическим воздействиям.
• Патология ферментных систем, обусловливающая повышение подверженности гемолизу под действием экзогенных факторов.
• Изменения формы эритроцитов, связанные с изменениями структуры цепей глобина (серповидноклеточная анемия) или нарушением синтеза одной из них (талассемии).
Микросфероцитарная гемолитическая анемия
Микросфероцитарная гемолитическая анемия (болезнь Минковского-Шоффара) - наследственное заболевание, характеризуемое гемолизом вследствие неполноценности структурного белка (спектрина) клеточной мембраны эритроцитов.
ЭПИДЕМИОЛОГИЯ
Микросфероцитарная гемолитическая анемия - одна из наиболее частых форм наследственных гемолитических анемий, распространена во всем мире.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Наследственные дефекты спектринов [тип I - дефект гена β-спектрина (*182870, 14q22-q23.2, ген SPTB, <R); тип II - дефект гена анкирина (*182900, 8p11.2, ген ANK1, Ʀ); тип III (IIIА) - дефект гена α-спектрина (*270970, 1q21, ген SPTA1, ρ)] обусловливают повышенную проницаемость мембраны для ионов натрия. Вследствие накопления избытка натрия и воды эритроциты приобретают сферическую форму и повреждаются при прохождении через синусы селезенки. Происходит захват поврежденных клеток макрофагами (внутриклеточный гемолиз). Постоянный избыточный распад гемоглобина приводит к непрямой гипербилирубинемии и желтухе.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Внутриклеточным распадом эритроцитов определяются клинические проявления болезни:
• желтуха;
• увеличение селезенки;
• анемия;
• склонность к образованию камней в желчном пузыре;
• ретикулоцитоз.
Хотя заболевание начинается с рождения, его клиническая манифестация возможна в любом возрасте. Персистирующий гемолиз сопровождается гиперплазией красного костного мозга, что приводит к нарушению формирования костей: деформации челюстей с неправильным расположением зубов, высокому нёбу, выступающему лбу и т.п. Во всех случаях увеличена селезенка. Вследствие выделения с желчью большого количества билирубина во многих случаях развивается желчнокаменная болезнь. Болезненность в области желчного пузыря и незначительное увеличение печени - частые явления при наследственном микросфероцитозе. При развитии механической желтухи (вследствие обтурации желчных протоков билирубиновыми камнями) свойственная гемолизу непрямая билирубинемия сменяется прямой.
В анализах крови выявляют микросфероцитоз, выраженный ретикулоцитоз (до десятков процентов), нормохромную анемию. Характерно
снижение осмотической резистентности эритроцитов. Во время гемолитического криза возможен нейтрофильный лейкоцитоз. В редких случаях возникает так называемый апластический криз (связанный с инфицированием парвовирусом В19), при котором усиленный гемолиз в течение нескольких дней не сопровождается стимуляцией эритропоэза, из периферической крови исчезают ретикулоциты, быстро нарастает анемия, а концентрация билирубина уменьшается.
Дифференциальная диагностика
Сфероцитоз эритроцитов возможен при аутоиммунных гемолитических анемиях. Однако в последнем случае отсутствуют костные изменения и семейный анамнез заболевания. В сомнительных случаях необходимо выполнение прямой пробы Кумбса, которая положительна в большинстве случаев аутоиммунных гемолитических анемий и отрицательна при наследственном микросфероцитозе.
ЛЕЧЕНИЕ
Радикальный метод лечения - спленэктомия, которая показана при выраженном гемолизе, анемии, желчнокаменной болезни. У детей спленэктомию желательно проводить после 7-8 лет, однако выраженную анемию и тяжелые гемолитические кризы считают прямым показанием к операции в любом возрасте. После операции у всех больных наступает клиническая ремиссия (хотя сфероцитоз эритроцитов и лабораторные признаки гемолиза сохраняются). При апластических кризах переливают эритроцитарную массу, в некоторых случаях назначают преднизолон в дозе 40-60 мг/сут.
Энзимопатические гемолитические анемии
Энзимопатические гемолитические анемии - группа заболеваний, характеризуемых недостаточностью ферментов эритроцитов, приводящей к постоянному гемолизу, или гемолитическими кризами. Наиболее распространенное заболевание этой группы - анемия при недостаточности глюкозо-6-фосфатдегидрогеназы.
ЭПИДЕМИОЛОГИЯ
Заболевание широко распространено в странах Азии, Африки, Средиземноморского бассейна. В России недостаточность глюкозо-6фосфатдегидрогеназы наблюдают преимущественно у выходцев из Закавказья, хотя спорадические случаи регистрируют повсеместно.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Ген глюкозо-6-фосфатдегидрогеназы (*305900, Xq28) расположен на X-хромосоме, поэтому болеют преимущественно мужчины. У женщин, гетерозиготных по патологическому гену, существуют 2 популяции эритроцитов - с нормальной активностью фермента (контролируемые нормальной X-хромосомой) и со сниженной активностью (контролируемые дефектной X-хромосомой). Ген глюкозо-6-фосфатдегидрогеназы отличается очень высоким полиморфизмом (известно >300 аллелей), что обусловливает значительную фенотипическую вариабельность: в одних случаях активность фермента лишь слегка ниже нормальной, в других - почти полностью отсутствует. Глюкозо-6-фосфатдегидрогеназа необходима для поддержания нормального внутриклеточного содержания глутатиона, защищающего сульфгидрильные группы гемоглобина и мембрану эритроцитов от окисления. При недостаточности фермента воздействие различных прооксидантных факторов приводит к повреждению клеточных мембран эритроцитов и выпадению в осадок цепей глобина, что способствует повышенному разрушению эритроцитов в селезенке.
Гемолиз при недостаточности глюкозо-6-фосфатдегидрогеназы могут провоцировать некоторые пищевые продукты (конские бобы) и многие ЛС - хинин, мепакрин, примахин, сульфаниламиды, нитрофураны, нитроксолин, налидиксовая кислота, изониазид, фтивазид, ацетилсалициловая кислота, менадиона натрия бисульфит (викасол♠) и пр.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Клинические проявления недостаточности глюкозо-6-фосфатдегидрогеназы весьма вариабельны: возможны как гемолитические кризы, возникающие при воздействии провоцирующих факторов, так и хроническое течение гемолиза с обострениями под влиянием тех же факторов. В зависимости от выраженности клинических симптомов и активности фермента в эритроцитах выделяют несколько вариантов течения заболевания.
• При снижении активности фермента до 0-10% нормальной развивается хроническая гемолитическая анемия, прием некоторых ЛС провоцирует развитие гемолитических кризов.
• При активности фермента 10-60% нормальных значений клинические проявления выражены слабо и связаны с приемом высоких
доз ЛС.
• При субнормальной активности фермента заболевание протекает практически бессимптомно.
Гемолитические кризы обычно развиваются на 2-3-й день приема ЛС, сопровождаются появлением в моче гемосидерина и свободного гемоглобина, небольшой желтухой. Продолжение приема ЛС приводит к тяжелому внутрисосудистому гемолизу с повышением температуры тела, болями в костях рук и ног, снижением АД, в тяжелых случаях возможно развитие анемической комы. В анализах крови обнаруживают нейтрофильный лейкоцитоз со сдвигом влево до миелоцитов, выраженную анемию, ретикулоцитоз, умеренное повышение концентрации билирубина за счет непрямой фракции. Тяжелый гемолитический криз может вызвать острый некротический нефроз с развитием ОПН.
В качестве отдельной клинической формы недостаточности глюкозо6-фосфатдегидрогеназы выделяют фавизм - острый гемолитический синдром при употреблении в пищу конских бобов или попадании пыльцы этого растения в дыхательные пути. Клинические проявления аналогичны таковым при лекарственном гемолитическом кризе, но развиваются через несколько часов после употребления конских бобов и обычно протекают тяжело, с формированием ОПН. Гемолиз, спровоцированный цветочной пыльцой конских бобов, развивается через несколько минут после контакта с ней и протекает легко.
Достоверное подтверждение диагноза недостаточности глюкозо-6фосфатдегидрогеназы возможно только при использовании методов, определяющих активность фермента в эритроцитах.
Дифференциальная диагностика
В дифференциальной диагностике с аутоиммунными гемолитическими анемиями определенную помощь оказывает проба Кумбса. Хронические формы гемолиза необходимо дифференцировать от талассемии и других гемоглобинопатий, зона распространения которых совпадает с регионами, эндемичными по недостаточности глюкозо-6фосфатдегидрогеназы.
ЛЕЧЕНИЕ
Основное терапевтическое мероприятие - отмена ЛС, вызвавшего гемолитический криз. Для уменьшения интенсивности гемолиза назначают рибофлавин по 0,015 г 2-3 раза в день внутрь. При тяжелом гемолитическом кризе для предупреждения развития ОПН проводят инфузии 5% раствора натрия гидрокарбоната (декстран противопоказан) и назначают фуросемид (внутривенно в дозе 40-60 мг и более) для стимуляции диуреза. При развитии анурии проводят плазмаферез, при
необходимости - гемодиализ. При тяжелой анемии с риском развития анемической комы проводят трансфузии отмытых эритроцитов.
Серповидноклеточная анемия
Серповидноклеточная анемия - тяжелое наследственное заболевание, связанное с патологией гемоглобина, вызывающей изменение формы и эластичности эритроцитов, клинически проявляющееся гемолитическими кризами, инфарктами внутренних органов и формированием костной и суставной патологии.
ЭПИДЕМИОЛОГИЯ
Серповидноклеточная анемия распространена преимущественно в тропических и субтропических регионах Африки, Америки, Средиземноморья, Ближнего Востока и странах Карибского бассейна. В России заболевание наблюдают редко.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Заболевание связано с мутацией гена HBB (*141900, 11p15.5, Ʀ), вследствие чего синтезируется аномальный гемоглобин S, в молекуле которого вместо глутаминовой кислоты в 6-м положении р-цепи находится валин. Носители патологического гена менее подвержены заболеванию малярией, что способствует сохранению мутации в популяции.
В условиях гипоксии гемоглобин S полимеризуется и откладывается в виде длинных цепей, изменяющих форму эритроцитов с формированием характерной серповидности. Серповидные эритроциты блокируют мелкие кровеносные сосуды, вызывая нарушения микроциркуляции, что приводит к развитию инфарктов, чаще локализующихся в селезенке, легких, почках и головном мозге. Дактилит пястных костей и проксимальных фаланг пальцев периоста обусловливается множественными микротромбозами в зонах. Микротромбозы также становятся причиной остеомиелита и аваскулярных некрозов бедренных костей. Нарушение кровоснабжения синовиальной оболочки приводит к накоплению реактивного выпота в суставах (обычно коленных и локтевых). Патологические изменения в почках связаны с образованием зон ишемии в корковом слое и общими трофическими нарушениями тубулярного аппарата, сопровождаемыми гипертрофией клубочков, гематурией, почечной АГ. Гиперплазия красного костного мозга проявляется образованием поднадкостничных очагов кроветворения в плюсневых и пястных костях, представленных в основном эритроидным ростком.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Основной фактор, определяющий тяжесть серповидноклеточной анемии, - генотип. Наиболее тяжело протекает гомозиготная форма заболевания. Гетерозиготная форма гемоглобинопатии протекает бессимптомно и проявляется лишь в условиях ишемии, провоцирующей тромбозы.
Наиболее типичный симптом серповидноклеточной анемии у детей - поражение костно-суставной системы:
• резкая болезненность суставов;
• припухлость стоп, голеней, кистей.
Больные обычно высокого роста, пониженного питания, с деформацией позвоночника. Нередко наблюдают формирование башенного черепа, изменение формы зубов. У детей раннего возраста селезенка увеличена, однако в дальнейшем ее размеры постепенно уменьшаются в результате фиброза, поэтому у больных старше 5 лет спленомегалии практически не бывает. Ишемия тканей приводит к образованию трофических язв в области голеней и лодыжек, асептическим некрозам костей (например, головки бедренной кости), остеомиелиту, кардиалгиям, аритмиям, нарушениям со стороны ЦНС. Гемолитические кризы могут быть спровоцированы инфекциями, лихорадкой, дегидратацией, гипоксией.
Выраженность тех или иных признаков болезни весьма вариабельна. У части пациентов в клинической картине доминируют гемолитические кризы, у других преобладают костно-суставная патология либо множественные инфаркты селезенки, легких, почек, головного мозга.
Диагноз подтверждают выявлением серповидных эритроцитов в мазке периферической крови или патологического гемоглобина S с помощью электрофореза. Также разработана ДНК-диагностика, позволяющая выявить ген гемоглобина S.
ЛЕЧЕНИЕ
Необходимо по возможности исключить факторы, провоцирующие гемолиз, - гипоксию, лихорадку, гиповолемию и пр. Прививки против гриппа типа B, менингококковой и пневмококковой инфекций следует проводить в раннем возрасте, до развития выраженного фиброза селезенки. Всем больным назначают фолиевую кислоту в дозе 1 мг/сут. Эффективность трансплантации аллогенного красного костного мозга сомнительна. В настоящее время разрабатываются методы генной терапии серповидноклеточной анемии.
ПРОГНОЗ
Продолжительность жизни пациентов с серповидноклеточной анемией в основном зависит от частоты гемолитических кризов: если они возникают чаще 3 раз в год, средняя продолжительность жизни - 35 лет, если 1 раз в год и реже - большинство пациентов доживают до 50 лет.
Талассемии
Талассемии - группа наследственных гипохромных микроцитарных гемолитических анемий, обусловленных нарушением синтеза α- или β-глобина (α- и β-талассемии соответственно).
ЭПИДЕМИОЛОГИЯ
Наиболее распространена β-талассемия, особенно в странах Средиземноморья, Юго-Восточной Азии, Африки, Китае. В России β-талассемию наблюдают преимущественно среди азербайджанцев, таджиков, грузин. Случаи гетерозиготной β-талассемии также зарегистрированы у русских, украинцев, армян, узбеков. α-Талассемию чаще всего наблюдают у жителей Нигерии, афроамериканцев, в Италии, Греции, Таиланде.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
В крови взрослого человека циркулируют эритроциты, содержащие разные типы гемоглобина, отличающиеся друг от друга составом цепей глобина, с преобладанием гемоглобина взрослого типа - гемоглобина A. Молекулы гемоглобина содержат четыре полипептидные цепи глобина, соединенные попарно:
• в гемоглобине A, составляющем 95% всего гемоглобина, - две αи две β-цепи;
• гемоглобине A2 (составляющем около 3,5%) - две α- и две δ-цепи;
• гемоглобине F (1-1,5%) - две α- и две γ-цепи.
Тетрамеры из одинаковых цепей глобинов нерастворимы, поэтому для образования нормального гемоглобина необходим сбалансированный синтез различных цепей. При нарушении этого баланса цепь, производимая в избыточном количестве, агрегирует и откладывается в эритрокариоцитах.
У человека 2 идентичных гена α-глобина на каждой хромосоме 16
и по одному гену β- и γ-глобина на хромосоме 11.
• α-Талассемия (*141800, 16p, дефекты генов HBAC, HBA1, HBA2, HBHR, Ʀ) связана с делецией одного или нескольких генов
α-глобина и сопровождается избыточным синтезом β-глобина (у детей и взрослых) с образованием гемоглобина H (β4-тетрамеры) или γ-цепей (у новорожденных) - гемоглобин Барта (γ4-тетрамер). Обе эти формы нестабильны, их связь с кислородом в 10 раз сильнее, чем у нормального гемоглобина, что делает их непригодными для транспорта кислорода. Повышенное содержание кислорода в эритроцитах и нарушение структуры мембраны приводят к их быстрому старению и разрушению. Выраженность гематологических нарушений зависит от количества образующихся патологических тетрамеров. α-Талассемия:
- тип 1 (α0) - характеризуется полным отсутствием синтеза α-цепей (делеция 4 генов);
- тип 2 (α+) - происходит уменьшение их синтеза, степень выраженности клинических проявлений определяется количеством генов (1, 2 или 3), которые подверглись делеции.
• β-Талассемия (*141900, 11p15.5, Ʀ) характеризуется снижением (тип β°) или отсутствием (тип β+) синтеза β-цепей. Синтез избыточного количества α-цепей при β-талассемии с образованием их агрегатов уже на уровне эритроидных предшественников приводит к разрушению эритрокариоцитов в красном костном мозге с возникновением признаков неэффективного гемопоэза. Накопление α-цепей в эритроцитах сопровождается изменением цитоскелета и структуры мембраны, что приводит к усиленному их разрушению в селезенке с накоплением в ее ткани белковых агрегатов и развитию значительной спленомегалии. При наличии 2 мутантных аллелей развивается большая β-талассемия (анемия Кули), у гетерозигот - малая β-талассемия.
• δβ-Талассемия возникает при одновременном снижении синтеза δ- и β-цепей c компенсаторным увеличением синтеза γ-цепей. Именно увеличение количества γ-цепей и определяет тяжесть заболевания. Количество гемоглобина H при гомозиготной δβталассемии достигает 100%.
• γδβ-Талассемия вызвана делецией или инактивацией всего комплекса генов γ-, δ-, β-цепей. Эта форма характеризуется тяжелым неонатальным гемолизом, постепенно прекращающимся с формированием клинической картины малой β-талассемии.
• Наследственное персистирование гемоглобина F связано со снижением синтеза δ- и β-цепей и увеличением количества γ-цепей, компенсируемым повышенным синтезом α-цепей. Данная форма протекает практически бессимптомно.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Степень тяжести клинических проявлений зависит от количества патологических гемоглобинов, т.е. от степени выраженности генных мутаций.
• Гомозиготная а-талассемия (делеция 4 генов) несовместима с жизнью. Дети рождаются с выраженной водянкой, смерть наступает либо внутриутробно, либо в первые часы после рождения.
• Гемоглобинопатия H (делеция 3 генов α-цепей) проявляется постоянным вялотекущим гемолизом (желтухой, увеличением селезенки и печени) с развитием умеренной анемии (концентрация гемоглобина - 70-100 г/л). В анализах крови выявляют повышение количества ретикулоцитов (5-10%), гипохромию, мишеневидность и базофильную зернистость эритроцитов, повышенную концентрацию непрямого билирубина. У новорожденных при гель-электрофорезе находят гемоглобин Барта, к концу первого года жизни - гемоглобин H.
• Малая α-талассемия (делеция 2 генов α-цепей) проявляется легкой микроцитарной анемией. Содержание гемоглобина Барта на 1-м году жизни - 2-10%. Делеция 1-го гена а-цепей протекает бессимптомно.
• Клинические проявления β-талассемии также весьма вариабельны - от тяжелой гемолитической анемии (при гомозиготности по патологическим генам) до практически бессимптомной. Изменения внешности при β-талассемии связаны с аномалиями скелета: квадратный череп, уплощение переносицы, выступающие скулы и пр. Истончение коркового слоя трубчатых костей приводит к развитию патологических переломов. Характерны отставание в росте, задержка полового созревания. Рентгенологически обнаруживают утолщение губчатых костей свода черепа, поперечную исчерченность на наружной пластинке лобной и теменной костей.
Для верификации диагноза, определения степени тяжести и, соответственно, прогноза заболевания используют исследование патологических гемоглобинов с помощью гель-электрофореза.
ЛЕЧЕНИЕ
Основной метод лечения при тяжелых формах талассемии - трансплантация красного костного мозга. Описаны несколько случаев внутриутробной трансплантации аллогенных кроветворных клеток.
При а-талассемии гемотрансфузии не показаны. Спленэктомия иногда облегчает течение гемоглобинопатии Н, уменьшая проявления анемии и гиперспленизма.
Адекватные гемотрансфузии при β-талассемии могут уменьшить проявления костной патологии и отставание в физическом развитии. Обязательно назначают дефероксамин (для уменьшения выраженности гемосидероза). Спленэктомия показана при выраженной спленомегалии, она может уменьшить потребность в гемотрансфузиях.
55.4. АПЛАСТИЧЕСКИЕ АНЕМИИ
Апластические анемии - гетерогенная группа заболеваний системы крови, характеризуемых панцитопенией в периферической крови вследствие угнетения кроветворной функции красного костного мозга.
ЭПИДЕМИОЛОГИЯ
В европейских странах заболеваемость апластической анемией составляет в среднем 2 на 1 млн населения. Заметной зависимости заболеваемости от пола и этнической принадлежности не обнаружено. Апластическая анемия может возникнуть в любом возрасте, однако чаще ее диагностируют у пациентов 20-40 лет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Принято считать, что подавление костномозгового кроветворения при апластической анемии связано с появлением в периферической крови и красном костном мозге активированных цитотоксических Т-лимфоцитов, продуцирующих γ-интерферон и ФНО, которые подавляют как нормальный гемопоэз, так и образование колоний кроветворных клеток in vitro.
В некоторых случаях апластическая анемия развивается на фоне приема некоторых ЛС (хлорамфеникола, препаратов золота, противосудорожных и др.) или при длительном контакте с химическими соединениями, например бензином. Патогенез лекарственной апластической анемии остается неясным: возможны как генетически обусловленная повышенная чувствительность к ЛС, так и прямое токсическое воздействие на стволовые кроветворные клетки и аутоиммунные реакции.
В качестве возможных этиологических факторов также рассматривают вирусы, в первую очередь гепатитов В, С и G. Апластическую анемию, развившуюся в течение 6 мес после перенесенного острого вирусного гепатита, принято называть постгепатитной. Патогенез постгепатитной
апластической анемии не изучен, но установлена возможность репликации вирусов в мононуклеарах периферической крови, красного костного мозга и в клетках иммунной системы. Именно поэтому подавление гемопоэза, возможно, связано с иммунным ответом, направленным на инфицированные и несущие на своей поверхности вирусные Аг-клетки красного костного мозга.
В большинстве случаев установить наличие конкретного этиологического фактора не представляется возможным (идиопатическая апластическая анемия).
Таким образом, апластическую анемию можно рассматривать как гетерогенную по своему происхождению группу аплазий кроветворения, для которых ведущими патогенетическими факторами являются поражение стволовых клеток (первичное или опосредованное иммунными реакциями) и аутоиммунная агрессия в отношении гемопоэза (первичная или в ответ на появление клона дефектных клеток).
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Основные клинические симптомы связаны с панцитопенией. Анемия обусловливает бледность кожных покровов и слизистых оболочек, общую слабость, повышенную утомляемость. Практически всегда развивается геморрагический синдром - кровоточивость слизистых оболочек полости рта, кожные геморрагические высыпания, маточные, носовые, желудочно-кишечные кровотечения. Вследствие глубокой гранулоцитопении у больных, как правило, развиваются инфекционные осложнения (стоматит, пневмония и пр.).
По степени выраженности тромбо- и гранулоцитопении различают апластическую анемию:
• тяжелую (гранулоцитов - 0,2-0,5х109/л, тромбоцитов - <20,0х109/л);
• очень тяжелую (гранулоцитов - <0,2х109/л, тромбоцитов -
<20,0х109/л).
Диагноз апластической анемии устанавливают на основании данных:
• гемограммы (тяжелая анемия, грануло- и тромбоцитопения);
• миелограммы (снижение общей клеточности, угнетение гранулоцитарного и эритроидного ростков, относительный лимфоцитоз, практически полное отсутствие мегакариоцитов).
При гистологическом исследовании трепанобиоптата выявляют преобладание жирового костного мозга над красным.
ЛЕЧЕНИЕ
Длительное время апластическую анемию считали фатальным заболеванием. Глубокая анемия, прогрессирующий геморрагический синдром, тяжелые инфекционные осложнения, обусловленные угнетением костномозгового кроветворения, остаются основными причинами летального исхода. Заместительная гемотрансфузионная терапия (переливания эритроцитов и тромбоцитов), использование ГК и анаболических гормонов сами по себе не решают проблему лечения апластической анемии. Достаточно широко применявшаяся спленэктомия позволила в свое время улучшить прогноз заболевания, но это прежде всего касалось больных нетяжелой апластической анемией, в целом же прогноз оставался неблагоприятным.
Значимым достижением в лечении апластической анемии стала трансплантация аллогенного красного костного мозга, которую начали применять в 70-х гг. XX в. Однако применение этого метода лечения весьма ограничено, что связано преимущественно с предшествующей массивной гемотрансфузионной терапией, значительно повышающей риск отторжения трансплантата в связи с аллосенсибилизацией.
Практически одновременно с трансплантацией аллогенного красного костного мозга в практику лечения внедрили иммуносупрессивные препараты, в первую очередь иммуноглобулин антитимоцитарный.
Иммуноглобулин антитимоцитарный обладает цитолитическим действием в отношении Т-лимфоцитов, подавляя продукцию ими лимфокинов. Препарат вводят в дозе 15-20 мг/(кгхсут) внутривенно (за 10-12 ч) в течение 5 дней. Поскольку при лечении развивается глубокая иммуносупрессия, больные должны находиться в асептических одноместных палатах. При гранулоцитопении до 0,2х109/л и эпизодах немотивированной лихорадки или наличии доказанных инфекционных осложнений с первого дня введения иммуноглобулина антитимоцитарного назначают антибиотики широкого спектра действия внутривенно на 2-3 нед, а в целях деконтаминации кишечника - ко-тримоксазол или ципрофлоксацин и противогрибковые препараты (кетоконазол или флуконазол) в течение 3-4 нед. Во время введения препарата проводят переливания тромбоцитарной массы (для поддержания количества тромбоцитов на уровне ≥20,0х109/л). В дальнейшем трансфузии эритроцитарной и тромбоцитарной массы проводят в зависимости от тяжести анемии и геморрагического синдрома. При рефрактерности к трансфузиям донорских эритроцитов и тромбоцитов в программу лечения включают плазмаферез.
В последнее десятилетие для лечения апластической анемии применяют циклоспорин. Препарат селективно и обратимо изменяет функцию лимфоцитов, способен подавлять продукцию лимфокинов и их связывание со специфическими рецепторами, обратимо блокирует образование ростковых факторов, что приводит к угнетению дифференцировки и пролиферации цитотоксических Т-лимфоцитов. Циклоспорин не подавляет способность нейтрофилов к хемотаксису или фагоцитозу. В дозах, вызывающих иммуносупрессию, препарат не токсичен, побочные эффекты (повышение концентрации креатинина в сыворотке крови, тремор, боли в костях, гиперплазия десен, АГ, гипертрихоз) обратимы. Начальная доза циклоспорина составляет 10 мг/(кгхсут), в дальнейшем ее корректируют в зависимости от концентрации препарата в крови. Курс терапии должен быть длительным (8-12 мес) для поддержания иммуносупрессии в течение всего периода, необходимого для стабилизации клинико-гематологического ответа.
Независимо от тяжести апластической анемии в момент установления диагноза лечение не должно ограничиваться одним методом. Необходимо проведение интенсивной иммуносупрессивной терапии, включающей иммуноглобулин антитимоцитарный, циклоспорин, в ряде случаев - спленэктомию.
55.5. ПАРЦИАЛЬНАЯ КРАСНОКЛЕТОЧНАЯ АПЛАЗИЯ
Парциальная красноклеточная аплазия - заболевание, характеризуемое тяжелой анемией с низким содержанием ретикулоцитов и отсутствием или значительным снижением количества эритрокариоцитов в красном костном мозге; в большинстве случаев имеет аутоиммунный генез.
ЭПИДЕМИОЛОГИЯ
Парциальная красноклеточная аплазия - редкое заболевание, наблюдаемое во всех регионах. Выявляют преимущественно у лиц старше 45 лет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Парциальная красноклеточная аплазия может быть как первичной (идиопатическая форма), так и вторичной - при миелопролиферативных заболеваниях, тимоме, аутоиммунной патологии, вирусных инфекциях. Также существует врожденная форма - анемия ДаймондаБлекфена (вариант анемии Фанкони; *205900, р).
В патогенезе приобретенной парциальной красноклеточной аплазии ведущее значение имеет угнетение кроветворения, связанное с аутоиммунными нарушениями - образованием аутоантител против Аг эритрокариоцитов и/или эритроцитов, эритропоэтина или его рецепторов.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
В клинической картине доминируют жалобы, связанные с анемией (общая слабость, повышенная утомляемость, одышка при физической нагрузке). Диагностика базируется:
• на обнаружении выраженного снижения содержания эритроцитов в крови, как правило, на фоне высокой концентрации сывороточного эритропоэтина;
• парциальной красноклеточной аплазии в красном костном мозге;
• выявлении иммунологических маркеров - антиэритроцитарных аутоантител и АТ против эритрокариоцитов.
ЛЕЧЕНИЕ
Основу лечения составляет иммуносупрессивная терапия - спленэктомия, ГК (преднизолон), цитостатики (циклофосфамид, азатиоприн, метотрексат). Описаны случаи достижения ремиссий на фоне терапии малыми дозами цитарабина и после применения циклоспорина. Кроме того, применяют плазмаферез. Больным необходимо обеспечить адекватное количество гемотрансфузий и введение дефероксамина. При вторичной парциальной красноклеточной аплазии необходимо лечение основного заболевания (например, удаление тимомы).
55.6. ПОРФИРИИ
Порфирии - группа заболеваний, включающая семь нозологических форм, каждая из которых обусловлена наследственным или приобретенным дефектом активности одного из ферментов цепи биосинтеза гема. В зависимости от ткани, в которой происходит преимущественное нарушение синтеза порфиринов (предшественников гема), порфирии подразделяют:
• на эритропоэтические (нарушение образования порфиринов эритробластами костного мозга);
• печеночные (дефект синтеза порфиринов в печени).
ЭПИДЕМИОЛОГИЯ
Порфирии более распространены на севере Европы, где заболеваемость ими составляет 7-12 на 100 000 населения. Бессимптомное носительство генетических дефектов встречается у 1 из 1000 человек.
ЭТИОЛОГИЯ
Порфирии наследуются по аутосомно-доминантному типу, кроме врожденной эритропоэтической порфирии, наследуемой аутосомнорецессивно.
К провоцирующим факторам, способным перевести латентно протекающую порфирию в острую форму, относят:
• голодание;
• бактериальные и вирусные инфекции (например, гепатиты);
• алкоголь;
• прием некоторых ЛС (НПВС, барбитуратов, некоторых антибиотиков, сульфаниламидов и др.);
• изменение гормонального профиля у женщин (менархе, беременность );
• инсоляцию.
Острая порфирия чаще развивается у женщин в период полового созревания. Атаки связаны с началом менструации.
ПАТОГЕНЕЗ
Патогенез клинических проявлений при острых печеночных порфириях обусловлен вовлечением вегетативной нервной системы. Поражение кожных покровов при порфириях связано с повышением чувствительности к солнечному излучению вследствие накопления в коже порфиринов. Воздействие солнечного света приводит к образованию метаболитов, повреждающих клетки базальной мембраны и способствующих высвобождению медиаторов тучных клеток, которые усиливают фототоксичность.
КЛИНИЧЕСКАЯ КАРТИНА
Наиболее частые симптомы печеночных порфирий - боли в животе (у 90% пациентов), сопровождаемые рвотой и запором (в 50-80% случаев). Последний связан с нарушением моторики кишечника и спазмом сосудов.
Тахикардия, обусловленная увеличением содержания в крови катехоламинов, возникает у 30-80% пациентов в момент острой атаки. Повышение АД наблюдается в 40-80% случаев острых приступов.
Боли в спине (у 60% больных), прогрессирующая слабость (у 4090%), нарушения кожной чувствительности - проявления полиневропатии. Симметричные парезы конечностей связаны с дегенерацией нейронов вследствие вторичной демиелинизации.
Энцефалопатия, эпилептиформные припадки, гемиплегия, интеллектуальные нарушения, галлюцинации, психозы (в 40-55% случаев) - признаки поражения ЦНС.
При порфириях, протекающих с поражением кожных покровов, пациенты жалуются на повышенную травматизацию кожи с вторичными воспалительными изменениями. Гиперпигментация и склеродермоподобные изменения локализуются на лице и руках. Под воздействием солнечных лучей на коже могут появляться эрозии, пузыри, глубокие трещины.
Во время приступов характерно появление специфического окрашивания мочи (от розоватого до красно-бурого). Окрашивание усиливается под действием солнечного света.
ДИАГНОСТИКА
Острую порфирию следует подозревать у любого больного с острой болью в животе, нарушениями психики, периферической нейропатией и типичными изменениями мочи. Для подтверждения диагноза необходимо провести специальное обследование, включающее исследование мочи на содержание порфобилиногена (при порфирии его количество повышается), определение активности порфобилиногендезаминазы и мутаций в гене порфобилиногендезаминазы (в гене PBGD).
Для исключения кожных порфирий определяют содержание порфиринов в эритроцитах, плазме крови, моче и кале, оценивают спектр поглощения порфиринов с помощью флюоресцентной спектроскопии.
ЛЕЧЕНИЕ
Лечение печеночных форм порфирий. Исключают воздействие провоцирующего фактора, у женщин - прерывают менструальный цикл назначением гормональных препаратов.
Лечение эритропоэтических порфирий. Для прерывания абсорбции порфиринов в кишечнике назначают активированный уголь по 60 г 3 раза в день, а для подавления собственного эритропоэза проводят
переливания избыточных количеств эритроцитарной массы. Избыток железа выводят с помощью комплексообразующих препаратов (дефероксамина). Подавления собственного эритропоэза достигают также приемом гидроксикарбамида по 1 г/сут под контролем анализа периферической крови.
Глава 56. ОПУХОЛИ СИСТЕМЫ КРОВИ
Гемобластозы (опухоли, возникающие из клеток кроветворной ткани) подразделяют:
• на лейкозы, т.е. опухоли с первичным поражением костного мозга;
• нелейкемические гемобластозы - опухоли, возникающие из клеток крови вне костного мозга (с возможным дальнейшим метастазированием в последний).
В зависимости от клеточного субстрата лейкозы подразделяют:
• на хронические;
• острые.
К острым лейкозам относят опухоли, представленные бластными клетками. Клеточный субстрат хронических лейкозов - зрелые дифференцированные клетки крови. Как и для опухолей других тканей, для гемобластозов характерны законы опухолевой прогрессии: клональность (происхождение из одной клетки) и увеличение количества хромосомных мутаций внутри первоначальной клеточной популяции, приводящее к появлению новых мутантных субклонов, определяющих изменчивость свойств опухоли.
ЭПИДЕМИОЛОГИЯ
Заболеваемость гемобластозами относительно невысока и в целом составляет приблизительно 10 на 100 000 населения (6-7% всех опухолевых заболеваний). В развитых странах на гемобластозы приходится приблизительно 1% общей смертности населения и около 6-10% случаев смерти от злокачественных новообразований (среди пациентов моложе 30 лет - 50%).
56.1. ОСТРЫЕ ЛЕЙКОЗЫ
Острые лейкозы - гетерогенная группа опухолевых заболеваний системы крови, характеризуемых первичным поражением красного костного мозга морфологически незрелыми кроветворными (бластными)
клетками с вытеснением его нормальных элементов и инфильтрацией различных тканей и органов.
ЭПИДЕМИОЛОГИЯ
На острый лейкоз приходится приблизительно 3% общего количества всех злокачественных опухолей человека. Это наиболее частая форма гемобластозов. Заболеваемость в среднем составляет 5 на 100 000 населения. В 75% всех случаев острый лейкоз регистрируют у взрослых. Среднее соотношение миелоидных и лимфоидных лейкозов составляет 6:1. В детском возрасте 80-90% всех случаев заболевания составляют лимфобластные формы (медиана возраста - 10 лет), а после 40 лет у 80% больных выявляют миелоидные варианты (медиана возраста - 60-65 лет).
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Острый лейкоз развивается вследствие мутации клоногенной кроветворной клетки, приводящей к нарушению контроля правильности клеточного цикла, изменению транскрипции и продукции ряда ключевых белков. В результате бесконтрольной пролиферации при отсутствии дифференцировки происходит накопление патологических клеток. В опухолевых клетках, как правило, выявляют различные хромосомные аберрации (транслокации, делеции, инверсии и т.д.). При нелимфобластных острых лейкозах более чем в 90% случаев выявляют хромосомные изменения. Клоны клеток с анеуплоидным количеством хромосом или их структурными изменениями исчезают во время ремиссии и вновь появляются при рецидиве заболевания. Существует четкая связь некоторых перестроек хромосом с формой острого лейкоза: t(8;21) и t(6;9) выявляют при миелобластном, t(15;17) - при промиелоцитарном, t(9;11) - при монобластном, t(4;11) - при лимфобластном лейкозе.
В большинстве случаев конкретная причина возникновения острого лейкоза остается неизвестной, тем не менее существует несколько предрасполагающих факторов, значительно повышающих риск развития заболевания.
• У детей с синдромом Дауна риск развития острого лейкоза в 20 раз выше, чем в популяции (то есть изменения, связанные с хромосомой 21, могут приводить к развитию лейкоза).
• Вероятность развития острого лейкоза повышена при врожденном агранулоцитозе, целиакии, анемии Фанкони, синдроме ВискоттаОлдрича, синдроме Клайнфелтера, вероятно, за счет повышенной хромосомной нестабильности.
• Прямые доказательства этиологической роли вирусов получены только для Т-клеточного лейкоза/лимфомы взрослых, наблюдаемой в Японии и странах Карибского бассейна (ассоциируется с вирусом человека, тропным к Т-лимфоцитам типа I).
• Доказана взаимосвязь между лучевой и химиотерапией, проводимых по поводу других опухолей, и повышенным риском развития острых лейкозов. При комбинированном применении лучевой и химиотерапии, включавшей мехлорэтамин*, риск заболевания острым лейкозом составляет 10%. Кроме мехлорэтамина* мутагенным эффектом обладают прокарбазин, хлорамбуцил, циклофосфамид, ломустин, этопозид. В 85% случаев вторичные лейкозы развиваются в течение 10 лет после окончания лечения.
• Достоверных данных, свидетельствующих в пользу увеличения частоты острого лейкоза после повторных воздействий ионизирующего излучения в малых дозах (в том числе применяемых с диагностическими целями в медицине), не получено.
• Доказано возможное лейкозогенное действие некоторых органических растворителей и ЛС (фенилбутазона, хлорамфеникола и
др.).
• Ряд исследователей предполагают, что приблизительно в 20% случаев острый миелобластный лейкоз может быть следствием курения.
• Существуют данные об учащении случаев острого миелобластного лейкоза у больных множественной миеломой, болезнью Вальденстрема, хроническим лимфолейкозом, получавших мелфалан, бусульфан, хлорамбуцил, азатиоприн, циклофосфамид.
КЛАССИФИКАЦИЯ
В настоящее время наиболее распространена классификация острых лейкозов франко-американо-британской группы (FAВ-классификация). Определение принадлежности лейкозных клеток к тому или иному типу проводят на основании морфологического, цитохимического исследований и иммунологического фенотипирования. В соответствии с FAВ-классификацией различают следующие основные формы острых лейкозов.
• Острый миелобластный недифференцированный (М0).
• Острый миелобластный без созревания (М1).
• Острый миелобластный с созреванием (М2).
• Острый промиелоцитарный (М3).
• Острый миеломонобластный (М4).
• Острый монобластный (М5).
• Острый эритробластный (М6).
• Острый мегакариобластный (М7).
• Острый лимфобластный Т-клеточный.
• Острый лимфобластный В-клеточный.
Основные постулаты современного учения о лейкозах следующие.
• Острые лейкозы клональны.
• Лейкозные клетки часто несут на своей поверхности маркеры, характеризующие определенные этапы дифференцировки нормальных гемопоэтических клеток.
• На нормальных клетках гемопоэза никогда не определяется аберрантная экспрессия Аг.
• Существует группа острых лейкозов, клетки которых несут маркеры разных линий кроветворения (миелопоэза и лимфопоэза) или уровня дифференцировки (так называемые ранние и поздние маркеры).
• В период морфологически доказанной ремиссии возможно обнаружение клеток с характерным лейкозным иммунологическим фенотипом или генотипом.
• В течении острых лейкозов выделяют первый острый период (дебют, или манифестацию), ремиссии и рецидивы.
• Клиническая манифестация опухоли происходит, когда ее масса составляет 1012 клеток и более. Продолжительность доклинического периода заболевания неизвестна. Отдельные симптомы (например, анемический синдром) могут появляться задолго до диагностики острого лейкоза.
• Полной ремиссией принято называть состояние кроветворения, при котором в красном костном мозге обнаруживают не более 5% бластных клеток при нормальном соотношении всех других ростков кроветворения, содержание гранулоцитов в периферической крови не менее 1х109/л, тромбоцитов - не менее 100х109/л, отсутствуют экстрамедуллярные очаги лейкозного поражения.
- Если у больного после 2 мес терапии не удается достигнуть ремиссии, констатируют первично-резистентную форму острого лейкоза.
- Минимальной остаточной болезнью принято называть наличие остаточной популяции лейкозных клеток, которая может быть выявлена лишь с помощью высокочувствительных методов в
тех случаях, когда подтверждена морфологическая ремиссия. Терапия острых лейкозов после достижения ремиссии (консолидации, поддерживающего лечения) направлена на удаление резидуального опухолевого клона.
• Рецидив острого лейкоза принципиально отличается от дебюта заболевания и рассматривается как появление (вследствие опухолевой прогрессии) и пролиферация нового, чаще всего устойчивого к проводимой терапии клона лейкозных клеток. Рецидивом острого лейкоза считают появление:
- более 10% бластных клеток в пунктате красного костного мозга независимо от изменений в анализе периферической крови;
- внекостномозговых поражений (нейролейкоза, поражения яичек, увеличения селезенки и т.д.), даже при отсутствии изменений в крови и красном костном мозге.
КЛИНИЧЕСКАЯ КАРТИНА
Клинические проявления различных форм острого лейкоза достаточно стереотипны и обусловлены:
• выраженным угнетением нормального кроветворения вследствие лейкемической инфильтрации красного костного мозга (анемией, тромбоцитопенией с геморрагическим синдромом, нейтропенией с различными инфекционными осложнениями);
• инфильтрацией опухолевыми клетками различных органов;
• продукцией цитокинов.
Дебют заболевания может быть внезапным, с высокой лихорадкой, выраженной слабостью и интоксикацией. Тем не менее нередко диагноз устанавливают случайно при профилактических осмотрах или госпитализации.
Особого внимания заслуживают признаки внекостномозговых поражений. Одно из наиболее частых - нейролейкоз, обусловленный метастазированием опухолевых клеток в оболочки головного и спинного мозга.
В ряде случаев наблюдают периферические невропатии, обусловленные лейкозной инфильтрацией нервов.
Клинически нейролейкоз проявляется менингеальным синдромом и признаками повышения внутричерепного давления:
• постоянной головной болью;
• рвотой;
• вялостью;
• отеком дисков зрительных нервов;
• признаками поражения черепных нервов.
При осмотре выявляют ригидность затылочных мышц, симптом Кернига. В части случаев нейролейкоз протекает бессимптомно и его диагностируют только на основании исследования ликвора (цитоз более 10 клеток, морфологически похожих на бластные). Нейролейкоз наиболее часто наблюдают при острых лимфобластных лейкозах. При отсутствии профилактики (интратекального введения цитостатических препаратов) нейролейкоз развивается у 30-50% больных. При миелоидных лейкозах нейролейкоз развивается реже, преимущественно при миеломоно- и монобластных вариантах (у 30% больных при отсутствии профилактики).
К экстрамедуллярным проявлениям острого лейкоза также относят поражения кожи в виде лейкемидов (багрово-синюшных уплотнений) сетчатки, десен, яичек, яичников, описаны случаи поражения лимфатических узлов, легких, кишечника, миокарда.
ПРОГНОСТИЧЕСКИЕ ФАКТОРЫ
Прогностические факторы, оцениваемые при диагностике острых лейкозов и в процессе лечения, могут считаться объективными только при условии проведения адекватной химиотерапии. Химиотерапию считают адекватной при соблюдении следующих условий.
• Применении программ химиотерапии, специально предусмотренных для данной конкретной формы острого лейкоза.
• Соответствии доз используемых ЛС, предусмотренных в программе.
• Соответствии интервалов между курсами и этапами терапии, предусмотренных в программе.
Другой важный прогностический фактор - возраст пациента в момент постановки диагноза. Большое значение имеют также данные лабораторных исследований, проводимых в дебюте заболевания, и время достижения ремиссии на фоне адекватной химиотерапии. Если полную ремиссию у больных достигают после первого курса индукционного лечения, долгосрочные результаты существенно лучше.
ДИАГНОСТИКА
Диагностика острых лейкозов базируется на оценке морфологических особенностей клеток красного костного мозга и периферической крови. Диагноз острого лейкоза устанавливают только на основании обнаружения в красном костном мозге или периферической крови бласт-
ных клеток, характеризуемых нежно-сетчатой структурой ядерного хроматина.
Цитохимическое исследование
Для определения принадлежности опухолевых клеток к миелоидной или лимфоидной линиям кроветворения обычной окраски по Романовскому-Гимзе недостаточно. Для точной идентификации необходимо цитохимическое исследование.
• Реакция на пероксидазу положительна в клетках миелоидного ряда (от миелобластов до зрелых нейтрофилов).
• Реакция на липиды положительна в клетках миелоидного ряда и моноцитах.
• ШИК-реакция (на гликоген) в клетках миелоидного ряда имеет диффузный вид, в моноцитах - диффузный или диффузногранулярный, в клетках лимфоидного ряда - гранулярный.
• Реакция на неспецифическую а-нафтилацетатэстеразу положительна в клетках моноцитарного ряда.
Иммунологическое фенотипирование
Иммунологическое фенотипирование проводят для подтверждения диагноза, установления варианта острого лейкоза в случаях, когда морфологический метод оказывается недостаточно информативным, для определения бифенотипических вариантов, характеристики аберрантного иммунного фенотипа в дебюте заболевания в целях дальнейшего мониторинга минимальной остаточной популяции клеток в период ремиссии и, наконец, для определения прогноза.
На поверхности и в цитоплазме гемопоэтических клеток выявлено более 150 специфических Аг, сгруппированных в так называемые кластеры дифференцировки (CD), к которым созданы моноклональные АТ. Хотя Аг, строго специфичных для лейкозных клеток, обнаружить не удалось, характеристика гемопоэтических клеток на основании совокупности Аг CD позволяет определять их линейную принадлежность и этап дифференцировки. При обнаружении одновременной экспрессии Аг, в норме вместе не наблюдаемой, констатируют аберрантный (лейкемический) иммунный фенотип. Бластные клетки считают позитивными по экспрессии того или иного Аг, если его выявляют более чем в 20% клеток. К Аг, определяемым на клетках лимфоидной принадлежности,
относят CD1, CD2, CD3, CD4, CD5, CD7, CD8, CD9, CD10, CD19, CD20, CD22, CD23, CD56, CD57, миелоидной - CD11, CD13, CD14, CD15,
CD33, CD36, CD41, CD42, CD65, HLA-DR.
С внедрением в практику метода иммунологического фенотипирования появилось понятие бифенотипического и/или билинейного острого лейкоза, при котором сосуществуют 2 популяции бластных клеток, иммунный фенотип которых принадлежит к различным линиям кроветворения. Бифенотипический острый лейкоз диагностируют в ситуациях, когда цитохимические и морфологические исследования не позволяют определить принадлежность клеток к той или иной линии кроветворения, а при иммунологическом фенотипировании выявляют принципиально значимые как лимфоидные, так и миелоидные маркеры. В настоящее время эффективные программы терапии бифенотипических острых лейкозов не разработаны и результаты лечения существенно хуже.
Цитогенетическая характеристика
На сегодняшний день практически у 90% больных с острыми лейкозами выявляют различные дефекты хромосом - транслокации, делеции, инверсии, исчезновение одной из пар хромосом и т.д. В настоящее время разработана цитогенетическая классификация острых лейкозов. Отдельные цитогенетические маркеры оказались принципиально важными в плане как терапии, так и прогноза острого лейкоза. Например, inv(16) часто выявляют у больных с острым миеломонобластным лейкозом с высокой (>3%) эозинофилией в красном костном мозге; t(15;17) - типичный маркер варианта М3; t(8;21) выявляют в 40% случаев варианта М2. Указанные выше транслокации характеризуют группу благоприятного прогноза, причем для острого миелоидного лейкоза с t(8;21) и t(15;17) созданы программы дифференцированного лечения, которые позволяют достичь ремиссии у 90-95% больных.
При остром лимфобластном лейкозе принципиальным считают обнаружение t(9;22) или t(4;11) - факторов крайне неблагоприятного прогноза (1-2% случаев у детей и 25-30% - у взрослых) и гиперплоидии, характерной для вариантов с благоприятным течением. К транслокациям, определяющим плохие результаты терапии, относят t(8;14),
t(2;8), t(8;22).
ЛЕЧЕНИЕ
Химиотерапия острых лейкозов имеет довольно короткую историю (приблизительно 30 лет), ее успехи связаны в основном с появлением эффективных цитостатических препаратов, совершенствованием терапии компонентами крови и разработкой новых принципов антибиотикотерапии. Использование в клинической практике стандарти-
зированных программ позволяет обеспечить однотипность терапии в любом гематологическом стационаре. Основные цели лечения острых лейкозов: эрадикация лейкозного клона, восстановление нормального кроветворения и, как следствие, достижение длительной безрецидивной выживаемости больных. Основополагающие принципы химиотерапии всех злокачественных опухолей человека, в том числе и острых лейкозов, следующие:
• принцип «доза-интенсивность»: необходимость использования адекватных доз цитостатических препаратов в сочетании с четким соблюдением временных межкурсовых интервалов (уменьшение доз цитостатиков на начальных этапах терапии на 20% приводит к уменьшению эффективности терапии на 50%);
• принцип использования сочетания цитостатических ЛС в целях получения наибольшего эффекта и уменьшения вероятности развития резистентности к химиотерапии;
• принцип этапности терапии.
При всех острых лейкозах терапия включает несколько основных этапов: индукцию ремиссии, консолидацию, поддерживающую терапию и при некоторых вариантах - профилактику нейролейкоза.
• Цель начального периода лечения - индукции ремиссии - максимально быстрое и существенное уменьшение массы опухоли, достижение аплазии кроветворения и создание условий для пролиферативного преимущества нормальных кроветворных клеток. В идеале после первого подобного курса интенсивного цитостатического воздействия достигается ремиссия.
• Второй этап терапии острых лейкозов - консолидация ремиссии (закрепление достигнутого противоопухолевого эффекта). В настоящее время в большинстве случаев консолидация является наиболее агрессивным этапом лечения с использованием максимальных доз химиопрепаратов. Задача этого периода - по возможности еще большее уменьшение количества остающихся после индукции лейкозных клеток.
• После консолидации (обычно 1-2 курса) следует период поддерживающего лечения. При разных вариантах острых лейкозов длительность и интенсивность поддерживающей терапии различны, но принцип ее одинаков - продолжение цитостатического воздействия в малых дозах на остающийся опухолевый клон.
• Принципиальный этап лечения некоторых вариантов острого лейкоза (лимфобластных, миеломоно- и монобластных) - про-
филактика или при необходимости лечение нейролейкоза. Этот этап распределяется на все периоды программного лечения - индукцию ремиссии, консолидацию и поддерживающую терапию. Основной метод - интратекальное введение метотрексата (15 мг), цитарабина (30-45 мг), дексаметазона (4 мг). При специфическом поражении оболочек и/или вещества головного мозга интратекальное введение препаратов сочетают с краниоспинальной лучевой терапией в дозе 18-24 Гр. • Необходимость полноценной терапии выхаживания больных в период миелотоксического агранулоцитоза, развивающегося под действием высоких доз цитостатических препаратов, - принципиальное положение лейкозологии. Вспомогательная терапия необходима для профилактики осложнений и их лечения. Основные мероприятия вспомогательной терапии следующие:
- обеспечение возможности полноценной цитостатической терапии (рациональный сосудистый доступ);
- обеспечение приемлемого уровня качества жизни пациентов на фоне проводимой цитостатической терапии (профилактика тошноты и рвоты, заместительные трансфузии эритроцитарной массы);
- профилактика осложнений основного заболевания и/или проводимой терапии, угрожающих жизни больного:
◊ полиорганной недостаточности на фоне массивного лизиса опухоли (водная нагрузка, форсированный диурез, аллопуринол);
◊ геморрагических осложнений (заместительные трансфузии тромбоцитов);
• гемокоагуляционных нарушений (свежезамороженная плазма и инфузии гепарина натрия при гиперкоагуляционных состояниях, менадиона натрия бисульфит на фоне длительного применения антибиотиков, угнетающих нормальную микрофлору кишечника и изменяющих метаболизм витамина К);
• электролитных нарушений, особенно на фоне применения выводящих калий ЛС;
• инфекционных осложнений (селективная деконтаминация кишечника, обработка полости рта).
Лечение осложнений, возникающих в период миелотоксической депрессии кроветворения, требует гораздо больших затрат, нежели их профилактика. К наиболее тяжелым осложнениям относят инфекции различной степени тяжести (лихорадку неясного генеза, локальные ин-
фекции, сепсис). Те или иные инфекционные осложнения в период индукции ремиссии возникают у 80-90% больных с острыми лейкозами. Главный принцип лечения любых инфекций - эмпирическая поэтапная антибиотикотерапия с обязательными бактериологическими исследованиями для коррекции спектра применяемых антибиотиков в соответствии с их результатами.
В качестве вспомогательных ЛС в программы терапии острых лейкозов включают ростковые гемопоэтические факторы. Они не влияют на пролиферацию лейкозных клеток и в то же время существенно уменьшают продолжительность периода миелотоксического агранулоцитоза, что приводит к снижению частоты инфекционных осложнений.
Острый лимфобластный лейкоз
Острый лимфобластный лейкоз - самая распространенная опухоль кроветворной ткани у детей. На него приходится приблизительно 30% всех случаев злокачественных опухолей детского возраста. У пациентов моложе 15 лет острый лимфобластный лейкоз диагностируют в 75% случаев всех острых лейкозов. Пик заболеваемости приходится на возраст 3-4 года, затем заболеваемость снижается. Второй подъем, не столь существенный, как первый, отмечают в возрасте 50-60 лет.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Клинические проявления весьма разнообразны. В части случаев заболевание начинается постепенно, но возможен и острый дебют. Наиболее частые симптомы: слабость, сонливость, лихорадка, не связанная с инфекцией, боли в костях и суставах. Инфекционные осложнения развиваются редко, преимущественно в тех случаях, когда количество нейтрофилов менее 0,2х109/л. В некоторых случаях единственной жалобой могут быть боли в костях и позвоночнике при отсутствии лимфаденопатии, увеличения внутренних органов и изменений в анализах крови. В таких случаях диагноз, как правило, устанавливают с существенной задержкой. Головные боли, тошнота, рвота чаще возникают при поражении ЦНС.
При объективном обследовании возможны бледность кожных покровов, петехиальные высыпания, кровоточивость десен, лимфаденопатия (в том числе увеличение нёбных миндалин), спленомегалия, гепатомегалия, увеличение размеров почек, болезненность при поколачивании костей. Поражение кожи наблюдают редко (обычно при пре-Виммунологическом фенотипе).
Гиперлейкоцитоз более 10х109/л выявляют у 60% пациентов, более 100х109/л - у 10%, тромбоцитопению менее 50х109/л - у 60%. В большинстве случаев гиперлейкоцитоз свыше 50х109/л сочетается со значительной лимфаденопатией и гепатоспленомегалией (чаще всего при Т-клеточном иммунологическом фенотипе). Гиперлейкоцитоз никогда не сопровождается церебральной или легочной недостаточностью (в отличие от острых миелоидных лейкозов).
При анализе пунктата красного костного мозга в большинстве случаев выявляют повышенную клеточность, тотальную бластную метаплазию; единичные эритроидные и миелоидные клетки выглядят нормальными; количество мегакариоцитов снижено. В 60-70% случаев выявляют хромосомные аберрации. При биохимическом анализе крови выявляют повышенную концентрацию ЛДГ, гиперурикемию, гиперфосфатемию, гиперкальциемию.
На рентгенограммах органов грудной полости у 5-10% больных обнаруживают увеличение размеров тени средостения (за счет вовлечения тимуса или внутригрудных лимфатических узлов). У 3-5% больных в ликворе обнаруживают лейкозные клетки.
Частота и прогноз иммунологических фенотипов острого лимфобластного лейкоза представлены в табл. 56-1.
Таблица 56-1. Частота и прогноз отдельных иммунологических фенотипов острого лимфобластного лейкоза
Дифференциальная диагностика
Дифференциальную диагностику острого лимфобластного лейкоза проводят в первую очередь с метастазами в костный мозг лимфосарком. В ряде случаев провести четкую грань между этими нозологиями бывает очень сложно, тем более что в настоящее время выделяют вари-
анты, объединяющие характеристики обоих и представляющие собой единую форму - лейкоз/лимфому. Это касается таких заболеваний, как В-клеточный лейкоз/лимфома (фенотип зрелых В-клеток), Т-клеточный лейкоз/лимфома (фенотип зрелых Т-клеток). Впрочем, такое объединение вполне допустимо, поскольку терапия их одинакова.
В ряде случаев острый лимфобластный лейкоз необходимо дифференцировать от метастазов в костный мозг нейробластомы, некоторых солидных опухолей (например, мелкоклеточного рака легкого), инфекционного мононуклеоза. В любом случае диагноз устанавливают только на основании комплексного анализа (морфологических, цитохимических, цитогенетических исследований, иммунологического фенотипирования бластных клеток).
ПРОГНОСТИЧЕСКИЕ ФАКТОРЫ
В настоящее время общепринятыми факторами принадлежности больного к прогностически неблагоприятной группе считают:
• возраст старше 50 лет;
• количество лейкоцитов в дебюте заболевания более 30х109/л для В-клеточного острого лимфобластного лейкоза и более 100х109/л - для Т-клеточного варианта;
• концентрацию ЛДГ в сыворотке крови более 1000 ЕД/л;
• достижение ремиссии более чем за 4 нед;
• иммунологический фенотип ранний пре-В- и ранний Т-варианты;
• цитогенетические маркеры t(9;22), t(4;11), множественные хромосомные аберрации.
ЛЕЧЕНИЕ
Существуют 2 принципиально различных подхода к лечению больных острым лимфобластным лейкозом: дифференцированный (в зависимости от наличия или отсутствия факторов риска) и унифицированный. Привлекательность унифицированного подхода состоит в возможности достаточно адекватного лечения независимо от иммунологического варианта болезни. Однако дифференцированное лечение, определяемое в соответствии с принадлежностью пациента к одной из прогностических групп, более прогрессивно. Перспективным считают включение в программы лечения всех больных из группы неблагоприятного прогноза трансплантации гемопоэтических клеток, особенно аллогенных. При отсутствии HLA-совместимого донора целесообразно выполнение аутологичной трансплантации гемопоэтических клеток.
Общие принципы терапии следующие:
• использование сочетания 5 цитостатических ЛС или более с обязательным применением ГК. Наиболее рациональная терапия индукции - сочетанное применение винкристина, преднизолона и даунорубицина. Различные комбинации также включают метотрексат, аспарагиназу, циклофосфамид, меркаптопурин, цитарабин;
• профилактика нейролейкоза;
• высокодозная консолидация;
• длительная поддерживающая терапия.
Для уменьшения большой опухолевой массы в дебюте и предупреждения цитолитического синдрома во многих лечебных протоколах предусмотрена «предфаза» индукции с использованием преднизолона и циклофосфамида. Продолжительность индукционных курсов обычно составляет 4-8 нед. Интенсификация индукционных курсов при ранних В- и Т-клеточных острых лимфобластных лейкозах позволяет значительно увеличить частоту полных ремиссий (до 80%).
Для консолидации и поддерживающей терапии применяют те же препараты, что и в период индукции ремиссии, с добавлением антиметаболитов (метотрексата, меркаптопурина). Циклофосфамид, высокие дозы цитарабина и другие препараты, включая этопозид (или другой ингибитор топоизомеразы), также включены во многие схемы лечения. Поддерживающую терапию проводят в течение 1-3 лет. Долгосрочные результаты химиотерапии у больных с резистентными формами и рецидивами остаются неудовлетворительными, продолжительность жизни от момента констатации рецидива или резистентности не превышает 3 лет.
Профилактика нейролейкоза - обязательный компонент лечебных протоколов. Профилактику целесообразно начинать еще в фазе индукции ремиссии в связи с возможностью раннего метастазирования лейкозных клеток в ЦНС. Применяют интратекальное введение химиопрепаратов (метотрексата, цитарабина, дексаметазона) и облучение черепа.
Нелимфобластные лейкозы
Термин «острые миелоидные лейкозы» объединяет группу острых лейкозов, возникающих из клетки-предшественницы миелопоэза и различающихся друг от друга по морфологическим и цитогенетическим признакам и иммунологическому фенотипу. Приблизительно в 10%
случаев клетки имеют признаки эритроидных или мегакариоцитарных предшественников, поэтому более адекватным представляется термин «острые нелимфобластные лейкозы».
ЭПИДЕМИОЛОГИЯ
Острые нелимфобластные лейкозы возможны в любом возрасте, заболеваемость несколько выше у пожилых (медиана возраста - 6065 лет). В среднем заболеваемость составляет 2 на 100 000 населения.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Развитию лейкоза может предшествовать предлейкемическая фаза (предлейкоз, миелодиспластический синдром), которую выявляют у 30-40% больных пожилого возраста.
В рамках острых миелоидных лейкозов рассматривают 3 подтипа заболевания, отличающихся друг от друга по степени дифференцировки лейкозных клеток: острый миелобластный недифференцированный лейкоз (М0) - 5% всех острых нелимфобластных лейкозов; острый миелобластный лейкоз без признаков созревания клеток (М1) - 15%; острый миелобластный лейкоз с признаками созревания (М2) - 25%.
• Для острого миелоидного лейкоза характерны острое начало и быстрое развитие клинических симптомов. Часто отмечают лихорадку, боли в костях. При глубокой гранулоцитопении возникают язвенно-некротические поражения слизистой оболочки полости рта, пищевода, кишечника, признаки общей интоксикации. Увеличение печени, селезенки, лимфатических узлов, экстрамедуллярные поражения нехарактерны, но возможны, особенно в случаях с t(8;21) (у 25% больных обнаруживают спленомегалию, у 20% - хлоромы). Для варианта с t(8;21) характерны выраженный эффект химиотерапии и хорошие долгосрочные результаты. При этой форме описан уникальный феномен персистенции в период полной клинико-гематологической ремиссии минимальной остаточной популяции лейкозных клеток.
• Острый промиелоцитарный лейкоз (М3) (10%) представляет собой четко очерченную нозологическую форму с характерными клинико-лабораторными признаками (типичной морфологией опухолевых клеток, тяжелым геморрагическим синдромом, гематомным типом кровоточивости, избыточно активированным фибринолизом, ДВС-синдромом, лейкопенией), поэтому нередко предварительный диагноз можно установить, основываясь лишь
на клинических проявлениях. Практически во всех случаях промиелоцитарного лейкоза обнаруживают t(15;17).
• Для острого миеломонобластного лейкоза (М4) (25-30% случаев) характерны увеличение размеров печени и селезенки, лимфаденопатия, гиперплазия десен, инфильтрация кожи; намного чаще, чем при других формах, наблюдают поражение ЦНС.
• Для острого монобластного лейкоза (М5) (10% случаев) довольно характерны выраженный лейкоцитоз, экстрамедуллярные поражения, включая инфильтрацию десен, кожи, нейролейкоз, гепатоспленомегалию, лимфаденопатию. Нередко развивается ДВСсиндром, который может прогрессировать на фоне химиотерапии. У 60% больных развивается почечная недостаточность. Прогноз неблагоприятный.
• Острый эритробластный лейкоз (М6) - редкая форма (<5% случаев у взрослых и 0,6% у детей). Нередко развитию заболевания предшествует довольно продолжительный предлейкемический период (снижение концентрации гемоглобина, не связанное с дефицитом железа и витамина B12, умеренная лейкопения до 2-4х109/л и тромбоцитопения до 100-160х109/л). В большинстве случаев острый эритробластный лейкоз развивается в возрасте старше 60 лет. Синдром Ди Гульельмо, характеризуемый наличием в красном костном мозге большого количества (>70% клеток) диспластичных эритробластов, выявляют в 10% случаев острого эритробластного лейкоза. При этом варианте также возможны гипергаммаглобулинемия, положительная проба Кумбса, положительные реакции на ревматоидный и антиядерный факторы. Иногда в клинической картине доминируют артралгии, серозиты, гемолитическая анемия.
• Острый мегакариобластный лейкоз (М7) - очень редкая форма (<1-3% случаев), болеют преимущественно дети первого года жизни. Характерны миелофиброз и остеосклероз, поэтому пунктат костного мозга получить довольно сложно. Иногда обнаруживают очаги остеолизиса. Прогноз крайне неблагоприятный.
ПРОГНОСТИЧЕСКИЕ ФАКТОРЫ
Наиболее важный прогностический фактор при острых нелимфобластных лейкозах - адекватность начальных этапов терапии. Неправильную тактику лечения на первом этапе зачастую не могут исправить ни интенсификация лечения на последующих этапах, ни трансплантация
аллогенных гемопоэтических клеток и связанный с ней иммунологический феномен «трансплантат против лейкоза». Факторы неблагоприятного прогноза также включают пожилой возраст, гиперлейкоцитоз или признаки 3-ростковой дисплазии в дебюте заболевания, предшествующую развитию лейкоза миелодисплазию, высокую концентрацию ЛДГ в сыворотке крови. Достаточно простой прогностический фактор - морфологический вариант. Моно-, эритро- и мегакариобластный варианты включены в группу неблагоприятного прогноза. К последней также относят всех больных с резистентным течением заболевания. Основные критерии включения пациентов в группу благоприятного прогноза:
• наличие inv(16), часто выявляют у больных с острым миеломонобластным лейкозом и выраженной эозинофилией в красном костном мозге;
• t(15;17) - типичный маркер варианта М3;
• t(8;21) - у 40% больных с вариантом М2.
ЛЕЧЕНИЕ
В настоящее время наиболее адекватной программой терапии индукции считают схему «7+3» (ежедневное введение цитарабина в течение 7 дней и даунорубицина в течение первых 3 дней). Для консолидации и поддерживающей терапии используют те же курсы, что и для индукционной терапии.
Особого внимания заслуживает лечение острого промиелоцитарного лейкоза. Одним из самых важных открытий в области гематологии за последние 10 лет стало обнаружение специфического эффекта на бластные клетки промиелоцитарного лейкоза производного ретиноевой кислоты - третиноина (полностью - трансретиноевой кислоты). Сочетанное применение третиноина и цитостатических ЛС позволяет достичь ремиссии в 90% случаев.
Лечение рецидивов и резистентных форм до настоящего времени остается нерешенной проблемой. Тем не менее при своевременной диагностике рецидива и активной терапевтической тактике удается получить повторную ремиссию у 30-35% больных. Большинство программ химиотерапии рецидивов и резистентных форм основано на использовании цитарабина в сочетании с митоксантроном и идарубицином.
Во всех случаях, когда удалось достичь второй полной ремиссии, следует рассмотреть возможность трансплантации аллогенных или аутологичных гемопоэтических клеток (с учетом возраста пациента - не старше 40-45 лет, соматического статуса, наличия донора и прочих факторов).
56.2. МИЕЛОДИСПЛАСТИЧЕСКИЙ СИНДРОМ
Миелодиспластический синдром (МДС) - гетерогенная группа клональных заболеваний системы крови, характеризуемых наличием признаков дисмиелопоэза в сочетании с цитопеническим синдромом, клональными изменениями и высокой частотой трансформации в острый лейкоз.
ЭПИДЕМИОЛОГИЯ
Миелодиспластический синдром в основном диагностируют у пожилых людей (медиана возраста - 70 лет). Несколько чаще болеют мужчины. В Европе заболеваемость МДС достаточно высока: 4-13 на 100 000 населения в год, что составляет ежегодно 20 000 новых случаев. В США при количестве больных 50-60 тыс. ежегодно диагностируют 12 000 новых случаев МДС. В группе старше 70 лет заболеваемость увеличивается до 15-50 новых случаев в год на 100 000 населения.
В мире частота диагностики МДС сопоставима с частотой диагностики хронического лимфолейкоза и хронического миелолейкоза вместе взятых и превышает частоту диагностики острого миелолейкоза.
Сведений о заболеваемости и распространенности МДС в Российской Федерации нет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
В 80-90% случаев МДС развивается как первичный и в 10-20% связан с предшествующим цитостатическим или радиологическим лечением, проводимым по поводу различных опухолевых заболеваний. Наиболее часто вторичный МДС развивается через 3-7 лет после использования в программах химиотерапии алкилирующих агентов (циклофосфана♠, мелфалана, мустаргена*), антрациклинов, ингибиторов топоизомеразы II.
Среди факторов, увеличивающих риск развития МДС, можно также выделить контакт с бензином, ароматическими полициклическими углеводородами, дериватами бензина, инсектицидами, пестицидами, удобрениями.
Изучение патогенеза МДС позволило выделить несколько основных этапов развития миелодисплазии. При воздействии повреждающих факторов на стволовую гемопоэтическую клетку в делящемся пуле появляются клетки с мутациями, которые приводят к появлению коммитированных клеток-предшественниц с высоким пролифератив-
ным потенциалом и способствуют формированию моноклонального гемопоэза. Дефектные клетки обнаружены во всех ростках кроветворения и среди В-лимфоцитов. Высокая пролиферативная активность способствует появлению новых мутаций и возникновению дополнительных клонов. Клетки с приобретенными мутациями получают преимущество роста, сохраняя способность к дифференцировке. Интенсивная внутрикостномозговая гибель опухолевых клеток сопровождается развитием панцитопении и определяет характерный для МДС симптомокомплекс:
• инфекционные осложнения;
• анемический синдром;
• геморрагический синдром.
Доказательством того, что при МДС исходно поражается стволовая кроветворная клетка, является обнаружение мутаций гена N-RAS в гранулоцитах, моноцитах, Т- и В-лимфоцитах, а также наличие одинаковых хромосомных аберраций в клетках миелоидной линии, моноцитах и эритроидных предшественниках.
КЛАССИФИКАЦИЯ
На основании количественных и качественных изменений гемопоэза, в частности, цитопенического синдрома, нормо- и/или гиперклеточности костного мозга, диспластических изменений в клетках кроветворной системы, клональных изменений кариотипа и вероятности трансформации в острый лейкоз, было выделено 5 клинических форм МДС:
• рефрактерная анемия;
• рефрактерная анемия с «кольцевыми сидеробластами» (РАКС);
• рефрактерная анемия с избытком бластов (РАИБ);
• рефрактерная анемия с избытком бластов в трансформации (РА-
ИБт);
• хронический миеломоноцитарный лейкоз (ХММЛ).
В России для описания заболеваний с бластозом в костном мозге от 5 до 30% используют термин «острый малопроцентный лейкоз».
В расширенной классификации МДС выделено 9 нозологических форм (табл. 56-2).
Рефрактерные анемии гетерогенны не только по количеству бластных клеток, но и по особенностям гистологической картины костного мозга, на основании чего выделяют МДС с гипоплазией кроветворения (преобладанием жировой ткани над деятельной) и МДС с миелофиброзом.
Таблица 56-2. Классификация миелодиспластического синдрома
В классификации ВОЗ группа РАИБт отсутствует в связи с тем, что у больных с острыми нелимфобластными лейкозами и рефрактерными анемиями с избытком бластов в трансформации одинаковы течение болезни, ответ на химиотерапию, частота достижения ремиссии и продолжительность жизни. Диагностически значимым принято считать количество бластных клеток в костном мозге более 20%, и РАИБт в классификации ВОЗ отнесена к острым лейкозам.
Вторичные МДС/острые миелолейкозы рассматривают в отдельной категории заболеваний и не классифицируют в рамках первичного
МДС.
В классификации ВОЗ также выделена группа миелодиспластических/миелопролиферативных заболеваний:
• хронический миеломоноцитарный лейкоз;
• атипичный хронический миелолейкоз, bcr-abH-негативный;
• ювенильный хронический миеломоноцитарный лейкоз;
• миелодиспластическое/миелопролиферативное заболевание неклассифицируемое, в том числе рефрактерная анемия с кольцевыми сидеробластами и тромбоцитозом.
КЛИНИЧЕСКАЯ КАРТИНА
Специфической клинической картины при МДС нет. Характерный симптомокоплекс обусловлен как количественными, так и качественными изменениями кроветворения.
Наиболее частой причиной обращения к врачу (в 60-80% случаев) являются анемические жалобы, которые в течение длительного периода времени носят скрытый характер и проявляются слабостью, быстрой утомляемостью, головокружением и сердцебиением как в покое, так и при физической нагрузке, учащением приступов стенокардии и прогрессированием сердечной недостаточности. Нередко (в 50-60% случаев) поводом обращения к врачу являются частые обострения уже имеющихся хронических заболеваний и/или вновь возникшие инфекционные осложнения, связанные с нейтропенией и функциональной неполноценностью нейтрофилов. Наиболее частыми инфекционными проявлениями МДС являются бактериальные пневмонии, абсцессы кожи и оппортунистические инфекции (вирусные, пневмоцистные, микобактериальные).
Угрожающее жизни осложнение МДС - геморрагический синдром (петехиальные высыпания на коже и слизистых оболочках, носовые, десневые, маточные, почечные и желудочно-кишечные кровотечения, кровоизлияния в склеры и головной мозг), который отражает не только глубину тромбоцитопении, но и нарушение адгезивно-агрегационных свойств тромбоцитов.
У больных с МДС описаны изменения иммунной системы: уменьшение активности NK-клеток, количества СD4+-Т-лимфоцитов, развитие моноклональной гаммапатии и появление аутоантител, изменение секреции иммуноглобулинов. Нередко МДС манифестирует как аутоиммунный процесс, проявляющийся системным васкулитом, серонегативным артритом, ревматической полимиалгией, перикардитом, плевритом .
ДИАГНОСТИКА И ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА
При обследовании больных с анемией и двухили трехростковой цитопенией в первую очередь необходимо исключить заболевания, сопровождаемые характерными для МДС изменениями периферической крови:
• солидные опухоли, протекающие с явными или скрытыми кровотечениями, в том числе метастазирующие в костный мозг;
• туберкулез;
• панмиелофтиз;
• лимфопролиферативные заболевания;
• системные заболевания соединительной ткани;
• вирусные инфекции, в том числе вирусные гепатиты;
• пароксизмальную ночную гемоглобинурию;
• острый эритробластный лейкоз;
• апластическую анемию;
• В12- и фолиеводефицитную анемии;
• хронические заболевания печени.
Все эти заболевания могут сопровождаться развитием диспластических изменений костного мозга (преимущественно в клетках эритроидного ряда) и характерных для МДС изменений периферической крови.
Диагноз МДС - диагноз исключения. Случаи цитопенического синдрома с характерной для МДС миелодисплазией, выявляемой в клетках крови и костного мозга, зачастую ошибочно, без уточнения этиологии, относят к МДС, тогда как, учитывая гетерогенность нозологии, достоверная диагностика возможна только на основании обнаружения сочетания качественных и количественных изменений кроветворной системы, выявляемых при цитологическом, цитогенетическом и гистологическом исследовании костного мозга.
Количественные и качественные изменения эритроидных предшественников костного мозга и эритроцитов периферической крови (в основном макроцитоз) - общая черта большинства вариантов МДС. Для клеток красного ряда характерны изменения формы, размеров и количества ядер, появление в цитоплазме вакуолей и базофильных гранул. Нередко в костном мозге встречаются эритрокариоциты с мегалобластоидным оттенком и расположенными вокруг ядра гранулами гемосидерина (кольцевые сидеробласты). Количественные изменения красного ростка могут проявляться и его расширением, и развитием парциальной красноклеточной аплазии.
Анемия при МДС может определяться гемолизом эритроцитов как аутоиммунного характера (наличие аутоантител к эритроцитам подтверждает положительная прямая проба Кумбса), так и клоном эритроцитов с дефектами белковых и липидных компонентов клеточной мембраны, аналогичным характерному для пароксизмальной ночной гемоглобинурии (положительные пробы Хема и сахарозная, обнаружение клона CD55-CD59-эритроцитов).
Изменения клеток гранулоцитарного ряда костного мозга и периферической крови могут проявляться как гиперсегментацией ядер, так и псевдопельгеровской аномалией. В цитоплазме возможно как увеличение или уменьшение зернистости до полного ее исчезновения, так и изменение размеров гранул.
В мегакариоцитарном ростке достаточно часто появление микромегакариоцитов, размеры которых при норме 60-120 m менее 25 m, с
одним или двумя ядрами и зрелой цитоплазмой. Возможно появление мегакариоцитов как с множественными отдельно расположенными ядрами разнообразных размеров и форм, так и с одним большим ядром. В периферической крови нередко определяются гигантские тромбоциты.
Для оценки выраженности миелодисплазии необходимо подсчитывать не менее 200 (500) ядросодержащих клеток и 20 мегакариоцитов. Диагностически значимым считают обнаружение признаков миелодисплазии не менее чем в 10% клеток того или иного ростка.
При гистоморфологическом исследовании кроветворной ткани наиболее часто выявляют увеличение клеточности костного мозга, в 20% случаев - гипоплазию кроветворения, в 20% - миелофиброз вне зависимости от степени клеточности.
Хромосомные аномалии выявляют у 40-70% больных с МДС. По мере увеличения количества бластных клеток вероятность обнаружения изменений кариотипа также увеличивается. К наиболее характерным относят: трисомию 8, моносомию 5 и 7 хромосомы, потерю Y-хромосомы, делецию длинного плеча 5 (5q-) и 7 (7q-) хромосом, транслокации
t(1;3), t(1;7), t(5;7), t(2;11), t(6;9), t(3;21), t(2;11), инверсию 3 хромосомы inv(3), изменения Х-хромосомы и некоторые другие.
ЛЕЧЕНИЕ
В связи с тем что МДС диагностируют преимущественно у людей старше 60 лет, терапевтические подходы варьируют от динамического наблюдения до трансплантации аллогенных стволовых гемопоэтических клеток от HLA-идентичного донора.
• Динамическое наблюдение, в том числе с сопроводительной терапией:
- заместительная терапия компонентами крови;
- эритропоэтин;
- хелаторы;
- антибиотическая терапия;
- витамины и микроэлементы.
• Цитостатическая терапия:
- монотерапия цитостатическими препаратами;
- комбинированное цитостатическое воздействие;
- трансплантация аллогенных стволовых гемопоэтических клеток.
• Эпигенетическая терапия:
- ингибиторы метилтрансфераз;
- ингибиторы гимстодеацетилаз;
- ингибиторы протеасом.
• Иммуносупрессивная терапия.
• Иммуномодулирующая и антиангиогенная терапия.
• Антиапоптотическая, дифференцирующая терапия.
• Сочетанное воздействие.
У одного больного на разных этапах болезни могут быть использованы различные терапевтические подходы, что может определяться как с эффективностью первой линии терапии, так и прогрессией заболевания.
У больных с МДС при выборе тактики терапии необходимо оценивать не только изменение кроветворения в момент диагностики, но и динамику процесса в течение определенного периода времени. Выделяют медленно- и быстропрогрессирующее течение. При первом возможны динамическое наблюдение и более детальное обследование, второе требует принятия решения в первые недели заболевания. По динамике основных показателей гемограммы и степени выраженности изменений костномозгового кроветворения выделяют 4 основных варианта течения болезни:
• стабильные показатели в течение 3-6 мес;
• медленное ухудшение показателей в течение 3-6 мес;
• быстрое изменение показателей в течение 1 мес;
• крайне неблагоприятные показатели в момент диагностики.
Единственным методом, позволяющим достичь биологического излечения больных с МДС, считают трансплантацию аллогенных гемопоэтических стволовых клеток, в силу соматического статуса пациента и наличия родственного донора применимую только у 3-5% больных. Использование кондиционирования в режиме пониженной интенсивности позволяет использовать этот метод у больных с МДС старше 40 лет, т.е. при отсутствии идентичных сиблингов для всех больных моложе 60 лет необходимо инициировать поиск HLA-идентичного неродственного донора, особенно при неблагоприятных факторах течения болезни.
При рефрактерных анемиях с концентрацией эндогенного эритропоэтина менее 200-500 и/л и незначительной (<2 раз в месяц) зависимостью от трансфузий донорских эритроцитов целесообразно применение эритропоэтинов, у больных c рефрактерными анемиями и кольцевыми сидеробластами - в сочетании с гранулоцитарным стимулирующим фактором.
При длительном трансфузионном анамнезе (>20 трансфузий эритроцитов), увеличении содержания ферритина в сыворотке более 10001500 мг/мл без признаков воспаления и/или с признаками вторичного гемохроматоза необходимо проводить терапию хелаторами.
При МДС с бластозом менее 5-10% в костном мозге, гипоплазией кроветворной ткани в момент диагностики и благоприятными хромосомными изменениями терапией выбора является иммуносупрессия: циклоспорин А, антитимоцитарный глобулин, при их неэффективности - спленэктомия с последующим возобновлением приема циклоспорина А.
У больных с изолированной делецией 5q препаратом выбора считают леналидомид (ревлимид*), который позволяет не только полностью отказаться от трансфузий донорских эритроцитов, но и достигнуть цитогенетической ремиссии.
При увеличении количества бластных клеток в костном мозге более 5%, при нормоили гиперклеточном костном мозге возможно применение нескольких алгоритмов лечения.
• Препаратов эпигенетического действия (децитабина, азацитидина, вальпроевой кислоты, вориностата), применение которых увеличивает возможность достижения эффекта за счет уменьшения токсичности, увеличивает продолжительность жизни и промежуток до трансформации в острый лейкоз. Эффект от проводимой эпигенетической терапии развивается через 1-4 курса. В случае достижения эффекта необходимо продолжать лечение до трансформации в острый лейкоз.
• Малых доз цитостатических препаратов (цитарабина, мелфалана, этопозида, 6-меркаптопурина), эффективность которых составляет 30-40%. Нередко этот метод лечения применяют у пожилых пациентов.
• Полихимиотерапии по программам лечения острого миелолейкоза, что позволяет достичь полной ремиссии в 30-75% случаев, однако сопровождается высокой ранней летальностью у 20% больных и относительно непродолжительным (от 4 до 18 мес с медианой 13 мес) эффектом. Подобный подход можно рекомендовать для молодых пациентов с наличием HLA-идентичного донора для трансплантации аллогенных гемопоэтических стволовых клеток.
При рефрактерной анемии, когда нецелесообразно применение эритропоэтина, рефрактерной цитопении с мультилинейной дисплазией и
гиперклеточным костным мозгом и нормальным кариотипом возможно применение препаратов а-интерферона.
Использование антиапоптотической терапии у больных с рефрактерной цитопенией и мультилинейной дисплазией позволяет уменьшить зависимость от гемотрансфузий и, соответственно, развитие гемохроматоза, а также улучшить показатели периферической крови, уменьшив количество осложнений.
У больных с МДС и миелофиброзом в качестве как монотерапии, так и в сочетании с другими препаратами возможно назначение даназола или преднизолона.
Таким образом, терапия МДС - специфическое, постоянное и длительное лечение, выбор которого зависит от варианта заболевания, динамики изменений кроветворения, возраста и соматического статуса пациента.
56.3. ХРОНИЧЕСКИЕ ЛЕЙКОЗЫ
При хронических лейкозах клеточный субстрат представлен зрелыми дифференцированными клетками крови. В большинстве случаев происходит неограниченная пролиферация преимущественно какого-либо одного или двух ростков кроветворения, хотя при некоторых формах возможна пролиферация всех трех линий гемопоэза. Наиболее частые заболевания этой группы - хронический миелолейкоз, истинная полицитемия (эритремия), сублейкемический миелоз.
Хронический миелолейкоз
Хронический миелолейкоз - опухоль, возникающая из ранних клеток-предшественниц миелопоэза, которые дифференцируются до зрелых форм. Клеточный субстрат опухоли - созревающие и зрелые гранулоциты, преимущественно нейтрофилы.
ЭПИДЕМИОЛОГИЯ
На хронический миелолейкоз приходится 7-15% общего количества гемобластозов. Заболеваемость в среднем составляет 1-1,5 на 100 000 населения. Болезнь одинаково часто развивается среди мужчин и женщин, преимущественный возраст заболевших - 30-40 лет. В детском и юношеском возрасте хронический миелолейкоз наблюдают редко.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Значительное увеличение заболеваемости среди пострадавших от атомной бомбардировки Хиросимы и Нагасаки подтверждает этиологическую роль ионизирующего излучения. Также существуют данные о возможной роли некоторых химических соединений и врожденных дефектов хромосом. У 85-98% больных в клетках гранулоцитарного, мегакариоцитарного, эритроидного рядов, моноцитах-макрофагах, а в последние годы и в В- и Т-лимфоцитах выявляют приобретенную хромосомную аномалию - так называемую филадельфийскую (Рh') хромосому. Ph'-хромосома образуется в результате сбалансированной транслокации между хромосомами 9 и 22 - t(9;22). При этом происходит перемещение c-ABL-протоонкогена с хромосомы 9 на хромосому 22 с образованием гибридного гена BCR-ABL, с которого считывается аномальная матричная РНК, кодирующая протеин p210 с повышенной тирозинкиназной активностью. Принято считать, что именно таким путем происходит превращение нормальных стволовых гемопоэтических клеток в опухолевые. Существуют и Рh'-негативные варианты хронического миелолейкоза, которые, тем не менее, почти в половине случаев являются BCR-ABL-позитивными вариантами хронического миелолейкоза с «маскированной» Рh'-хромосомой. Рh'-хромосома часто сочетается с трисомией 8, 9, 19, 21, делецией хромосомы 3 и другими дефектами.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Заболевание закономерно проходит 2 стадии:
• развернутую доброкачественную (моноклоновую);
• терминальную злокачественную (поликлоновую).
В течении хронического миелолейкоза выделяют 3 основные фазы, которые отражают характерную для данного заболевания опухолевую прогрессию.
• Хроническую фазу. Характеризуется гиперплазией гранулоцитарного ростка кроветворения в красном костном мозге. При этом способность клеток к дифференцировке и созреванию сохранена. В пределах этой фазы выделяют начальный период, когда опухолевая масса еще невелика и заболевание проявляется только умеренным лейкоцитозом в крови со сдвигом лейкоцитарной формулы до миелоцитов, увеличением содержания зрелых и созревающих гранулоцитов в красном костном мозге; эритро- и тромбоцитопоэз сохранены, селезенка нормальных размеров или незначитель-
но увеличена. По мере увеличения опухолевой массы нарастают лейкоцитоз и содержание молодых форм гранулоцитов, значительно увеличиваются размеры селезенки и печени, появляются умеренно выраженные симптомы интоксикации. Этот период известен как развернутая стадия заболевания. В хронической фазе, как правило, эффективны все виды стандартной цитостатической монохимиотерапии, с помощью которой обычно удается добиться максимального уменьшения опухолевой массы.
• Фазу акселерации. Наступает обычно через 3-3,5 года от начала заболевания, ассоциируется с началом терминальной стадии (бластной трансформации). Эта фаза характеризуется не только дальнейшим увеличением опухолевой массы, но и качественным ее преобразованием - нарастающими признаками нарушения дифференцировки на уровне стволовой клетки. В крови появляются бластные клетки, развиваются анемия, тромбоцитопения (или, наоборот, тромбоцитоз); в красном костном мозге (а в ряде случаев и в селезенке) наряду со зрелыми и созревающими гранулоцитами обнаруживают очаги бластной пролиферации. Нарастают симптомы интоксикации: лихорадка, повышенное потоотделение, слабость, снижение массы тела. Важный признак фазы акселерации - резистентность к успешно применяемой ранее химиотерапии. Введение в схемы лечения цитостатических препаратов, действие которых проявляется на уровне коммитированных предшественников миелопоэза (цитарабина, даунорубицина, меркаптопурина и др.), в некоторых случаях оказывает кратковременный эффект с возвратом процесса в хроническую фазу.
• Властный криз. Терминальная стадия заболевания. Происходит резкое ухудшение общего состояния (выраженные признаки интоксикации, лихорадка, боли в костях, увеличение размеров печени и селезенки с возможным развитием инфарктов последней). В периферической крови высокое (>30%) содержание бластных клеток. Развиваются тяжелые анемия, тромбоцитопения вследствие бластной инфильтрации красного костного мозга с подавлением эритро- и мегакариоцитопоэза. Течение заболевания осложняется присоединением инфекций. О приближении бластного криза свидетельствуют развитие резистентности к химиотерапии и изменение кариологического профиля лейкозных клеток (анеуплоидия, большие, уродливые ядра клеток). Моноклоновая популяция клеток с Рh'-хромосомой заменяется поликлоновой,
отличающейся резкой анаплазией клеток. Летальный исход в фазе бластного криза практически неизбежен, ремиссия или возврат во 2-ю фазу - явление чрезвычайно редкое. Непосредственной причиной смерти обычно являются инфекционные осложнения или кровотечения.
Стадии течения хронического миелолейкоза устанавливают на основании комплекса клинических признаков и данных гемограммы, миелограммы, гистологического исследования красного костного мозга. Большое значение для диагностики имеют обнаружение Ph'-хромосомы в гранулоцитах, моноцитах, эритро- и мегакариоцитах красного костного мозга (важно помнить о вариантах хронического миелолейкоза без Ph'хромосомы) и выявление гибридного («химерного») гена BCR-ABL.
ЛЕЧЕНИЕ
Основная задача проводимой терапии - уменьшение (в идеале - полное удаление) опухолевого клона. Основной метод определения эффективности терапии - цитогенетическое исследование. Цитогенетический ответ оценивают по содержанию Ph'-положительных клеток в пунктате красного костного мозга:
• полный - отсутствие Ph'-позитивного клона;
• частичный - менее 35% Рh'+-клеток;
• минимальный - Рh'+-клеток от 35 до 95%;
• отсутствие ответа - сохранение на фоне терапии исходного количества Рh'+-клеток.
Бусульфан, в течение многих лет составлявший основу монотерапии хронического миелолейкоза, в настоящее время не используют в связи с недостаточной эффективностью (продолжительность эффективного применения не превышает 3,0-4,5 г, медиана выживаемости - 45 мес). Гидроксикарбамид обеспечивает нормализацию состава крови у 70-80% пациентов, медиана продолжительности жизни - 56 мес. Дозы препарата подбирают индивидуально в зависимости от показателей периферической крови. Начальная доза - 20-50 мг/(кгхсут) с постепенным ее снижением под контролем содержания лейкоцитов. Доза препарата при поддерживающей терапии - 10 мг/кг. При отмене гидроксикарбамида количество лейкоцитов быстро увеличивается.
В настоящее время препаратом выбора в лечении хронического миелолейкоза считают препараты интерферона-альфа. Медиана продолжительности жизни при терапии интерферонами - 66-89 мес. Преимущественно используют рекомбинантные препараты
интерферона-альфа-2а и интерферона-альфа-2b в дозе от 35х106 ЕД/м2 3 раза в неделю до 5х106 ЕД/м2 1 раз в день.
Больным молодого возраста при наличии HLA-идентичного сиблинга необходимо провести трансплантацию красного костного мозга. На сегодняшний день это единственный метод, позволяющий добиться излечения. Для уменьшения риска осложнений трансплантацию предпочтительно выполнять в ранней хронической фазе. Трансплантация реально выполнима не более чем у 12-20% больных. Ранняя летальность при этом методе лечения составляет 20-30%, у 15% пациентов развивается рецидив. Если родственного донора нет, молодым больным может быть выполнена трансплантация от HLA-совместимого неродственного донора.
В последние годы разрабатывают протоколы лечения ингибиторами тирозинкиназы (иматинибом), которые вызывают остановку пролиферации или индукцию апоптоза в клетках, экспрессирующих BCR-ABL-тирозинкиназу. Препарат назначают в дозе 300 мг/сут, эффект развивается обычно на 2-4-й неделе лечения. Применение иматиниба часто позволяет достичь полных цитогенетических ремиссий во всех стадиях болезни.
Проблема лечения в терминальной стадии остается нерешенной. При миелоидном бластном кризе применяют программы химиотерапии, аналогичные таковым при остром миелоидном лейкозе (например, «7+3»; см. раздел «Острые лейкозы»). При лимфобластном варианте бластного криза иногда применяют сочетания антрациклинов (даунорубицина, идарубицина, митоксантрона), циклофосфамида, аспарагиназы, винкристина и преднизолона. При лимфобластном варианте бластного криза хронического миелолейкоза, так же как и при остром лимфобластном лейкозе, нередко развивается нейролейкоз, поэтому необходимы соответствующие профилактические мероприятия.
ПРОГНОЗ
В дебюте заболевания наиболее достоверными, прогностически неблагоприятными факторами считают снижение концентрации гемоглобина менее 100 г/л, наличие в крови и/или красном костном мозге более 3% бластных клеток, увеличение размеров селезенки более +5 см из-под края реберной дуги, эозинофилия периферической крови более 4%. К категории низкого риска относят больных, не имеющих ни одного неблагоприятного признака, промежуточного - при наличии 1-2 признаков, высокого - 3 и более.
Один из основных прогностических факторов, помимо определения групп риска, - эффективность терапии. Достижение гематологической ремиссии в течение 3-6 мес и цитогенетического ответа к 12-му месяцу свидетельствует о благоприятном прогнозе и возможности полного восстановления нормального кроветворения.
Истинная полицитемия
Истинная полицитемия (эритремия, болезнь Вакеза) - форма хронического лейкоза с поражением клетки-предшественницы миелопоэза, сохраняющей способность к дифференцировке по 4 росткам, преимущественно эритроидному.
ЭПИДЕМИОЛОГИЯ
Заболеваемость истинной полицитемией составляет 0,6-1,6 на 100 000 населения. Заболевание наблюдают преимущественно в пожилом и старческом возрасте.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Истинная полицитемия развивается в результате клональной экспансии трансформированной мультипотентной стволовой клетки. Преимущественная дифференцировка по эритроидной линии, вероятно, связана с гиперчувствительностью клеток-предшественниц аномального клона к эритропоэтину. Также доказана повышенная чувствительность клеток-предшественниц к ИЛ-3, гранулоцитарно-моноцитарному колониестимулирующему фактору, что, возможно, и объясняет гиперплазию 3 ростков гемопоэза. В отличие от хронического миелолейкоза, характерного цитогенетического маркера истинной полицитемии нет, хотя к моменту постановки диагноза хромосомные аномалии выявляют в 17-26% случаев.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Выделяют несколько стадий течения истинной полицитемии.
• I - начальная стадия, продолжается до 5 лет и более, характеризуется минимальными клиническими проявлениями.
• IIA - эритремическая развернутая стадия без миелоидной метаплазии селезенки продолжительностью 10-20 лет и более.
• IIБ - эритремическая развернутая стадия с миелоидной метаплазией селезенки.
• III - стадия миелоидной метаплазии (анемическая стадия) с миелофиброзом или без такового; возможен исход в острый лейкоз.
В целом истинная полицитемия характеризуется длительным и относительно доброкачественным течением. Начало заболевания довольно вариабельно. У многих больных в анамнезе присутствуют указания на кровотечения после экстракции зубов, кожный зуд, связанный с водными процедурами, несколько повышенные показатели гемоглобина. Кожа лица, ушей, кончика носа, дистальных отделов конечностей и видимые слизистые оболочки имеют красно-цианотичную окраску различной степени выраженности. Увеличение массы циркулирующих эритроцитов приводит к повышению вязкости крови, стазам в микроциркулярном русле, повышению периферического сосудистого сопротивления. АГ выявляют у 35-40% больных на момент установления диагноза. Нарушение обмена мочевой кислоты (гиперурикемия и урикозурия) осложняет течение IIIБ и III стадии. К висцеральным осложнениям истинной полицитемии относят язвы желудка и двенадцатиперстной кишки (10-17% случаев).
Сосудистые осложнения представляют наибольшую опасность для больных. Одновременная склонность как к тромбозам, так и к кровотечениям - уникальная особенность истинной полицитемии. Тромботические осложнения (инфаркт миокарда, ишемический инсульт, тромбоэмболия легочной артерии) - самая частая причина смерти больных. Геморрагический синдром проявляется кровоточивостью десен, носовыми кровотечениями, экхимозами.
Увеличение селезенки в IIА стадии незначительное, связано с усиленным депонированием и секвестрацией форменных элементов крови. В стадии IIБ спленомегалию вызывает прогрессирующее развитие миелоидной метаплазии. Для обеих стадий характерно развитие фиброза печени. Течение постэритремической стадии вариабельно. У некоторых больных оно вполне доброкачественно, увеличение размеров селезенки и печени происходит медленно, показатели красной крови нормальны. В других случаях наблюдают быстрое нарастание анемии, прогрессирование спленомегалии, появление в крови бластных клеток. Развитие острого лейкоза возможно как в эритремической стадии заболевания, так и в стадии постэритремической миелоидной метаплазии.
Критерии диагностики истинной полицитемии следующие.
• Увеличение количества циркулирующих эритроцитов у мужчин выше 36 мл/кг, у женщин - 32 мл/кг.
• Увеличение сатурации кислорода в артериальной крови до 92% и более.
• Спленомегалия.
• Тромбоцитоз более 400х109/л.
• Лейкоцитоз более 12х109/л.
• Повышение активности ЩФ лейкоцитов.
• Повышение концентрации витамина B12 в сыворотке более 900 пг/мл.
• Снижение плазменной концентрации эритропоэтина.
Дифференциальная диагностика
Дифференциальную диагностику чаще всего приходится проводить с вторичными эритроцитозами, обусловленными тканевой гипоксией (врожденными «синими» пороками сердца, пневмосклерозом, адаптацией к условиям высокогорья и т.д.) и изменением содержания эндогенного эритропоэтина (например, при опухолях почки). Среди всех случаев вторичной полицитемии сочетания гиперплазии всех 3 ростков, как правило, не бывает, и в развернутых стадиях заболевания диагностика обычно достаточно проста.
На ранних стадиях истинную полицитемию достаточно трудно дифференцировать от хронического миелолейкоза, основной диагностический критерий в таких случаях - исследование на наличие Рh'-хромосомы. В диагностически затруднительных случаях необходимо провести культуральное исследование, выявляющее спонтанный рост эритроидных колоний, характерный только для истинной полицитемии.
ЛЕЧЕНИЕ
Современная терапия эритремии состоит в использовании эксфузий крови, цитостатических препаратов, радиоактивного фосфора и интерферона-альфа.
• Кровопускания могут быть самостоятельным методом лечения или дополняют цитостатическую терапию. В начальной стадии истинной полицитемии, протекающей лишь с увеличением содержания гемоглобина и эритроцитов, проводят 2-3 кровопускания по 500 мл крови с интервалом 3-5 дней с последующим введением адекватных количеств реополиглюкина♠ или 0,9% раствора натрия хлорида. Вместо кровопускания можно использовать эритроцитофорез. Процедура обычно хорошо переносится и вызывает нормализацию показателей красной крови на срок от 8 мес до 1 года. Ни кровопускания, ни эритроцитоферез не оказывают влияния на содержание лейкоцитов и тромбоцитов, более того, они иногда могут вызывать реактивный тромбоцитоз.
• Цитостатическая терапия показана при эритремии, протекающей с лейкоцитозом, тромбоцитозом и спленомегалией, кожным зу-
дом, висцеральными и сосудистыми осложнениями, а также при недостаточной эффективности предшествующих кровопусканий. Эффект от цитостатической терапии следует оценивать через 3 мес после начала лечения, хотя снижение количества лейкоцитов и тромбоцитов наступает значительно раньше. Применяют алкилирующие производные (бусульфан, мелфалан, циклофосфамид) и антиметаболиты (меркаптопурин, тиогуанин). Алкилирующие препараты оказывают несомненный терапевтический эффект, однако в настоящее время их применяют редко из-за лейкозогенного действия.
• В последние годы широко применяют препараты интерферонаальфа, которые угнетают миелопролиферацию, значительно снижают количество тромбоцитов и сопутствующих сосудистых осложнений. Максимальный терапевтический эффект развивается через 3-8 мес. Лечение проводят в течение многих лет (при хорошей переносимости). Одно из несомненных достоинств терапии интерферонами - отсутствие лейкозогенного действия.
• У очень пожилых больных эффективно применение радиоактивного фосфора (32Р). Полная ремиссия развивается в 98% случаев после однократной инъекции препарата в дозе 2,8-3,5 мКю/м2. В дальнейшем препарат вводят с интервалом 3-6 мес, что позволяет длительно поддерживать гематологическую ремиссию, сохраняя при этом удовлетворительное качество жизни пациента. Радиоактивный фосфор обладает выраженными тератогенным и мутагенным эффектами, что ограничивает возможность его применения у пациентов относительно молодого возраста.
Симптоматическое лечение включает назначение аллопуринола (при гиперурикемии), антиагрегантов и низкомолекулярных гепаринов (при тромбозах сосудов и выявлении гиперкоагуляции), антигистаминных препаратов (при кожном зуде) и др.
56.4. ЛИМФОМЫ И ХРОНИЧЕСКИЕ ЛИМФОЛЕЙКОЗЫ
В классификации ВОЗ выделены 3 большие группы лимфом - В-клеточные, T-клеточные и NK-клеточные, а также болезнь Ходжкина (лимфогранулематоз). В свою очередь, классификация В-клеточных и Т-/NK-клеточных опухолей основана на анализе предшественников опухолевых клеток и в дальнейшем определяется стадиями их дифференцировки. Лимфомы также группируются в соответствии с клиниче-
скими признаками: преимущественно диссеминированные, первичные экстранодальные и преимущественно нодальные (то есть поражающие лимфатические узлы).
Стандартная классификация опухолей TNM для лимфом малоприменима, поскольку в большинстве случаев им свойственна исходно генерализованная манифестация, что объясняется распространенностью лимфатической ткани в организме.
Выделяют лимфомы низкой степени злокачественности (хронические, вялотекущие, зрелоклеточные) и агрессивные лимфомы.
В США и Европе 70-80% всех лимфом составляют B-клеточные опухоли, а T- и NK-клеточные - 20-30%. В странах Восточно-Тихоокеанского региона складывается обратное соотношение. В пределах каждого иммунологического варианта условно выделяют агрессивные и хронические опухоли. К первым относят преимущественно крупноклеточные опухоли (лимфосаркомы), характеризуемые высокой пролиферативной активностью и бурным клиническим течением, ко вторым - зрелоклеточные лимфомы. Клетки таких опухолей активно не пролиферируют, а накопление клеток обусловлено дефектными механизмами апоптоза. Со временем хронические лимфатические опухоли могут трансформироваться в агрессивные, что всегда прогностически неблагоприятно.
Лимфомы возникают в лимфатических узлах и экстралимфатических областях (желудке, кишечнике, мышечной ткани, костях, ЦНС, яичке и др.). Одинаковые по морфологии, основным иммунологическим и цитогенетическим характеристикам, но разные по локализации лимфатические опухоли различаются по характеру и особенностям роста, поэтому при выделении нозологических форм необходимо учитывать первичные очаги их возникновения.
Часть опухолей диагностируют при случайном обследовании, и годами они не требуют лечения. Для других характерен быстрый рост, сопровождаемый симптомами интоксикации. В этом случае лечение откладывать нельзя.
В классификации ВОЗ перечислены 36 нозологических форм, но встречаемость их далеко не одинакова. Более 50% лимфом составляют диффузная B-крупноклеточная и фолликулярная лимфомы. T-клеточные опухоли редки, полиморфны по морфологическим и клиническим проявлениям и представлены в основном периферическими T-клеточными лимфомами.
Лимфомы низкой степени злокачественности
Хронические (вялотекущие, или зрелоклеточные) лимфомы цитологически имеют лимфоцитарный состав.
Лимфомы низкой степени злокачественности составляют следующую группу.
• Фолликулярные лимфомы (22% случаев).
• Лимфомы из малых В-лимфоцитов (7%).
• Экстранодальные лимфомы из В-клеток маргинальной зоны (8%).
• Нодальные лимфомы из В-клеток маргинальной зоны (2%).
• Лимфоплазмоцитарные лимфомы (макроглобулинемия Вальденстрема ) (1%).
• Лимфомы из В-клеток маргинальной зоны селезенки (<1%).
Фолликулярная лимфома
Фолликулярная лимфома - опухоль из В-клеток фолликулярного центра, имеющая хотя бы частично фолликулярную структуру роста. В большинстве случаев находят иммунологический маркер болезни CD10 и перестройку или амплификацию гена BCL2, приводящие к нарушению апоптоза. Этиология заболевания неизвестна.
ЭПИДЕМИОЛОГИЯ
В США фолликулярная лимфома составляет 35% всех лимфом, в мире - 22%. Наименьшую заболеваемость отмечают в Европе, Азии и развивающихся странах. Страдают преимущественно взрослые, средний возраст пациентов - 55 лет. Женщины заболевают в 1,5 раза чаще мужчин. Дети болеют крайне редко.
КЛИНИЧЕСКАЯ КАРТИНА
У 70% пациентов уже в дебюте заболевания выявляют генерализованную лимфаденопатию и умеренную спленомегалию, у половины - поражение костного мозга. Лимфатические узлы обычно имеют эластическую или тестоватую консистенцию, безболезненны, не спаяны между собой и окружающими тканями. В ряде случаев наблюдают слияние лимфатических узлов в конгломераты, обычно в паховых, подвздошных областях и забрюшинно. Симптомы интоксикации (повышенное потоотделение, гипертермия, похудение, слабость) беспокоят только 10% больных.
При генерализованной стадии болезни продолжительность жизни составляет 8-10 лет. Локальные формы фолликулярной лимфомы (по-
ражение 1-2 групп лимфатических узлов) находят у 20-30% больных. В таких случаях облучение пораженных областей у ряда пациентов может привести к выздоровлению. В крайне редких случаях диагностируют изолированные экстранодальные опухоли - кожи, яичка, кишечника.
У 25-40% больных по клиническим данным и у 70% по данным секционного материала может возникнуть саркомная трансформация. Клинически она проявляется прогрессирующим похудением, повышенным потоотделением, фебрильной лихорадкой, быстрым ростом и уплотнением одной или нескольких групп лимфатических узлов или селезенки. Трансформация висцеральных лимфатических узлов может привести к сдавлению и прорастанию опухоли в окружающие ткани и органы, появлению плеврита или асцита. Продолжительность жизни таких больных обычно измеряется неделями или месяцами. Саркомная трансформация подтверждается биопсией опухолевого очага.
Массивная спленомегалия также нередко бывает признаком крупноклеточной трансформации, но прогностически это всегда более благоприятная ситуация, нежели саркомная трансформация. Проведение спленэктомии с последующей консолидирующей химиотерапией могут дать продолжительный хороший результат. К другим клиническим признакам крупноклеточной трансформации относят значительное повышение активности лактатдегидрогеназы (более чем в 1,5 раза), концентрации фибриногена и мочевой кислоты в крови, гиперкальциемию, увеличение СОЭ более 30 мм/ч.
Лабораторные и инструментальные исследования
• При цитологическом исследовании крови обнаруживают мелкие и крупные клетки с расщепленными ядрами (центроциты) и крупные клетки с нерасщепленными ядрами (центробласты).
• Гистологическая картина биоптата лимфатического узла в большинстве случаев представлена многочисленными фолликулоподобными структурами: более 75% среза лимфатического узла - фолликулярными, 25-75% - фолликулярными с зонами диффузного роста, менее 25% - частично фолликулярными. В зонах диффузного роста может быть выражен фиброз ткани.
• Проводят исследование иммунофенотипа опухоли.
• Цитогенетическое исследование позволяет диагностировать транслокацию t(14;18)(q32;q21) (кроме случаев у детей), вариантные транслокации [t(2;18)(p12;q21)], в редких случаях - трисомию
хромосомы 18. В результате транслокации ген BCL2 гиперэкспрессируются и белок Bcl-2 блокирует апоптоз в опухолевых клетках. Таким образом, увеличение опухоли обусловлено не пролиферацией, а накоплением опухолевых клеток. Обычно определяют комплексный кариотип (3 цитогенетических нарушения и более). При трансформации можно обнаружить делеции локуса 17p13 (содержащие ген TP53, супрессирующий опухоль), t(8;14), характерные для лимфомы Беркитта.
ЛЕЧЕНИЕ И ПРОГНОЗ
Лечение зависит от стадии болезни.
• На I и II стадии болезни при удалении единственного опухолевого очага без последующей консолидирующей терапии рецидивы развиваются у 70% больных в среднем через 6 лет. В большинстве случаев они возникают в той же группе лимфатических узлов, а у 25% пациентов при рецидиве диагностируют генерализованный опухолевый процесс.
- Облучение вовлеченных зон можно проводить только при полном исключении поражения других групп лимфатических узлов по данным УЗИ, КТ или позитронно-эмиссионной томографии. В результате лучевой терапии в дозе 30-36 Гр на вовлеченные зоны у 40-50% пациентов удается получить длительную ремиссию заболевания.
- Химиотерапию после облучения вовлеченных областей (аддитивную терапию) применяют для консолидации облучения, а также в тех случаях, когда использование только лучевой терапии не приводит к полному исчезновению очага болезни. Имеются данные, что применение схем СОР (циклофосфамид, винкристин, преднизолон) или CHOP (циклофосфамид, доксорубицин, винкристин, преднизолон) позволяет увеличить частоту полных ремиссий до 65-70%.
• Тактика ведения больных с III и IV стадией заболевания зависит от наличия или отсутствия симптомов интоксикации, возраста, общего состояния больного. В данной ситуации проводят:
- симптоматическую сдерживающую терапию, направленную на исчезновение симптомов интоксикации, уменьшение массы опухоли и улучшение качества жизни;
- оптимально эффективное (нередко опасное) антилимфомное лечение, которое, возможно, увеличит продолжительность жизни.
Показания к началу терапии перечислены ниже.
• Нарастание симптомов интоксикации.
• Быстрое прогрессирование и генерализация заболевания.
• Потенциально опасное для жизни поражение органов, в том числе признаки поражения почек и печени.
• Костномозговая недостаточность (гемоглобин <100 г/л, лейкоциты <3,0х1012/л, тромбоциты <100х109/л).
При появлении показаний проводят стандартную химиотерапию или лучевое лечение (в зависимости от распространенности процесса).
• Стандартная терапия генерализованных фолликулярных лимфом включает алкилирующие ЛС (хлорамбуцил и циклофосфамид) и ГК. Основное свойство всех схем химиотерапии - продолжительное лечение и длительное ожидание эффекта. Именно поэтому в некоторых случаях менее длительная и интенсивная комбинированная внутривенная химиотерапия (курсы COP или CHOP) может иметь в большей степени тактические, а не терапевтические преимущества. Использование комбинации CHOP и этанерцепта (моноклональных АТ к В-клеточному Аг CD20) достоверно приводит к большей общей выживаемости. Добавление моноклональных CD20-AT этанерцепта к курсу FCM (флударабин, циклофосфамид, митоксантрон) также достоверно увеличивает общую выживаемость больных. В большинстве исследований в качестве поддерживающей терапии использовали интерферон-альфа. Этот препарат увеличивает продолжительность ремиссии, но только у тех пациентов, которые в качестве индукционной терапии получали курсы лечения антрациклиновым ЛС. Отрицательная сторона такой терапии заключается в необходимости частых (2-3 раза в неделю) внутримышечных инъекций препарата на протяжении нескольких лет и побочных эффектах интерферонов (гриппоподобный синдром, в редких случаях - иммунокомплексный синдром).
• Лучевую терапию назначают на область массивных очагов (в качестве дополнительного лечения) или, при частичной ремиссии заболевания, на резидуальные опухолевые локусы.
Роль трансплантации аутологичных гемопоэтических клеток. Трансплантация аутологичных гемопоэтических клеток в качестве консолидирующей терапии первой линии, равно как и при первом рецидиве, не привела к увеличению выживаемости больных.
Роль трансплантации аллогенных гемопоэтических клеток. Смертность при данном виде лечения составляет 30-40%. Рецидивы, по
сравнению с аутологичной трансплантацией, возникают значительно реже. Часть больных излечивается от фолликулярной лимфомы. Опасное осложнение операции - реакция «трансплантат против хозяина» - значительно снижает качество жизни больных.
В настоящее время доступным и часто применяемым методом лечения при лимфомах низкой степени злокачественности становится трансплантация аллогенных гемопоэтических клеток после кондиционирования пониженной интенсивности (трансплантация в немиелоблативном режиме). При посттрансплантационных рецидивах опухоли эффективны инфузии донорских лимфоцитов.
Экстранодальная лимфома из клеток маргинальной зоны
Экстранодальная лимфома из клеток маргинальной зоны [MALTлимфома (Mucosal Associated Lymphoid Tissue - лимфоидная ткань, ассоциированная со слизистыми оболочками)] - экстранодальная В-клеточная опухоль. Она первично поражает слизистую оболочку желудка и мочевого пузыря, слюнные железы, легкие, кожу, щитовидную и молочную железы.
MALT-лимфома желудка
ЭТИОЛОГИЯ
У ряда больных MALT-лимфома желудка развивается на фоне хронической инфекции Helicobacter pylori, слюнных желез - на фоне длительного течения синдрома Шёгрена, щитовидной железы - у пациентов с тиреоидитом Хасимото.
ЭПИДЕМИОЛОГИЯ
По данным ВОЗ, MALT-лимфома желудка составляет 8% всех лимфом.
КЛИНИЧЕСКАЯ КАРТИНА
Специфических клинических симптомов болезни нет. Больные могут длительное время страдать атрофическим гастритом, вызванным H. pylori, предъявлять жалобы диспептического характера.
Проводят следующие лабораторные и инструментальные исследования.
• Обычно опухоль обнаруживают при фиброэзофагогастродуоденоскопии, чаще в антральном отделе желудка, и она имеет вид банальной язвы, очагового воспаления или утолщения складки желудка.
• В связи с трудностью макроскопической оценки размеров опухоли при повторной гастроскопии проводят множественные биопсии близлежащих отделов слизистой оболочки, эндосонографическое исследование (УЗИ с эзофагогастральным датчиком), позволяющие оценить реальные размеры опухоли (глубину ее распространения в стенку желудка и размеры парагастральных лимфатических узлов).
• Гистологическая картина MALT-лимфомы желудка имеет отличительные особенности: опухоль растет в пределах слизистой и подслизистой оболочек желудка, представлена гетерогенными мелкими клетками (моноцитоидными клетками, малыми лимфоцитами), иногда с плазмоклеточной дифференцировкой и примесью более крупных клеток. В слизистой оболочке опухоль инфильтрирует эпителий, формируя специфический лимфоэпителиальный дефект.
• Проводят иммунотипирование опухоли.
• При цитогенетическом исследовании и методами молекулярной биологии выявляют, что опухоль представлена постгерминальными B-клетками памяти. Патогенетической считается транслокация t(11;18)(q21;q21) (гены AP 12 и MLT), в ряде случаев находят транслокации t(1;14), t(14;18), трисомию хромосомы 3. При отсутствии клональных цитогенетических нарушений, особенно t(11;18)(q21;q21), дифференциальный диагноз MALT-лимфомы EI стадии и хронического атрофического гастрита, вызванного H. pylori (в ряде случаев которого может отмечаться моноклональная B-клеточная инфильтрация слизистой оболочки желудка), крайне затруднителен.
Помимо вышеописанных методов исследования показаны УЗИ и КТ органов брюшной полости и средостения, трепанобиопсия, клинический, биохимический и иммунохимический анализы крови. В соответствии с полученными результатами определяют стадию опухолевого процесса.
Метастазирование при MALT-лимфоме обычно ограничено внутрибрюшными лимфатическими узлами. Редко обнаруживают отдаленные метастазы или другие экстранодальные поражения.
ЛЕЧЕНИЕ
• При единичном или множественном поражении, ограниченном стенкой желудка, и невозможности доказать неопластическую
природу имеющихся изменений, т.е. отсутствует t(11;18)(q21;q21), назначают антибактериальную терапию против H. pylori.
• Только после антибактериальной терапии (или отсутствии ее эффекта) назначают хлорамбуцил в дозе 6 мг/м2 в сутки в течение 14 дней с последующим перерывом в течение 28 сут. Всего проводят 6 курсов. Возможно проведение лучевой терапии в суммарной очаговой дозе 30-36 Гр.
• Другими вариантами химиотерапии могут быть курсы COP или терапия малыми дозами циклофосфамида в дозе 200 мг/сут 5 дней с проведением повторных курсов каждые 21 сут.
Необходимо длительно наблюдать за больными, особенно за получавшими только антибактериальную терапию. Следует выполнять повторные фиброэзофагогастродуоденоскопии с множественными биопсиями (как минимум 6: 2 - из тела желудка; 2 - из дна; 2 - из привратника). Эндосонография - оптимальный метод контрольного обследования пациента с MALT-лимфомой желудка.
MALT-лимфомы других локализаций
MALT-лимфомы могут возникать в конъюнктиве, коже, слюнных железах, легком, щитовидной железе, мочевом пузыре, простате, молочной железе. В некоторых случаях такие опухоли связывают с хронической антигенной стимуляцией (например, Borrelia burgdorferi, дерматиты, хламидийная инфекция, конъюнктивиты, синдром Шёгрена и тиреоидит Хасимото). Хотя данных об исчезновении лимфомы после успешного лечения инфекции (например, хламидиоза) пока нет, первоочередным мероприятием в таких случаях должно быть лечение того заболевания, с которым, возможно, связана лимфома. Это касается тех случаев, когда патогенный фактор определен, а клиническое течение лимфомы не представляется угрожающим.
Локальные формы
Экстранодальные лимфомы из клеток маргинальной зоны в 60-75% случаев обнаруживают на ранних (I-II) стадиях. Если выявить связь с какой-либо хронической инфекцией не удается или лечение инфекции не приводит к исчезновению лимфомы, показано облучение вовлеченной области. Суммарная доза меняется в пределах 25-35 Гр в зависимости от локализации и объема лучевой нагрузки на окружающие нормальные ткани. Полную ремиссию удается получить в 95-100% случаев.
Рецидивы заболевания регистрируют у 20-30% больных, и возникают они обычно в контралатеральных областях. Безрецидивная выживаемость в течение 5-10 лет составляет 75%.
Диссеминированная опухоль
В настоящее время, даже используя современные терапевтические средства, добиться излечения при диссеминированных стадиях (III и IV) практически невозможно. Больные в этих случаях живут 7-10 лет. Если заболевание протекает бессимптомно, за пациентами наблюдают, не проводя лечения. Различий в общей выживаемости при применении монотерапии алкилирующими препаратами (хлорамбуцилом, циклофосфамидом) или более агрессивной комбинированной химиотерапии по программам COP и CHOP не выявлено. Добавление эпирубицина к монотерапии хлорамбуцилом также не привело к увеличению числа полных ремиссий и общей выживаемости.
Агрессивные лимфомы
Агрессивные лимфомы - гетерогенная группа опухолей. Для некоторых из них (например, для лимфомы из клеток мантийной зоны и периферических Т-клеточных лимфом) свойственны прогрессирующее течение, изначальная резистентность к терапии, невозможность получения длительной ремиссии и быстрая гибель больного. Особенностью других служит высокая пролиферативная активность: быстрый рост опухоли с прорастанием и сдавлением органов, сосудов, нервов, возникновением тяжелой интоксикации. Такие опухоли обычно очень химиочувствительны, что позволяет вылечить значительную часть больных.
К наиболее распространенным агрессивным опухолям относят следующие.
• В-клеточные лимфомы:
- диффузная В-крупноклеточная лимфома (30%);
- лимфома из клеток мантийной зоны (6-10%);
- медиастинальная (тимическая) В-крупноклеточная лимфома (2,4%);
- лимфома Беркитта и беркиттоподобная лимфома (1-2%).
• Т-клеточные лимфомы:
- периферическая Т-клеточная лимфома (7,6%);
- анаплазированная крупноклеточная лимфома Т- или О-типа
(2,4%).
ЭПИДЕМИОЛОГИЯ
Агрессивные лимфомы составляют половину всех лимфом. Возрастной разброс заболеваемости широк и зависит от конкретной нозологической формы. В целом заболеваемость увеличивается с возрастом: в основном опухоль диагностируют у 60-70-летних.
КЛИНИЧЕСКАЯ КАРТИНА
В большинстве случаев обнаруживают быстрорастущую нодальную или экстранодальную опухоль. Поражение экстранодальных областей наблюдают в 40% случаев (главным образом это различные сегменты ЖКТ). Экстранодальное поражение любой локализации (кожа, кости, ЦНС, яичко, молочная железа и другие органы) может быть изолированным.
Обследование больного включает следующие мероприятия.
• Сбор анамнестических и физикальных данных.
• Рентгенографию грудной клетки, КТ грудной, брюшной полостей, малого таза.
• Выполнение клинического и биохимического анализов крови с обязательным определением активности ЛДГ и концентрации мочевой кислоты.
• Трепанобиопсию.
• Диагностическую люмбальную пункцию (при поражении носоглотки, первичной лимфоме яичка, поражении костного мозга, значении международного прогностического индекса >2).
• Радионуклидные исследования (с галлием или позитронноэмиссионную томографию).
• ЭхоКГ.
• Определение стадии в соответствии с риском.
Больных в соответствии с риском разделяют на группы, каждая из которых имеет определенный прогноз (табл. 56-3). Выделяют 5 факторов, влияющих на риск:
• возраст (моложе или старше 60 лет);
• стадия или III-IV);
• количество экстранодальных очагов болезни;
• общее состояние больного;
• активность ЛДГ в крови.
Таблица 56-3. Группы риска
Диффузная B-крупноклеточная лимфома
Диффузная B-крупноклеточная лимфома - гетерогенная группа B-клеточных опухолей, характеризуемых пролиферацией крупных клеток с размерами ядер, соответствующими или превышающими размеры ядра макрофага или более чем в 2 раза больше ядра лимфоцита.
ЭТИОЛОГИЯ
В ряде случаев диффузная B-крупноклеточная лимфома развивается в результате трансформации лимфом низкой степени злокачественности и у больных с первичным или вторичным иммунодефицитом. У пациентов с иммунодефицитами возможную патогенетическую роль играет вирус Эпстайна-Барр.
ЭПИДЕМИОЛОГИЯ
Распространенность диффузной B-крупноклеточной лимфомы составляет 30-40% всего количества лимфом. Средний возраст больных на момент диагностики - 60-70 лет. Мужчины болеют незначительно чаще женщин.
КЛИНИЧЕСКАЯ КАРТИНА
Диффузная B-крупноклеточная лимфома - быстрорастущая нодальная или экстранодальная опухоль, сопровождаемая изменением конфигурации и размеров лимфоидных органов и симптомами интоксикации (лихорадкой, проливными ночными потами, похудением, слабостью).
При гистологическом исследовании обнаруживают полное нарушение структуры лимфатического узла или экстранодальной ткани диффузной пролиферацией крупными, средними и небольшими лим-
фоидными клетками. Крупные клетки могут быть резко атипичными - многоядерными, гигантскими. В ряде случаев определяют значительную примесь реактивных Т-клеток или тканевых макрофагов, опухолевые клетки составляют меньшинство. Этот гистологический вариант называют В-клеточной лимфомой с избытком Т-клеток/гистиоцитов. Возможно обнаружение выраженного фиброза.
При поражении костного мозга могут быть мелкоклеточные, обычно очаговые пролифераты. Опухолевые клетки в периферической крови выявляют крайне редко.
При исследовании иммунофенотипа в основном определяют поверхностный IgM (реже IgG и IgA) и В-клеточные Аг CD19, CD20, CD22, CD79a. В ряде случаев, при анаплазированной морфологии, один или несколько В-клеточных Аг могут отсутствовать и экспрессируется Аг CD30. В 10% случаев обнаруживают CD5, что прогностически неблагоприятно. Наличие Аг CD10 в 25-50% случаев служит благоприятным признаком. Протоонкоген BCL-2 выявляют в 30-50% случаев (свидетельствует о плохом прогнозе). Пролиферативная активность, определяемая по Аг Ki-67, высока и составляет 40-90%.
При цитогенетическом исследовании и методами молекулярной биологии в 20-30% случаев определяют транслокацию t(14;18), в 30% - транслокацию локуса 3q27 (протоонкоген BCL-6).
ЛЕЧЕНИЕ
Около 40% больных выздоравливают после проведения терапии первой линии.
• Терапия первой линии при локальных стадиях. Проведение курсов химиотерапии с доксорубицином с последующей консолидирующей лучевой терапией на вовлеченные области приводит к ремиссиям заболевания практически у 100% больных, а 5-летняя общая выживаемость составляет 80%. В группе больных с I-II стадией заболевания без массивных (<10 см) очагов поражения комбинированная терапия (3 курса CHOP и облучение в дозе 45-50 Гр) более эффективна, чем только химиотерапия (8 курсов CHOP) как в отношении 5-летней безрецидивной, так и общей выживаемости.
• Терапия первой линии при генерализованной лимфоме.
Наиболее эффективна полихимиотерапия. Первое поколение схем полихимиотерапии включает CHOP и CHOP-подобные курсы, в которых доксорубицин заменяли другим антрациклином или ан-
трацендионом (митоксантроном). Схемы 2-го и 3-го поколений включают большее количество ЛС (обычно 6-8) в максимально переносимых дозах. • Этанерцепт и стандартный CHOP:
- монотерапия этанерцептом позволяет достичь ремиссии у 31% больных с рецидивами диффузной В-крупноклеточной лимфомы;
- сочетанное применение курсов СНОР с этанерцептом (R-CHOP) позволило получить ремиссии у 76% пациентов. Преимущество R-CHOP отмечалось во всех группах риска, максимально - в группе низкого риска. Наибольший эффект курсов R-CHOP был отмечен в случаях, характеризуемых повышенной экспрессией гена BCL-2 (фактор плохого прогноза).
Лимфогранулематоз
Лимфогранулематоз, или лимфома Ходжкина, - опухоль лимфатической системы, субстратом которой служат характерные гигантские опухолевые клетки, а также полиморфноклеточная гранулема, образованная реактивными лимфоцитами, нейтрофилами, эозинофилами и плазматическими клетками.
ЭПИДЕМИОЛОГИЯ
По данным американских исследований, лимфогранулематозом болеют лица всех возрастов. Кривая заболеваемости имеет 2 пика: в возрасте 16-30 и после 50 лет. Каждый пик имеет свои особенности: первый приходится на возраст 20-30 лет, женщины заболевают чаще; второй - на возраст старше 60 лет, чаще болеют мужчины. В разных этнических группах заболеваемость отличается: наиболее часто заболевают представители европеоидной расы (1,1 среди мужчин и 0,7 среди женщин на 100 000 населения), наиболее редко - японцы (0,1 среди мужчин на 100 000). Эпидемиологических данных по России в настоящее время нет.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Долгое время роль этиологического агента при лимфогранулематозе отводилась инфекционным возбудителям, в том числе микобактериям туберкулеза и вирусам. Была выявлена связь между заболеваемостью лимфогранулематозом и инфицированностью вирусом Эпстайна-Барр. Риск развития лимфогранулематоза в 3 раза выше в популяции людей, инфицированных вирусом Эпстайна-Барр или перенесших инфекцион-
ный мононуклеоз. В семьях больных ВИЧ-инфицированных пациентов риск возрастает параллельно углублению иммунодефицита.
Иммунофенотипический анализ подтвердил лимфатическую природу клеток Березовского-Штернберга и Ходжкина. Клетки Березовского-Штернберга в большинстве случаев происходят из В-клеток зародышевого центра фолликула в результате различных мутаций, обладают высокой секреторной активностью и обусловливают характерную гистологическую картину.
Количественные изменения хромосом в клетках БерезовскогоШтернберга были выявлены практически в 100% случаев, что свидетельствует о нестабильности кариотипа этих клеток. Характерных хромосомных аномалий не обнаружено. Изменения в опухолевых клетках чаще (в 54% случаев) затрагивали короткое плечо хромосомы 2.
КЛАССИФИКАЦИЯ
Согласно Международной клинической классификации, лимфогранулематоз по распространенности процесса подразделяют на 4 стадии (табл. 56-4).
Таблица 56-4. Стадии лимфогранулематоза
КЛИНИЧЕСКАЯ КАРТИНА
Пациенты предъявляют жалобы на увеличение периферических лимфатических узлов, одышку, кашель, слабость, быструю утомля-
емость, проливные поты по ночам, кожный зуд, субфебрилитет или изматывающую лихорадку неправильного типа, беспричинное снижение массы тела.
Увеличенные лимфатические узлы имеют плотноэластическую консистенцию, как правило, не спаяны с кожей и безболезненны. У 10-25% больных после приема алкоголя появляются боли в увеличенных лимфатических узлах. Обычно в опухолевый процесс вовлекаются надключичные, медиастинальные, подмышечные лимфатические узлы, но чаще в первую очередь увеличиваются шейно-надключичные лимфатические узлы справа. Поражение лимфатических узлов только выше диафрагмы в момент диагностики обнаруживают у 90% больных, только ниже диафрагмы - у 10%. При значительном увеличении медиастинальных лимфатических узлов у больных появляется сухой кашель. Синдром сдавления, боли в грудной клетке возникают только при очень больших конгломератах узлов средостения. Такие конгломераты могут прорастать в плевру, перикард, легкие, бронхи, пищевод, нередко вовлекается селезенка.
Из нелимфоидных органов чаще всего поражаются легкие. Поражение легочной ткани может носить как очаговый, так и инфильтративный характер, иногда с распадом и формированием полостей. При выявлении полостей заболевание следует дифференцировать от туберкулеза, бактериальной пневмонии, актиномикоза. Значительное увеличение забрюшинных лимфатических узлов сопровождается болями в пояснице.
Примерно у 10-20% больных в процесс вовлекается костный мозг, что можно предположить при стойкой панцитопении. Тем не менее часто подтвердить поражение костного мозга можно только при трепанобиопсии.
Поражение печени редко обнаруживают в начале болезни, однако при прогрессировании заболевания метастазы в нее находят более чем у половины больных. Заподозрить вовлечение в патологический процесс печени можно в случае увеличения ее размеров, выявления в ней очаговых теней в сочетании с повышением более чем в 4 раза активности ЩФ в крови.
Лабораторные и инструментальные исследования
• Общий анализ крови. У части больных отмечают повышение СОЭ, нейтрофильный лейкоцитоз и лимфопению.
• Биохимический анализ крови. Определяют активность ЩФ, ЛДГ, АЛТ, АСТ, концентраций билирубина, креатинина, мочеви-
ны, выявляют признаки биологической активности (содержание α2-глобулина, фибриногена, гаптоглобина и церулоплазмина).
• Рентгенографическое исследование органов грудной клетки. Позволяет уточнить состояние лимфатических узлов средостения, ткани легких и плевры.
• КТ грудной клетки. Проводят обязательно. Позволяет выявить невидимые на рентгенограммах медиастинальные лимфатические узлы и мелкие очаги в легочной ткани.
• УЗИ печени, селезенки, забрюшинных и внутрибрюшных лимфатических узлов, почек. Целесообразно также проведение УЗИ тех зон периферических лимфатических узлов, которые при пальпации кажутся сомнительными.
• КТ брюшной полости. Показана в сомнительных случаях.
• Методы радиоизотопной диагностики. Позволяют выявить субклиническое поражение костной системы (все зоны патологического накопления изотопа следует подтвердить при рентгенографическом исследовании). Сканирование лимфатических узлов с галлия цитратом - ценный метод, имеющий наибольшее значение после завершения лечения. У больных с исходно большими размерами средостения накопление индикатора в остаточных медиастинальных лимфатических узлах свидетельствует о высокой степени риска раннего рецидива.
• Биопсия лимфатического узла с последующими цитологическим и гистологическим исследованиями полученного материала. Проводят обязательно. При изолированном увеличении медиастинальных или внутрибрюшных лимфатических узлов следует прибегнуть к парастернальной медиастинотомии или лапаротомии.
• Двусторонняя трепанобиопсия подвздошной кости.
• Диагностическая лапаротомия со спленэктомией, ревизией внутрибрюшных и забрюшинных лимфатических узлов и краевой биопсией печени. Используют лишь в диагностически трудных ситуациях.
ДИАГНОСТИКА
Диагноз лимфогранулематоза устанавливают морфологически. Для морфологического исследования применяют цитологические отпечатки и гистологические препараты, полученные при биопсии опухолевого образования, чаще всего лимфатического узла. Основное условие
гистологической диагностики - выявление характерных многоядерных опухолевых клеток. Субстрат лимфогранулематоза составляет полиморфноклеточная гранулема, образованная реактивными лимфоцитами, нейтрофилами, эозинофилами, плазматическими клетками, характерными опухолевыми клетками. Клетки Березовского-Штернберга и клетки Ходжкина расположены преимущественно в центре гранулемы.
ЛЕЧЕНИЕ
Основные положения противоопухолевой химиотерапии:
• применение ЛС в максимально переносимых дозах;
• проведение не одного, а нескольких курсов лечения даже при достижении полного эффекта после первого.
Программа MOPP (мустарген, винкристин, преднизолон, прокарбазин)/ABVD (доксорубицин, блеомицин, винбластин, дакарбазин) - общепризнанный стандарт лечения лимфогранулематоза. Лучевую терапию применяют для консолидации ремиссии после химиотерапевтического лечения.
Использование гранулоцитарных колониестимулирующих факторов (ленограстима, филграстима) позволяет проводить более агрессивную химиотерапию. Программа BEACOPP предполагает 2 различающихся по дозам режима: BEACOPP I (базовый) и BEACOPP II (усиленный). Базовый ВЕАСОРР при ИВ-IV стадии лимфогранулематоза позволяет получить полную ремиссию у 92% больных.
В зависимости от распространенности опухолевого процесса проводят 4-8 курсов химиотерапии. После 2, 4, 6 и 8-го курса проводят рестадирование, оценивают противоопухолевый эффект примененной схемы химиотерапии. Через 1 мес после наступления ремиссии и завершения химиотерапии с консолидирующей целью показана лучевая терапия на области, исходно вовлеченные в опухолевый процесс.
Множественная миелома
Множественная миелома - злокачественная опухоль из группы В-клеточных лимфопролиферативных заболеваний, характеризуемых пролиферацией и накоплением плазматических клеток в красном костном мозге и избыточной продукцией моноклональных Ig (М-компонента) и белка Бенс-Джонса (легких цепей Igk и λ). Определение множественной миеломы как плазмоклеточной опухоли с парапротеинемией не совсем точно, так как существует вариант заболевания, при котором патологические клетки синтезируют Ig, но не секретируют их в кровь (так называемая несекретирующая миелома).
ЭПИДЕМИОЛОГИЯ
На множественную миелому приходится до 1% всех злокачественных новообразований и примерно 10% всех гемобластозов. Заболеваемость увеличивается с возрастом, достигая пика на 7-м десятилетии жизни (в целом - 4 на 100 000 населения). Множественная миелома несколько чаще развивается у мужчин. Средняя продолжительность жизни больных после постановки диагноза составляет 3 года, 10-летняя выживаемость не превышает 5%.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Причины развития множественной миеломы неизвестны. В основе заболевания лежит злокачественная трансформация отдельной В-клетки-предшественницы, что приводит к бесконтрольному синтезу моноклонального Ig (c идентичным типом легкой цепи и эпитопом). От опухолевой трансформации клетки-предшественницы до появления клинических проявлений проходит 20-30 лет. Малигнизированная плазматическая клетка приобретает опухолевые свойства - способность прорастать в костную ткань, почки, формируя клиническую картину распространенной многоочаговой опухоли, что и отражено в названии болезни.
КЛАССИФИКАЦИЯ
Существуют следующие варианты парапротеинемических гемобластозов:
• солитарная плазмацитома;
• тлеющая миелома;
• множественная миелома;
• плазмобластный лейкоз.
Согласно иммунохимической классификации, выделяют 5 основных форм множественной миеломы: G, А, D, E и Бенс-Джонса, к редким формам относят несекретирующую и М-миелому. Частота распределения иммунологических типов миеломы примерно соответствует концентрации различных классов нормальных Ig в сыворотке крови: на G-миелому приходится 60% случаев, A-миелому - 20%, D-миелому - 2%, Е-миелому - менее 0,1%, миелому Бенс-Джонса - 18%.
• Миелома Бенс-Джонса. Отличается от других форм множественной миеломы по клинической картине и лабораторным признакам (М-компонента в крови не обнаруживают, в моче определяют белок Бенс-Джонса). Концентрация белка в сыворотке крови нор-
мальная, поэтому симптомы, обусловленные гиперпротеинемией, отсутствуют.
• Несекретирующая миелома. Встречается в 2% случаев. Характеризуется отсутствием парапротеинов в сыворотке крови и моче. В 85% моноклональный белок выявляют в цитоплазме плазматических клеток при электронной микроскопии. В 15% парапротеин обнаружить не удается - в таких случаях говорят о так называемой истинной несекретирующей миеломе.
• Плазмоклеточный лейкоз. Выделен в самостоятельную нозологическую форму, отличающуюся от множественной миеломы быстрым прогрессированием, наличием значительного количества плазматических клеток в крови, тромбоцитопенией с выраженным геморрагическим синдромом, обширной плазматической инфильтрацией различных органов.
• Плазмоцитома. Редкая форма плазмоклеточной пролиферации, ее частота не превышает 5%. Красный костный мозг при плазмоцитоме не поражается, парапротеин образуется в незначительном количестве либо вообще отсутствует. В течение 3 лет у 30% больных развивается множественная миелома, через 10 лет этот показатель достигает 60%.
КЛИНИЧЕСКАЯ КАРТИНА
Поражения скелета. Связаны с инфильтрацией костной ткани злокачественными плазматическими клетками, выделяющими цитокины, способствующие образованию остеокластов и разрушению кости. Боли в костях в начальных стадиях заболевания непостоянные, затем они становятся более интенсивными и продолжительными. Нередко возникают патологические переломы. В момент постановки диагноза деструкции костей выявляют у 70% больных. Патологические переломы костей при локальной опухоли зачастую позволяют диагностировать заболевание примерно за 5 лет до появления других клинических и лабораторных признаков. Рентгенологически в плоских и коротких костях (ребрах, грудине, черепе, тазе, позвоночнике), а также в проксимальных эпифизах бедренных и плечевых костей обнаруживают очаги деструкции округлой или овальной формы, с четкими границами, диаметром до 2-3 см. При диффузной форме множественной миеломы развивается общий остеопороз без очагов деструкции.
Гиперкальциемия. Результат деструкции костной ткани развивается у одной трети больных множественной миеломой. Основные проявле-
ния - тошнота, рвота, жажда, слабость, запор, полиурия, артериальная гипотензия, укорочение интервала Q-T на ЭКГ.
Патология почек (миеломная нефропатия). Развивается у 6090% больных и характеризуется протеинурией, отеками и гематурией. Почечная недостаточность возникает приблизительно у 20% больных. Поражение почек при множественной миеломе связано с многими факторами. В основе почечной недостаточности лежит восходящий нефросклероз, обусловленный большой реабсорбционной нагрузкой на проксимальные отделы канальцев вследствие фильтрации больших количеств белка Бенс-Джонса. Гиперурикемия, обусловленная повышенным клеточным распадом, приводит к отложению кристаллов мочевой кислоты в дистальных отделах канальцев, мочеточниках, уретре. Накопление легких цепей Ig или их фрагментов в клубочках приводит к формированию нефротического синдрома.
Синдром гипервязкости. Обусловлен повышенной концентрацией белков в крови. Наблюдают преимущественно при G- и A-миеломах, может проявляться ухудшением зрения, нарушением сознания, носовыми кровотечениями и пр.
Геморрагический синдром. Обусловлен связыванием парапротеинов с мембранами тромбоцитов, факторами свертывания V, VII, VIII, протромбином, фибриногеном с нарушением их функций.
Нарушения гемопоэза. Связаны с инфильтрацией плазматическими клетками красного костного мозга, также имеют значение дефицит эритропоэтина при миеломной нефропатии, дефицит железа вследствие частых кровотечений. Как правило, наблюдают анемию, которая обычно сопровождается уменьшением содержания и других форменных элементов крови.
Иммунодефицит. Связан со снижением концентрации нормальных Ig, уменьшением количества гранулоцитов и нарушением их функций, что в совокупности обусловливает предрасположенность больных к инфекциям.
Амилоидоз. Развивается у 10% больных множественной миеломой.
Криоглобулинемия. Встречается в 2-5% случаев, клинически она проявляется симптомами холодовой аллергии, акроцианозом и синдромом Рейно. Иногда присоединяются трофические изменения конечностей, гангрена пальцев, геморрагии, тромбозы крупных сосудов.
При длительном течении процесса увеличиваются селезенка (в 1531% случаев), печень (в 18-46% случаев), лимфатические узлы, реже поражаются щитовидная железа, надпочечники, яичники, легкие, перикард и ЖКТ. Стадии множественной миеломы приведены в табл. 56-5.
Таблица 56-5. Стадии множественной миеломы
ДИАГНОСТИКА
Лабораторные и инструментальные исследования
Необходимые исследования при множественной миеломе.
• Клинический анализ крови.
• Биохимический анализ крови: концентрация общего белка, кальция, креатинина, калия, мочевой кислоты.
• Электрофорез белков в сыворотке крови.
• Концентрация нормальных Ig в сыворотке крови.
• Общий анализ мочи.
• Определение суточной протеинурии.
• Электрофорез белков в моче.
• Пункция красного костного мозга и трепанобиопсия.
• Рентгенография костей.
Для постановки диагноза множественной миеломы необходимо получить морфологическое подтверждение плазмоклеточной природы опухоли и выявить парапротеин.
• В подавляющем большинстве случаев выявляют типичный М-компонент и/или белок Бенс-Джонса. Структурная идентификация М-компонента возможна с использованием методов иммуноэлектрофореза.
• Типичные миеломные клетки округлой или овальной формы, с круглыми, эксцентрично расположенными ядрами и одним или несколькими ядрышками. Иногда плазматические опухолевые клетки могут быть большого диаметра, с 2-3 ядрами и базофильной цитоплазмой.
Диагностические критерии Большие критерии:
• более 30% плазматических клеток в красном костном мозге;
• концентрация парапротеина в крови - более 35 г/л (для IgG) или более 20 г/л (для IgA);
• протеинурия Бенс-Джонса - более 1 г/сут. Малые критерии:
• количество плазматических клеток в красном костном мозге 1030%;
• наличие литических костных очагов;
• концентрация парапротеина в крови - менее 35 г/л (для IgG) или менее 20 г/л (для IgA) и/или протеинурия Бенс-Джонса менее 1 г/сут;
• снижение концентрации нормальных Ig в крови.
Диагноз множественной миеломы устанавливают только при наличии не менее 2 из 3 больших диагностических критериев (обязательно наличие первого из них).
Дифференциальная диагностика
Наибольшие сложности возможны при дифференциальной диагностике с моноклональной гаммапатией невыясненного значения, тлеющей множественной миеломой.
• При моноклональной гаммапатии невыясненного значения клинические проявления отсутствуют, концентрация М-компонента в сыворотке крови - менее 30 г/л, плазматических клеток в красном костном мозге - не более 10%, нет костных деструкций, анемии, гиперкальциемии.
• При тлеющей множественной миеломе клинические проявления отсутствуют, в красном костном мозге более 10% плазматических клеток, концентрация М-компонента в сыворотке крови - более 30 г/л.
ЛЕЧЕНИЕ
Лечение множественной миеломы включает:
• химиотерапию;
• лучевую терапию;
• купирование развивающихся осложнений (гиперкальциемии, синдрома гипервязкости, анемии, патологических переломов и пр.).
При компенсированной множественной миеломе (стадия I), тлеющей миеломе, моноклональной гаммапатии невыясненного значения
от цитостатической терапии воздерживаются и ограничиваются наблюдением (клиническим анализом крови и определением концентрации патологического Ig 1 раз в месяц).
Основание для начала цитостатической терапии - появление признаков прогрессирования процесса (нарастание анемии, гиперкальциемии, появление почечной недостаточности, болевого синдрома, анемии, увеличение концентрации патологических белков в крови и моче). Базовые препараты - ГК. Схема МР (мелфалан по 8 мг/м2 в сутки и преднизолон по 100 мг/сут в течение 4 дней внутрь) оказывает эффект у 50-60% больных, однако полной ремиссии достигают не более чем в 5% случаев, а общая выживаемость не превышает 3 лет. Эта программа показана пациентам старше 70 лет в случае небольшой опухолевой массы, а также больным с тяжелой сопутствующей патологией. Разработаны множественные модификации схемы МР с добавлением винкристина, кармустина, доксорубицина, циклофосфамида и дексаметазона. Альтернативная схема - VAD (винкристин, доксорубицин и дексаметазон). Хотя интенсификация терапии с использованием аутологичной трансплантации гемопоэтических клеток приводит к увеличению частоты полных ремиссий и общей выживаемости больных, основной проблемой остаются рецидивы заболевания. Наиболее распространенный способ поддержания ремиссии после аутотрансплантации - применение интерферона-альфа, иногда в сочетании с дексаметазоном.
Лучевую терапию проводят при крупных очагах костной деструкции, резко выраженных локальных болях, обусловленных переломами, корешковом синдроме при компрессии тел позвонков, а также в случае солитарной плазмоцитомы.
56.5. ИНФЕКЦИОННЫЙ МОНОНУКЛЕОЗ
Инфекционный мононуклеоз - заболевание, вызываемое вирусом Эпстайна-Барр и проявляющееся лихорадкой, болями в горле, генерализованной лимфаденопатией с преимущественным увеличением заднешейных лимфатических узлов.
ЭПИДЕМИОЛОГИЯ
Вирусом Эпстайна-Барр инфицировано до 80% взрослого населения. Инфекционный мононуклеоз наиболее часто развивается у детей в возрасте до 10 лет. Второй пик заболеваемости приходится на 30 лет.
В настоящее время исследуют роль вируса Эпстайна-Барр в развитии саркомы миндалин, Т-клеточной лимфомы, лимфосаркомы ЦНС, тимомы, саркомы желудка. Высокий титр АТ к этому вирусу определяют при лимфоме Беркитта, назофарингеальной карциноме, смешанно-клеточном варианте лимфогранулематоза, синдроме хронической усталости.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Вирус Эпстайна-Барр - герпес-вирус, состоящий из 2-нитевой молекулы ДНК, окруженной нуклеокапсидом.
Заражение происходит воздушно-капельным путем. В детстве первый контакт с вирусом обычно протекает бессимптомно. При первичном инфицировании взрослых развивается типичная клиническая картина. В ротовой полости вирус размножается в эпителии, слюнных железах и криптах миндалин. Из эпителиальных клеток вирус проникает в В-лимфоциты, на которых представлен рецептор для вируса (CD21, рецептор C3d-компонента комплемента). Пролиферация вируса Эпстайна-Барр в лимфоцитах приводит к увеличению объема лимфоидной ткани (в первую очередь, миндалин и лимфатических узлов).
Инфицирование вирусом Эпстайна-Барр приводит к поликлональной активации лимфоцитов с выработкой АТ, направленных как против инфицированных клеток, так и против вирусного капсида. В дальнейшем вырабатывается генерация цитотоксических Т-лимфоцитов, распознающих специфические белки вируса. Вирус может длительно сохраняться в В-лимфоцитах.
КЛИНИЧЕСКАЯ КАРТИНА
Заболевание может протекать как бессимптомно - в стертой, абортивной форме, так и с развернутой клинической картиной.
Инкубационный период длится 4-6 нед. Основные симптомы заболевания в продромальный период (за 1-2 нед до появления фарингита, лихорадки и генерализованной лимфаденопатии) - утомляемость и миалгии. Лихорадка может продолжаться от 1-2 нед до 1 мес. Увеличение лимфатических узлов, особенно заднешейных, и фарингит, часто осложняющийся стафилококковой ангиной, характерны для первых 2 нед. Увеличение селезенки и печени наиболее выраженно на 2-3-й неделе заболевания.
Наиболее угрожающим в первые 2 нед заболевания является поражение ЦНС, особенно характерное для детей. Развивается клиническая картина менингита и энцефалита с церебральной атаксией, гемиплегиями, поражением лицевого нерва.
К редким проявлениям инфекционного мононуклеоза относят гепатит (возможно, с фульминантным течением), перикардит, пневмонию с вовлечением плевры, интерстициальный нефрит и васкулит.
ДИАГНОСТИКА
При инфекционном мононуклеозе обнаруживают гранулоцитопению, иногда тромбоцитопению, сохраняющиеся в течение месяца. Повышение активности трансаминаз и ЩФ отмечают в 90% случаев, билирубин повышен у 40% больных.
Содержание лейкоцитов в первые 2 нед заболевания повышается до 10-20х109/л, в основном за счет лимфоцитов, до 10% из числа которых составляют атипичные формы. Это большие лимфоциты с широкой, вакуолизированной цитоплазмой и неровными, морщинистыми очертаниями. Среди атипичных лимфоцитов преобладают CD8+-клетки.
Из серологических тестов проводят определение титра АТ:
• IgM к вирусному капсиду обнаруживаются рано и исчезают через 4-6 нед от момента инфицирования;
• IgG к вирусному капсиду образуются в острой фазе и сохраняются в течение всей жизни;
• IgG к раннему Аг появляются в острой фазе и исчезают через 3-6 мес;
• IgG к ядерному Аг определяются через 2-4 мес после начала заболевания и сохраняются в течение всей жизни.
Дифференциальная диагностика
Дифференциально-диагностическими критериями инфекционного мононуклеоза служат морфологические особенности лимфоцитов. Учитывая связь инфицирования вирусом Эпстайна-Барр с развитием лимфопролиферативных заболеваний, обнаружение у больных серологических маркеров инфекции не гарантирует исключения опухолевого процесса.
ЛЕЧЕНИЕ
Терапия направлена на профилактику или на лечение инфекционных осложнений. Применение ГК не оказывает влияния на течение вирусного процесса и при этом увеличивает риск развития инфекций. Назначение 40-60 мг преднизолона на 2-3 дня показано при выраженной гиперплазии миндалин с высоким риском обструкции дыхательных путей, при доказанном аутоиммунном гемолизе и выраженной тромбоцитопении. Эффективность противовирусных препаратов, действующих подобно ацикловиру, не подтверждена.
Глава 57. ГЕМОРРАГИЧЕСКИЕ СОСТОЯНИЯ
Геморрагический синдром (склонность к появлению кожных геморрагий и кровоточивости слизистых оболочек) развивается вследствие изменений в одном или нескольких звеньях гемостаза - при поражениях сосудистой стенки, уменьшении количества тромбоцитов или расстройстве их функций, нарушениях свертывания крови. Из наследственных нарушений гемостаза в терапевтической практике наиболее часто наблюдают тромбоцитопатии, гемофилии А и В, болезнь Виллебранда, а из сосудистых форм - телеангиэктазии. Наиболее частые приобретенные формы геморрагического синдрома - вторичные тромбоцитопении и тромбоцитопатии, ДВС-синдром, дефицит факторов протромбинового комплекса и геморрагический васкулит. Другие формы наблюдают редко. Следует учитывать, что в последние годы нарушения гемостаза все чаще связаны с приемом ЛС (особенно антиагрегантов и антикоагулянтов).
ТИПЫ КРОВОТОЧИВОСТИ
В клинической практике выделяют 5 типов кровоточивости.
• Гематомный тип. Характерен для гемофилий А и В. Возникают болезненные напряженные кровоизлияния в мягкие ткани, а также в суставы с постепенным нарушением функции опорнодвигательного аппарата.
• Петехиально-пятнистый (синячковый) тип. Наблюдают при тромбоцитопениях, тромбоцитопатиях, некоторых нарушениях свертывающей системы, гипо- и дисфибриногенемиях, дефиците факторов протромбинового комплекса (VII, X, V и II).
• Смешанный синячково-гематомный тип. Развивается при выраженном дефиците факторов протромбинового комплекса и фактора XIII, болезни Виллебранда, ДВС-синдроме, передозировке антикоагулянтов и тромболитических препаратов, появлении в крови иммунных ингибиторов факторов VIII и IX. Проявляется сочетанием петехиально-пятнистых кожных геморрагий с от-
дельными большими гематомами в забрюшинном пространстве, стенке кишки. В отличие от гематомного типа кровоточивости кровоизлияния в суставы возникают крайне редко.
• Васкулитопурпурный тип. Наблюдают при инфекционных и иммунных васкулитах (см. главу «Системные васкулиты»), легко трансформируется в ДВС-синдром и характеризуется геморрагиями в виде сыпи или эритемы, возможным присоединением нефрита и кишечных кровотечений.
• Ангиоматозный тип. Развивается в зонах телеангиэктазий, ангиом, артериовенозных шунтов и характеризуется упорными локальными геморрагиями, связанными с зонами сосудистой патологии. С определенной долей вероятности предположить патологию сосудисто-тромбоцитарного или коагуляционного звеньев гемостаза можно по особенностям геморрагических проявлений (табл. 57-1).
Таблица 57-1. Признаки нарушения сосудисто-тромбоцитарного и коагуляционного гемостаза
57.1. ТРОМБОЦИТОПЕНИИ
Тромбоцитопения - уменьшение содержания тромбоцитов в крови ниже 150х109/л. Наиболее частые, а во многих случаях единственные клинические проявления тромбоцитопении - кровоточивость из слизистых оболочек и кожный геморрагический синдром.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Выделяют наследственные и приобретенные формы тромбоцитопений. Наследственные тромбоцитопении всегда сопровождаются нарушением функциональных свойств тромбоцитов, поэтому рассматриваются в группе тромбоцитопатий (см. ниже). Приобретенные тромбоцитопении могут быть обусловлены нарушением тромбоцитопоэза, повышенным разрушением тромбоцитов в кровеносном русле или изменением их распределения в организме. Основные факторы, обусловливающие нарушение продукции тромбоцитов, следующие:
• Аплазии кроветворения и нарушение дифференцировки по всем направлениям гемопоэза (апластическая анемия).
• Гибель клеток в красном костном мозге, приводящая к панцитопении (миелодиспластические синдромы).
• Вытеснение всех ростков нормального кроветворения в результате пролиферации мутантной кроветворной клетки (острые лейкозы).
• Вытеснение кроветворной ткани опухолевыми клетками некостномозгового происхождения (метастазы злокачественных новообразований).
• Подавление синтеза ДНК и нарушение деления всех клеток, в том числе и мегакариоцитов (дефицит витамина B12 и фолиевой кислоты).
• Образование вследствие мутации тромбоцитов с неполноценной мембраной, легко разрушающихся в периферической крови под влиянием комплемента (пароксизмальная ночная гемоглобинурия).
• Прием цитостатических ЛС.
• Прием ЛС, являющихся гаптенами и приводящих к образованиям АТ к мегакариоцитам (гидрохлоротиазида, гидралазина, эстрогенов, диэтилстилбэстрола).
Кровоточивость при тромбоцитопениях, связанных с нарушенным тромбоцитопоэзом, обусловлена дистрофическими изменениями эндотелиальных клеток вследствие выпадения трофической функции тромбоцитов (в норме до 15% циркулирующих тромбоцитов используется для поддержания нормального функционирования сосудистой стенки). Нарушение структуры и формы эндотелиальных клеток приводит к диапедезному проникновению эритроцитов в окружающие ткани и повышению ломкости капилляров.
Повышенное разрушение тромбоцитов в кровеносном русле чаще всего связано с иммунными нарушениями, приводящими к появлению в крови антитромбоцитарных АТ. В частности, при идиопатической тром-
боцитопенической пурпуре происходит синтез АТ класса IgG, которые, фиксируясь к мембране тромбоцитов, нарушают их функцию и способствуют их разрушению в селезенке и печени (срок жизни тромбоцитов уменьшается с 2-3 дней до нескольких минут). Развитие тромбоцитопении при приеме хинидина, дигитоксина, сульфаниламидов, рифампицина, солей золота происходит в связи с выработкой АТ против комплекса, образующегося при фиксации ЛС на мембране тромбоцита.
Тромбоцитопения при ДВС-синдроме связана с избыточным потреблением тромбоцитов (см. ниже «Синдром диссеминированного внутрисосудистого свертывания»). При тромботической тромбоцитопенической пурпуре уменьшение количества циркулирующих тромбоцитов обусловлено интенсивной спонтанной агрегацией тромбоцитов в сосудистом русле.
Возможные патогенетические факторы тромбоцитопении при вирусных инфекциях следующие:
• уменьшение образования тромбоцитов на фоне репликации вируса в мегакариоцитах (при геморрагических лихорадках, цитомегаловирусной инфекции);
• разрушение циркулирующих тромбоцитов при контакте с вирусом;
• повреждение тромбоцитов под действием АТ, направленных против вирусов, фиксированных к их мембране.
КЛИНИЧЕСКАЯ КАРТИНА
Клинические проявления возникают при снижении содержания тромбоцитов до 20-30х109/л. Тромбоцитопенический геморрагический синдром характеризуется кожными кровоизлияниями и кровотечениями из слизистых оболочек ротовой полости, носа, половых путей. Кожные кровоизлияния могут быть представлены экхимозами (чаще локализуются на конечностях, туловище) и петехиями (чаще возникают на нижних конечностях). Кровотечения из ЖКТ, кровохарканье и гематурия при тромбоцитопении наблюдают редко. Кровотечения при экстракции зубов возникают не всегда, начинаются сразу после вмешательства и продолжаются несколько часов или дней. После остановки они, как правило, не возобновляются.
Идиопатическая тромбоцитопеническая пурпура обычно развивается после перенесенной инфекции. Заболевание обычно начинается остро, с появления обильной яркой сыпи на конечностях и туловище. Геморрагии на лице и шее появляются редко, только после кашля. Отдельные элементы геморрагической сыпи не сливаются между собой, не
поднимаются над поверхностью кожи, не исчезают при надавливании. При увеличении количества циркулирующих тромбоцитов геморрагии исчезают в течение одного дня. Подкожные гематомы возникают чаще в местах незначительного надавливания и на задней поверхности бедер. Часто наблюдают кровотечения из носа, десен, ЖКТ (мелена), гематурию, в 1% случаев происходит кровоизлияние в головной мозг. Продолжительность заболевания обычно не превышает 3-4 нед, спонтанные ремиссии развиваются у 93% пациентов. Иногда заболевание приобретает хроническое циклическое течение. В анализах крови иногда обнаруживают гигантские тромбоциты. В красном костном мозге выявляют увеличение количества и размеров мегакариоцитов. Количественных изменений эритроцитов и лейкоцитов чаще всего не бывает, коагуляционные тесты - в пределах нормы. Диагностическим критерием считают обнаружение антитромбоцитарных АТ.
Тромбоцитопения, связанная с ЛС, чаще начинается через 3-4 дня после начала приема препарата и проходит самостоятельно через 3-5 дней после его отмены. Основные проявления: петехиальные высыпания, носовые и маточные кровотечения.
Тромбоцитопения, связанная с подавлением гемопоэза, обычно бывает достаточно тяжелой, сопровождается выраженной петехиальной сыпью и кровотечениями из слизистых оболочек, наслаивающимися на клиническую симптоматику основного заболевания.
Тромботическая тромбоцитопеническая пурпура в большинстве случаев начинается остро после перенесенной вирусной инфекции. Нередко отмечают продромальный период, характеризуемый слабостью, повышенной утомляемостью, головной болью, потерей аппетита, тошнотой, рвотой, болями в животе, лихорадкой, нарушением зрения. Для развернутой стадии заболевания характерно сочетание лихорадки, петехиальных высыпаний, носовых и желудочно-кишечных кровотечений, кровоизлияний в сетчатку, кровоточивости десен, реже - кровохарканья и разнообразной неврологической симптоматики (дезориентации, атаксии, гемипарезов, судорог, психических расстройств, в тяжелых случаях - коматозного состояния). В анализах крови, помимо тромбоцитопении различной степени выраженности, обнаруживают уменьшение концентрации гемоглобина до 50-80 г/л, ретикулоцитоз, повышение концентрации непрямого билирубина и свободного гемоглобина. Диагностика основывается на выявлении в биоптатах участков кожи в местах кровоизлияний тромбоцитарно-гиалиновых тромбов в мелких артериях и артериолах без признаков воспаления.
Гемолитико-уремический синдром также развивается после перенесенной инфекции и по патогенезу близок к тромботической тромбоцитопенической пурпуре, однако отличается от последней преимущественным поражением почек. Основные клинические проявления: ОПН, острый гемолиз, тромбоцитопения, коагулопатия потребления и геморрагический синдром. Заболевание чаще наблюдают у детей.
ЛЕЧЕНИЕ
• У 90% детей с впервые диагностированной идиопатической тромбоцитопенической пурпурой в течение 4-6 нед происходит нормализация состояния без дополнительной терапии. При стойкой тромбоцитопении с геморрагическим синдромом назначают преднизолон в дозе 1-3 мг/(кгхсут). Продолжительность терапии ГК не должна превышать 6 мес. При отсутствии эффекта показана спленэктомия, позволяющая достигнуть ремиссии у 50-90% пациентов. В последние годы также применяют введение иммуноглобулина в высоких дозах (иммуноглобулина человеческого нормального по 400 мг/кгхсут) в течение 5 дней).
• При тромботической тромбоцитопенической пурпуре единственный метод лечения - сеансы плазмафереза с замещением не менее 1,5 л плазмы в течение как минимум 4-5 дней. Спленэктомия обычно неэффективна.
• При тромбоцитопении, связанной с приемом ЛС, необходима срочная отмена соответствующего препарата. Исключение составляет химиотерапия злокачественных опухолей, при которой тромбоцитопения - неизбежный побочный эффект, для ее купирования проводят инфузии тромбоцитарной массы.
• При тромбоцитопении, связанной с инфекционными заболеваниями, необходимости в дополнительном лечении, как правило, не возникает: содержание тромбоцитов нормализуется по мере купирования инфекционного процесса. В тяжелых случаях, сопровождаемых развитием ДВС-синдрома, необходимо введение свежезамороженной плазмы.
57.2. ТРОМБОЦИТОПАТИИ
Термин «тромбоцитопатия» объединяет все нарушения гемостаза, обусловленные качественной неполноценностью или дисфункцией тромбоцитов. Выделяют врожденные (наследственные) и приобретенные тромбоцитопатии.
ЭПИДЕМИОЛОГИЯ
Заболеваемость наследственными тромбоцитопатиями составляет приблизительно 3-5 на 100 000 населения. Приобретенные нарушения функции тромбоцитов наблюдают значительно чаще, причем распространенность их в последние годы увеличивается, что в первую очередь связано с бесконтрольным применением ЛС, особенно при попытках самолечения.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Врожденные тромбоцитопатии связаны с генетически обусловленными нарушениями адгезии, агрегации или дегрануляции тромбоцитов вследствие дефектов гликопротеидов мембраны, цитоплазменных ферментов и пр. Содержание тромбоцитов в крови при некоторых формах (тромбастении Гланцманна) нормальное, при других (синдромах Бернара-Сулье, Вискотта-Олдрича, Хегглина и пр.) - пониженное, главным образом в связи с уменьшением продолжительности жизни дефектных тромбоцитов.
Основные причины приобретенных тромбоцитопатий.
• Нарушения адгезии/агрегации тромбоцитов и снижение доступности тромбоцитарного фактора 3 при уремии, циррозе печени, опухолях и паразитарных заболеваниях.
• Повышенное потребление и структурные повреждения тромбоцитов при заболеваниях, сопровождаемых развитием ДВСсиндрома.
• Блокада рецепторов тромбоцитов протеинами при парапротеинемических гемобластозах.
• Прием ЛС. Механизмы нарушения функций тромбоцитов под действием ЛС многообразны и недостаточно изучены. Чаще всего дисфункцию тромбоцитов вызывают НПВС, особенно ацетилсалициловая кислота (ингибируют синтез тромбоксана А2), антибактериальные препараты (ампициллин, карбенициллин, нитрофураны), антидепрессанты (амитриптилин, имипрамин), адреноблокаторы (дигидроэрготамин, фентоламин, пропранолол), антигистаминные препараты, декстраны, нитраты, блокаторы медленных кальциевых каналов, витамин Е, а также этанол.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Основное клиническое проявление тромбоцитопатий - геморрагический синдром, протекающий как тромбоцитопенический (кожные
кровоизлияния в виде петехий и экхимозов, чаще на конечностях, туловище, кровоточивость слизистых оболочек ротовой полости, носа, половых путей).
Для некоторых врожденных тромбоцитопатий также характерны сопутствующие симптомы, не связанные с нарушением гемостаза, - нарушения пигментации кожи при болезни Херманского-Пудлака, аномалии скелета при синдроме TAR (Thrombocytopenia-Absent Radius - тромбоцитопения и отсутствие лучевой кости), патология лейкоцитов при аномалии Мая-Хегглина, иммунодефицит при болезни Вискотта-Олдрича.
• При тромбастении Гланцманна (тип A: *273800, дефекты генов GP2B, GP3A, р или Ʀ; тип B: * 173470, дефекты генов ITGB3, GP3A,
нарушается преимущественно агрегация тромбоцитов вследствие аномалии гликопротеина IIb/IIIа, характерны увеличение времени капиллярного кровотечения по Дьюку, отсутствие или резкое ослабление ретракции кровяного сгустка при нормальном или почти нормальном содержании тромбоцитов в крови. Клинические проявления: петехии и экхимозы на коже, повторные носовые и маточные кровотечения, кровотечения после порезов, экстракции зубов. Геморрагический синдром более выражен в детском и юношеском возрасте и у женщин.
• При синдроме Бернара-Сулье (тип А: GP1BA, 231200, р; тип B: GP1BB, 138720, полигенное наследование; тип C: GP9, 173515) нарушается адгезия тромбоцитов к коллагену вследствие отсутствия гликопротеина Ib, обеспечивающего взаимодействие между фактором Виллебранда и мембраной тромбоцита. В крови выявляют гигантские тромбоциты, процентное содержание которых коррелирует с тяжестью геморрагического синдрома.
• Синдром Вискотта-Олдрича (*301000, Xp1 1.23-p1 1.22, дефекты генов WAS, IMD2, THC, К (наиболее частая форма); 277970, ρ; *600903, Ʀ) характеризуется сочетанием геморрагического синдрома, обусловленного тромбоцитопенией и тромбоцитопатией, иммунодефицита вследствие нарушения синтеза IgM, экземы (в раннем возрасте).
• Синдром Чедика-Хигаси (*214500, 1q42.1-q42.2, ген CHS1, внутрилизосомный регулятор транспорта, ρ) характеризуется сочетанием геморрагического синдрома, связанного с тромбоцитопенией и дисфункцией тромбоцитов, с альбинизмом и иммунодефицитом вследствие нейтропении и нарушения фагоцитарной активности нейтрофилов.
• При синдроме TAR (Thrombocytopenia-Absent Radius - тромбоцитопения и отсутствие лучевой кости; *270400, р) умеренно выраженный геморрагический синдром сочетается с двусторонним отсутствием лучевой кости с укорочением предплечий, деформацией позвоночника, лопаток, дисплазией тазобедренных суставов, незаращением нёба, косоглазием, гипоплазией легких и другими пороками развития.
• Синдром Хегглина (155100, 99) характеризуется нарушением созревания и аномалией мегакариоцитов, тромбоцитов и нейтрофилов. Геморрагический синдром обычно умеренный, степень его выраженности коррелирует с тяжестью тромбоцитопении.
ЛЕЧЕНИЕ
Верифицированный диагноз наследственной тромбоцитопатии следует рассматривать как своеобразную пожизненную характеристику больного, которую необходимо учитывать при назначении ЛС по поводу любых других заболеваний. При приобретенных тромбоцитопатиях основу лечения составляет терапия основного заболевания.
Особое внимание следует уделять устранению всех воздействий, провоцирующих или усиливающих кровоточивость. Запрещают прием алкоголя, употребление уксусной кислоты. Из ЛС особенно опасны салицилаты, НПВС, карбенициллин, хлорпромазин, антикоагулянты непрямого действия, фибринолитики. Также нежелательно применение пиридоксина в связи с его способностью ингибировать функции тромбоцитов. Следует избегать тугой тампонады носа (после удаления тампона кровотечение часто возобновляется с еще большей силой) и выскабливаний полости матки. Противопоказаны прижигания слизистых оболочек.
Переливания крови и ее компонентов при большинстве тромбоцитопатий бесполезны и могут усугубить нарушения функции тромбоцитов. Трансфузии эритроцитов и плазмы рекомендуют только при массивных кровопотерях для коррекции анемии и возмещения объема. Переливание донорских тромбоцитов необходимо при кровотечениях, возникающих на фоне оперативных вмешательств при состояниях, характеризуемых сочетанием тромбоцитопатии и тромбоцитопении.
Специфической терапии, кардинально улучшающей функции тромбоцитов, в настоящее время не разработано (ГК, этамзилат, трифосаденин неэффективны). Традиционно назначают витамины (аскорбиновую кислоту, ретинол и пр.). При массивных маточных и носовых кровотече-
ниях необходимо внутривенное введение 5-6% раствора аминокапроновой кислоты (до 100 мл). Гемостатическим эффектом также обладает карбазохром.
57.3. КОАГУЛОПАТИИ
Коагулопатии (нарушения свертываемости крови) могут быть наследственными и приобретенными.
• Наиболее распространенные наследственные коагулопатии - гемофилии и болезнь Виллебранда. На них приходится до 96% случаев всех врожденных нарушений свертываемости крови (гемофилия A - 68-78%, гемофилия B - 6-13%, болезнь Виллебранда - 9-18%). Значительно реже наблюдают дефицит факторов XI, VII и V (2-3%). Все остальные формы относятся к клинической казуистике, в структуре наследственных коагулопатий на них приходится менее 1%.
• Приобретенные коагулопатии могут быть связаны с дефицитом витамина К, заболеваниями печени, приемом непрямых антикоагулянтов, появлением в крови патологических ингибиторов свертывания. Коагулопатия, наряду с другими нарушениями гемостаза, также развивается в рамках ДВС-синдрома.
Гемофилии и болезнь Виллебранда
Гемофилия - наследственная коагулопатия, связанная с дефицитом фактора VIII (гемофилия А) или IX (гемофилия В). Болезнь Виллебранда обусловлена наследственным дефицитом одноименного фактора.
ЭПИДЕМИОЛОГИЯ
Частота гемофилии А составляет 1 на 10 000 новорожденных мальчиков, гемофилии В - 1 на 50 000. Болезнь Виллебранда наблюдают значительно чаще, заболеваемость составляет 1 на 30 000 населения.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Патологические гены, обусловливающие развитие гемофилии A (306700, Xq28, дефекты генов F8C) и B (болезнь Кристмаса, 306900, Xq27.1-q27.2, дефекты генов F9, HEMB), расположены на Х-хромосоме, поэтому заболевание регистрируют почти исключительно у мужчин. В 70-85% случаев гемофилии А и в 90% случаев гемофилии В положите-
лен семейный анамнез. До 30% случаев гемофилии А связаны со спорадическими мутациями.
Факторы VIII и IX принимают участие в трансформации протромбина в тромбин, при их дефиците нарушается формирование тромба в местах повреждения сосудов, что клинически проявляется гематомным типом кровоточивости.
Болезнь Виллебранда (*193400, 12pter-p12, дефекты генов VWF, F8VWF, Ʀ; *277480, ρ, типы ПС, III; *177820, дефект рецептора фактора Виллебранда) связана с врожденным отсутствием мультимерных форм фактора VIII Виллебранда.
Фактор Виллебранда опосредует адгезию тромбоцитов к эндотелию, а также участвует в стабилизации фактора VIII и защите его от инактивации противосвертывающими системами плазмы. Дефицит фактора Виллебранда сопровождается преимущественно микроциркуляторной кровоточивостью, реже (преимущественно в тяжелых случаях) наблюдают смешанный тип.
КЛИНИЧЕСКАЯ КАРТИНА
По клинической симптоматике гемофилии А и В практически идентичны. Тяжесть геморрагического синдрома напрямую связана со степенью дефицита фактора:
• тяжелая форма - концентрация фактора ниже 2%;
• среднетяжелая - 2-5%;
• легкая - более 5-8%.
В клинической картине преобладают кровоизлияния в крупные суставы конечностей (95% пациентов), глубокие подкожные, межмышечные и внутримышечные гематомы (93%), обильные и длительные кровотечения при травмах (91%). Реже встречаются кровотечения из носа и десен (56%), после экстракции зубов (38%), макрогематурия (28%), забрюшинные гематомы (16%), кровоизлияния в брыжейку и стенку кишки (7%), внутричерепные гематомы (14%).
При гемофилии прослеживается отчетливая возрастная эволюция клинической симптоматики. Обычно диагноз устанавливают к 1-2му году жизни. В более раннем периоде геморрагические проявления обычно выражены слабо (хотя при тяжелых формах у новорожденных возможны образование кефалогематом, поздние кровотечения из пупочной ранки). Иногда болезнь проявляется при первых внутримышечных инъекциях (при вакцинации). Кровотечения при прорезывании зубов обычно незначительные. В период, когда ребенок учится ходить,
часто возникают обширные гематомы в области черепа. Затем на первый план выступают гемартрозы, возникающие тем раньше, чем тяжелее гемофилия. С возрастом тяжесть и распространенность суставных поражений неуклонно прогрессируют, особенно при недостаточной заместительной терапии.
• Гемартрозы обычно развиваются после незначительной травмы либо спонтанно: возникают острые боли и нарушение подвижности в суставе, он увеличивается в объеме, появляется гиперемия кожных покровов над ним. При обширных кровоизлияниях возможна флюктуация. С наибольшей частотой поражаются коленные суставы, затем локтевые и голеностопные. Кровоизлияния в лучезапястные и тазобедренные суставы наблюдают редко.
• Подкожные, межфасциальные и забрюшинные гематомы могут достигать огромных размеров. Они очень болезненны, напряжены, их возникновение сопровождается лихорадкой. За счет сдавления сосудисто-нервных пучков возможны ишемия тканей, параличи, контрактуры, атрофия мышц. Весьма характерны сгибательная контрактура бедра при кровоизлиянии в область подвздошнопоясничной мышцы и появление псевдоопухолей и ложных суставов на фоне деструкций кости при поднадкостничных гематомах.
• Почечные кровотечения могут возникать как спонтанно, так и после незначительных травм и нередко приводят к выраженной анемии. Гематурия часто сопровождается дизурическими явлениями, приступами почечной колики.
• Кровоизлияния в головной и спинной мозг всегда имеют травматический генез. Между моментом травмы и появлением клинических симптомов может пройти от 1 до 24 ч. Любую, даже незначительную травму головы рассматривают как показание к профилактическому введению антигемофильных препаратов.
• Кровоизлияния в брыжейку, сальник и стенку кишки протекают с лихорадкой, нейтрофилезом, выраженными симптомами раздражения брюшины и часто имитируют острую хирургическую патологию.
• Количество вирусных и иммунологических осложнений растет по мере увеличения продолжительности жизни больных гемофилией и с интенсификацией заместительной трансфузионной терапии. До 95% больных гемофилией инфицированы вирусами гепатитов, а в 20% случаев у них появляются ингибиторы факторов VIII и IX, снижающие эффект проводимой заместительной терапии.
ДИАГНОСТИКА
Диагностика гемофилий А и В основывается на определении концентрации факторов VIII и IX в плазме крови. Разработана молекулярногенетическая диагностика гемофилии A (выявление патологического гена с помощью ПЦР), в том числе пренатальная (материал для исследования в последнем случае - ворсины хориона или амниотическая жидкость).
Интенсивность геморрагического синдрома при болезни Виллебранда весьма вариабельна - от незначительных кожных высыпаний до частых тяжелых длительных кровотечений. В период полового созревания даже на фоне благоприятного течения болезни нередко возникают крайне тяжелые носовые и маточные кровотечения. Подкожные кровоизлияния в легких случаях поверхностны, безболезненны, но по мере нарастания тяжести болезни становятся все более похожими на гематомы, возникающие при гемофилии. Иногда возникают тяжелые кровотечения из ЖКТ. Маточные кровотечения иногда продолжаются до 15-25 дней и настолько плохо поддаются терапии, что приходится прибегать к экстирпации матки. При тяжелой форме болезни Виллебранда в редких случаях возможны кровоизлияния в крупные суставы. Гематурию и внутричерепные кровоизлияния наблюдают редко.
Диагностика болезни Виллебранда основывается на определении активности соответствующего фактора в крови (например, с помощью ристоцетин-кофакторного теста).
ЛЕЧЕНИЕ
Терапия наследственных коагулопатий направлена на обеспечение минимально необходимой концентрации дефектных факторов свертывания.
• При гемофилии А вводят криопреципитат, концентрат фактора VIII, полученный от многих доноров, рекомбинантный фактор VIII. Профилактическая доза фактора VIII, позволяющая предотвратить инвалидизацию у детей раннего возраста, - 20 ЕД/кг 3 раза в неделю. При тяжелых кровотечениях необходимо введение фактора VIII в дозе до 50 ЕД/кг. При оперативных вмешательствах концентрация фактора VIII должна составлять не менее 1000
ЕД/л.
• При гемофилии B дефицит фактора IX восполняют свежезамороженной плазмой, плазмой после отделения криопреципитата, концентратами фактора IX. Доза фактора IX для первичной про-
филактики составляет 40 ЕД/кг 2 раза в неделю. При тяжелых кровотечениях концентрацию фактора IX необходимо поддерживать на уровне не менее 300 ЕД/л, при оперативных вмешательствах - не менее 500 ЕД/л в момент операции и 400 ЕД/л в течение всего послеоперационного периода.
• Для лечения болезни Виллебранда применяют препараты, содержащие большое количестоа фактора VIII, - криопреципитат♠, концентраты фактора свертывания крови VIII. Терапевтический эффект указанных препаратов зависит от содержания в них высокомолекулярных мультимеров фактора. При тяжелых кровотечениях необходимо введение фактора свертывания крови VIII в дозе 10-20 ЕД/кг каждые 8-12 ч.
Синдром диссеминированного внутрисосудистого свертывания
ДВС-синдром - неспецифический универсальный патологический процесс, связанный с поступлением в кровоток активаторов свертывания крови, характеризуемый образованием множественных тромбов в сосудах микроциркуляторного русла различных органов и тканей с последующим развитием коагулопатии и тромбоцитопении потребления, сопровождаемых многочисленными кровоизлияниями. В течении ДВСсиндрома различают фазы гипер- и гипокоагуляции, продолжительность и чередование которых зависят от причины, вызвавшей развитие заболевания.
ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
Диссеминированное внутрисосудистое свертывание (ДВС) - один из наиболее распространенных клинических синдромов, развивающийся при всех формах шока, сепсисе, терминальных состояниях, остром внутрисосудистом гемолизе, злокачественных новообразованиях, массивных некротических процессах в органах и тканях и пр.
В основе развития ДВС-синдрома, несмотря на многообразие этиологических факторов, всегда лежат следующие механизмы:
• активация свертывающей системы крови и тромбоцитарного гемостаза эндогенными (тканевым тромбопластином, продуктами распада тканей и клеток крови, поврежденным эндотелием) либо экзогенными (бактериями, вирусами, ЛС, околоплодными водами, ядами змей и т.д.) факторами;
• системное поражение эндотелия, истощение его антитромботического потенциала;
• диффузное внутрисосудистое свертывание крови и агрегация тромбоцитов с образованием множества микротромбов, блокада кровообращения в органах и тканях;
• выраженные дистрофические и деструктивные изменения в органах с нарушением их функций;
• нарушения микроциркуляции, гипоксия тканей и ацидоз, связанные с развитием сладж-синдрома.
КЛИНИЧЕСКАЯ КАРТИНА И ДИАГНОСТИКА
Течение ДВС-синдрома может быть острым, затяжным, рецидивирующим, хроническим и латентным. Зачастую формы, принимаемые за острые и даже молниеносные, в действительности оказываются финальным этапом латентно или стерто протекавшего затяжного процесса.
• Острое течение характерно для инфекционно-септического, акушерского (кроме внутриутробной гибели плода), посттравматического, хирургического, токсического (укусы змей) и шокового ДВС-синдрома.
• Острое и подострое течение ДВС-синдрома наблюдают при деструктивных процессах в легких (стафилококковых и анаэробных инфекциях), дистрофии печени вирусного или токсического происхождения, панкреонекрозе, остром внутрисосудистом гемолизе.
• Подострое течение ДВС-синдрома наблюдают при затяжном септическом процессе, при острых лейкозах, повторных массивных трансфузиях консервированной крови, воспалительных процессах (пневмонии), отравлениях уксусной кислотой, при некоторых видах акушерской патологии.
• Затяжное течение ДВС-синдрома характерно для большинства онкологических, иммунокомплексных и миелопролиферативных заболеваний, сердечной недостаточности, циррозов печени, а также на фоне хронического гемодиализа, при использовании аппаратов искусственного кровообращения, протезировании сосудов и клапанов сердца. Чем более остро протекает ДВС, тем меньше продолжительность фазы гиперкоагуляции и тем тяжелее фаза выраженной гипокоагуляции и кровоточивости. Клиническая картина ДВС-синдрома складывается из симптомов заболевания, ставшего причиной его развития, признаков гемокоагуляционного или смешанного шока, глубоких нарушений всех звеньев системы
гемостаза, тромбозов и геморрагий, гиповолемии и анемии, дисфункции и дистрофических изменений внутренних органов и метаболических нарушений. Одно из основных проявлений ДВС-синдрома - геморрагический синдром, развивающийся как результат коагулопатии и тромбоцитопении потребления. Наиболее часто кровоточивость возникает при остром ДВС. На коже появляются петехии и экхимозы, возникают кровотечения из мест инъекций, десен, полости носа, матки, ЖКТ.
Гемокоагуляционный шок сопровождается падением АД и центрального венозного давления, развитием ОПН и гепаторенальной недостаточности, шокового легкого. При присоединении профузных кровотечений гемокоагуляционный шок трансформируется в геморрагический. Шок при ДВС-синдроме также усугубляется острой надпочечниковой недостаточностью, возникающей из-за кровоизлияний в надпочечники. Спазм артериол обусловливает формирование полиорганной недостаточности. Наиболее часто органами-мишенями становятся легкие (развитие острой сердечно-легочной недостаточности, интерстициального отека легких), почки (возникновение ОПН), печень (развитие паренхиматозной желтухи), желудок и кишечник (формирование очаговой дистрофии слизистой оболочки с образованием острых эрозий). Нарушение церебральной гемодинамики приводит к спутанности сознания.
Затяжное течение ДВС-синдрома чаще проявляется гиперкоагуляцией с развитием венозных тромбозов с тромбоэмболиями и ишемией внутренних органов. В терминальный период затяжного течения ДВС-синдрома возможны множественные тромбозы органных и магистральных вен с развитием тромбоэмболии легочной артерии или трансформация тромботического процесса в терминальную стадию острой гипокоагуляции с кровотечениями, преимущественно из ЖКТ.
Волнообразное течение ДВС-синдрома проявляется временными ремиссиями, сменяющимися повторными острыми нарушениями гемостаза.
Лабораторные исследования
Нарушения гемостаза при ДВС-синдроме проходят разные фазы - от гиперкоагуляции до более или менее глубокой гипокоагуляции вплоть до полного отсутствия свертывания крови.
• Характерные признаки гиперкоагуляционной фазы - уменьшение АЧТВ и протромбинового времени, снижение концентрации факторов свертывания, фибриногена, антитромбина III, протеи-
на C. Если в таких случаях обнаруживают спонтанную агрегацию тромбоцитов и фрагментацию эритроцитов в мазках, диагноз ДВС-синдрома вполне достоверен.
• Характерные признаки гипокоагуляционной фазы - увеличение АЧТВ, протромбинового времени, уменьшение содержания тромбоцитов, дальнейшее снижение концентрации факторов свертывания, фибриногена, антитромбина III, протеина С, усиление фибринолиза, повышение концентрации продуктов деградации фибрина.
Алгоритм диагностики
При наличии у пациента заболевания, при котором существует риск развития ДВС-синдрома, можно использовать следующий алгоритм диагностики, основанный на балльной оценке результатов коагуляционных тестов.
• Содержание тромбоцитов:
- более 100х109/л - 0 баллов;
- 50-100х109/л - 1 балл;
- менее 50х109/л - 2 балла.
• Концентрация мономеров фибрина или продуктов деградации фибрина:
- не повышена - 0 баллов;
- повышена незначительно - 2 балла;
- повышена значительно - 3 балла.
• Увеличение протромбинового времени:
- менее 3 с - 0 баллов;
- 3-6 с - 1 балл;
- больше 6 с - 2 балла.
• Концентрация фибриногена:
- более 1 г/л - 0 баллов;
- менее 1 г/л - 1 балл.
Если сумма баллов больше 5, диагностируют ДВС-синдром, если меньше 5 - необходимо дальнейшее наблюдение с повторными исследованиями.
ЛЕЧЕНИЕ
Основные компоненты комплексного лечения ДВС-синдрома
• Этиотропная терапия.
• Противошоковая терапия и поддержание необходимого объема и состава циркулирующей крови.
• Инфузии свежезамороженной плазмы, плазмаферез.
• Введение гепарина (только в фазу гиперкоагуляции).
• Возмещение потерь эритроцитов на фоне выраженного геморрагического синдрома (поддержание гематокрита на уровне не менее 22%).
• При выраженной гипокоагуляции, кровотечениях на фоне тромбоцитопении - введение тромбоцитарной массы.
• Коррекция органных поражений. Трансфузионная терапия ДВС-синдрома направлена:
• на коррекцию нарушений гемостаза, в первую очередь на возмещение компонентов, поддерживающих антитромботический потенциал крови (антитромбина III, плазминогена, протеина С, фибронектина) и нормализующих процесс свертывания;
• восстановление антипротеазной активности плазмы;
• возмещение дефицита объема циркулирующей крови и восстановление нормального центрального венозного давления, нарушенных вследствие шока и/или кровопотери.
На поздних стадиях ДВС-синдрома для остановки кровотечений, особенно маточных, наряду с инфузиями свежезамороженной плазмы необходимо введение ингибиторов фибринолиза. Плазмаферез проводят при подострых и хронических ДВС-синдромах. За одну процедуру удаляют до 500-1000 мл плазмы с ее замещением донорской.