Лекция 6ЭНДОГЕННЫЕ ПИГМЕНТАЦИИ•
Эндогенные пигментации — разновидность смешанных дистрофий. В основе их лежат нарушения эндогенных пигментов.
Эндогенные пигменты — окрашенные вещества различной химической природы, которые синтезируются в самом организме, придавая органам и тканям различную окраску. По своей структуре они являются
хромопротеидами (от греч. chroma — цвет, окраска + протеиды), т.е. окрашенными белками. Хромопротеиды широко распространены в живой природе и выполняют разнообразнейшие биологические функции: перенос и депонирование кислорода для осуществления окислительно-восстановительных процессов в клетках, в том числе и дыхания (гемоглобин, цитохромы, миоглобин, липофусцин), рецепция света и защита от действия ультрафиолетового излучения (меланин), синтез биологически активных веществ (пигмент гранул энтерохромаффинных клеток), секретов (желчь), доставка и регуляция обмена микроэлементов (церулоплазмин, ферритин, гемосидерин), витаминов (липохромы) и др.
Классификация. Эндогенные пигменты разделяют, согласно их формальному генезу, на 3 группы:
▲ гемоглобиногенные, представляющие собой различные производные гемоглобина;
▲ протеиногенные, или тирозиногенные, связанные с обменом тирозина;
▲ липидогенные, или липопигменты, образующиеся при обмене жиров.
Продукты нарушенного обмена эндогенных пигментов обычно откладываются как в паренхиме органов, так и вне ее, в строме. При нарушении обмена пигментов учитывают следующие особенности:
количество пигмента. Оно может быть увеличено или, наоборот, уменьшено вплоть до полного исчезновения;
распространенность процесса (общий или местный характер процесса);
характер наследования. Этиологические факторы, вызывающие нарушение обмена хромопротеидов, являются генетически обусловленными или же приобретаются в течение жизни; в связи с этим различают наследственные и приобретенные нарушения обмена пигментов.
Пигментный обмен может нарушаться при многих болезнях и патологических состояниях, т.е. возникает вторично; однако иногда нарушения обмена хромопротеидов возникают первично и являются морфологическим субстратом самостоятельных заболеваний. В большинстве случаев патологические пигментации возникают в связи с избыточным накоплением пигментов, которые встречаются и в норме, но иногда накапливается пигмент, который возникает только в условиях патологии.
ГЕМОГЛОБИНОГЕННЫЕ ПИГМЕНТЫСхема 8. Циклы железа в организме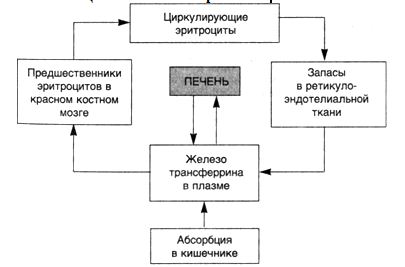
Гемоглобиногенные пигменты получили свое наименование вследствие того, что их образование связано с метаболизмом гемоглобина. При этом часть пигментов образуется в физиологических условиях. Это гемосидерин, ферритин и билирубин. Часть пигментов — гематоидин, гематины и порфирин, образуются только в условиях патологии. Некоторые из этих пигментов (ферритин, гемосидерин) синтезируются, помимо гемоглобина, из железа, всасывающегося в кишечнике (схема 8). Поэтому определение "гемоглобиногенные пигменты" является для них весьма условным.
Гемоглобин — хромопротеид, который в качестве простетической группы содержит железопорфириновый комплекс гем. Белковая часть молекулы гемоглобина состоит из двух пар полипептидных (а и Ь) цепей, содержащих по 140 аминокислот. Своим
огромным значением гемоглобин обязан содержащемуся в нем железу, с которым филогенетически связана функция дыхания. Обмен гемоглобина тесно связан с эритроцитами, в которых он содержится, с их состоянием, старением, разрушением. Физиологический гемолиз происходит в основном в костном мозге, реже — в селезенке и печени, в клетках макрогистиоцитарной системы этих органов образуются ферритин, гемосидерин и билирубин.
Ферритин — железопротеид, содержащий белок апоферритин и трехвалентный атом железа в составе фосфатного гидроксида. Ферритин неоднороден, известно до 20 изоферритинов. Это разнообразие обусловлено различием вариантов входящего в его состав апоферритина (Н-, L- и HL-субъединицы), различием способов происхождения пигмента ("анаболический" — из железа, всасывающегося в кишечнике, "катаболический" — из железа гемолизированных эритроцитов), разной локализацией (в сыворотке крови — HL-ферритин, в печени и селезенке — L-ферритин). Наконец, важное значение имеет кислород: ферритин синтезируется из двухвалентного железа в присутствии кислорода и содержит много SS-групп. При гипоксии образуется SH-ферритин, обладающий вазопаралитическим действием. Значение ферритина трудно переоценить. Он является главным участником метаболизма железа. Известно, что свободные атомы железа токсичны для организма. Именно в форме ферритина депонируется железо (до 30 %, хотя расходуется только 0,1 %). Ферритин содержится практически во всех органах и тканях и является акцептором железа в клетках, которые в нем нуждаются (эритробласты). Он также осуществляет перенос железа в кишечнике и плаценте, т.е. является медиатором при соединении железа с трансферрином и в переносе его от матери к плоду.
Ферритин выявляют в тканях с помощью сульфата кадмия по методу Клочкова, а также иммуногистохимически с использованием специфических антисывороток. На практике чаще всего используется гистохимический метод — реакция образования берлинской лазури (железистосинеродистое железо) или реакции Перльса — реакция на выявление солей оксида железа (III) с помощью железосинеродистого калия и хлороводородной (соляной) кислоты.
Гемосидерин — это продукт полимеризации ферритина. По химической структуре он является коллоидным гидроксидом железа, соединенным с мукопротеидами клетки. В норме гемосидерин образуется в ретикулярных и эндотелиальных клетках селезенки, лимфатических узлов, печени и костного мозга. При окраске гематоксилином и эозином гемосидерин выявляется в виде зерен бурого цвета в цитоплазме этих клеток, а при реакции Перльса — в виде гранул зеленовато-синего цвета (берлинская лазурь). Гемосидерин — внутриклеточный пигмент. Синтез его происходит в клетках, которые называют сидеробластами, в специализированных органеллах — сидеросомах. Иногда в сидеробластах накапливается такое большое количество гемосидерина, что клетки разрушаются и гемосидерин оказывается свободно лежащим в строме органов. В этих случаях он обычно захватывается макрофагами, которые принято называть сидерофагами. В цитоплазме этих клеток сидеросомы не выявляются.
Билирубин — конечный продукт гемолиза. Билирубин образуется, когда от гемоглобина отщепляется гем, а затем от гема отщепляется железо и разворачивается тетрапиррольное кольцо. Этот процесс начинается в клетках ретикуломакрофагальной системы костного мозга, селезенки, лимфатических узлов и печени. Затем продукт, соединяясь с альбумином, с током крови поступает в печень. В печени синтез пигмента завершается — гепатоциты, обладая специфическими рецепторами, захватывают его и с помощью ферментов специфической глюкуронилтрансферазной системы осуществляют его конъюгацию. Конъюгаты билирубина поступают в желчные капилляры. Таким образом, билирубин становится основным пигментом желчи.
Обычно билирубин находится в виде кристаллов красновато-желтого цвета. Он легко окисляется, образуя при этом продукты различного цвета. Именно это происходит при выявлении его по методу Гмелина — при окислении его азотной кислотой образуются продукты сначала зеленого, а затем синего или пурпурного цвета.
Гематоидин — пигмент, не содержащий железа. По химической структуре близок к билирубину и также дает положительную реакцию Гмелина. Гематоидин формирует ярко-оранжевые кристаллы в виде ромбических пластинок, иголок или зерен. Образуется при распаде эритроцитов и гемоглобина, как и гемосидерин, внутриклеточно, но в клетках не остается и при их гибели оказывается свободно лежащим среди некротических масс.
Гематины образуются при гидролизе оксигемоглобина и представляют собой окисленную форму гема, содержащую трехвалентный атом железа в связанном состоянии. Имеют вид темно-коричневых кристаллов или зерен. К гематинам относят малярийный пигмент (гемомеланин), солянокислый гематин и формалиновый пигмент.
Малярийный пигмент (гемомеланин) образуется из гема в теле малярийного плазмодия, который, как известно, паразитирует в эритроцитах. Пигмент построен из буровато-черных аморфных гранул и синтезируется обычно в ретикулярных и эндотелиальных клетках печени, костного мозга, селезенки и лимфатических узлов.
Солянокислый гематин (гемин) образуется исключительно в желудке при взаимодействии гемоглобина, ферментов желудочного сока и соляной кислоты. Пигмент откладывается в виде ромбовидных или игловидных кристаллов.
Формалиновый пигмент образуется в тканях при фиксации их кислым формалином (рН<5,6), имеет вид бурых зерен или кристаллов, расположенных, как правило, в просвете венозных сосудов.
Порфирины — предшественники гема, которые имеют строение замкнутых тетрапиррольных колец, лишенных железа. Пигменты повышают чувствительность кожи к ультрафиолетовому облучению, являются антагонистами меланина. Обычно метаболизм порфиринов в организме человека заканчивается на стадии уропорфириногена III, который затем принимает участие в реакциях синтеза гема. При отсутствии фермента уропорфириноген Ш-косинтетазы появляются предшественники уропорфириногена III — уропорфириноген I, порфобилин, порфобилиногены. В норме они в минимальных количествах определяются в тканях, крови и моче: они дают оранжевую флюоресценцию в ультрафиолетовом свете.
Нарушения обмена гемоглобиногенных пигментов ГемосидерозНарушения обмена гемосидерина, ферритина и билирубина происходят при усиленном гемолизе эритроцитов, возникающем в результате действия различных патогенных факторов. В этих случаях обычно говорят о гемосидерозе, хотя одновременно происходит накопление некоторого количества ферритина и билирубина. Гемосидероз может возникать в результате усиления как внутрисосудистого гемолиза (общий гемосидероз), так и При развитии внесосудистого гемолиза (местный гемосидероз).
Общий гемосидероз. Развивается при болезнях системы кроветворения (анемии, гемобластозы), интоксикациях гемолитическими ядами (бертолетова соль, сульфаниламиды, хинин, свинец), при некоторых инфекциях (сепсис, малярия, бруцеллез, возвратный тиф), при переливании иногрупной крови и резус-конфликте (гемолитическая болезнь новорожденных). В этих случаях гемосидерин в избыточном количестве накапливается в ретикулярных, эндотелиальных клетках и макрофагах селезенки, костного мозга, лимфатических узлов, печени. Кроме того, сидеробластами становятся эпителиальные клетки печени, потовых и слюнных желез, легких, почек. Микроскопически в них выявляются гранулы бурого цвета. Внешний вид органов характерен: они приобретают ржавый оттенок. В далеко зашедших случаях
гемосидерин накапливается и в строме органов, и в стенках сосудов. Появляется большое количество сидерофагов, которые не успевают утилизировать гемосидерин, загружающий межклеточное вещество. Одновременно накапливаются "катаболический" ферритин и билирубин. Последний образуется в таком большом количестве, что печень не успевает его утилизировать, и развивается гемолитическая желтуха.
Местный гемосидероз. Развивается при внесосудистом гемолизе в очагах кровоизлияний. Сидеробластами становятся лейкоциты, гистиоциты, ретикулярные клетки, эндотелий и эпителиальные клетки. Из продуктов гемолиза в органах, где возникают кровоизлияния, в цитоплазме этих клеток синтезируются ферритин и гемосидерин. В крупных кровоизлияниях, помимо гемосидерина, образуется еще гематоидин. При этом гемосидерин обычно располагается на периферии кровоизлияний, а гематоидин, для образования которого кислород не нужен, откладывается в центре, в очагах аутолиза. В мелких, чаще диапедезного характера кровоизлияниях обычно образуется только гемосидерин. В участках бывших кровоизлияний сидерофаги сохраняются очень долго, отчего ткани приобретают бурый оттенок.
В клинике большое значение имеет гемосидероз легких, который развивается в результате хронического венозного застоя у больных с заболеваниями сердца на стадии декомпенсации (пороки сердца, кардиосклероз и др.). В легких развиваются многочисленные диапедезные кровоизлияния, в клетках альвеолярного эпителия и в гистиоцитах синтезируются гемосидерин и ферритин. Сидеробласты и сидерофаги "заболачивают" полости альвеол, гипоксия нарастает и в этих условиях начинает синтезироваться SH-ферритин, обладающий, как было сказано выше, вазо-паралитическим действием. Это приводит к еще большему повышению сосудистой проницаемости, нарастанию диапедеза и соответственно накоплению гемосидерина и SH-ферритина. Порочный круг замыкается, и у больных развивается ферритиновый коллапс или шок. Гипоксия, помимо того, стимулирует коллагеносинтетическую активность фибробластов — в легких нарастает склероз, они становятся плотными на ощупь и бурыми за счет накопления гемосидерина. Подобные изменения принято называть "бурая индурация легких" (от лат. induratio — затвердение, уплотнение). Сидеробласты и сидерофаги нередко обнаруживают и в мокроте, которой они придают ржавый оттенок. В таких случаях их называют клетками сердечных пороков.
Гемосидероз является морфологическим субстратом самостоятельного заболевания, которое называется "идиопатический гемосидероз легких", или "синдром Делена — Геллерстедта". Он встречается у детей в возрасте 3—8 лет и характеризуется повторяющимися кровоизлияниями в легочную паренхиму с последующим массивным гемосидерозом и склерозом, кровохарканьем и развитием вторичной железодефицитной анемии. В легких имеется типичная картина бурой индурации, но поражение сердца у больных отсутствует. Причина заболевания до конца неясна. В настоящее время имеется большое количество данных, подтверждающих, что в основе процесса лежит аутоагрессивное поражение легких, при котором реакция антиген — антитело реализуется на сосудах микроциркуляторного русла легких. Иммунологическая природа заболевания подтверждается тем, что при идиопатическом гемосидерозе легких могут поражаться и почки с развитием синдрома Гудпасчера, а в крови больных нередко обнаруживают антитела к ткани легкого и к коровьему молоку.
Гемохроматоз• Гемохроматоз — избыточное накопление гемосидерина, обусловленное нарушением всасывания пищевого железа в тонкой кишке. Таким образом, сущностью гемохроматоза является избыточное содержание гемосидерина, являющегося полимером анаболического ферритина. В то же время морфологически гемохроматоз и общий гемосидероз имеют много общего. Различают гемохроматоз первичный (идиопатический гемохроматоз) и вторичный (сидероз, гемосидероз).
Первичный гемохроматоз. Это самостоятельное заболевание из группы тезаурисмозов (наследственные болезни накопления), обусловленное дефектом ферментов, обеспечивающих всасывание железа в тонкой кишке. Заболевание передается по аутосомно-рецессивному типу. Всасывание пищевого железа повышено и количество его (обмен железа в эритроцитах не нарушен) возрастает в десятки раз. Развивается гемосидероз печени, поджелудочной железы, слюнных и потовых желез, сетчатки глаза, кожи, миокарда, слизистой оболочки кишечника и синовиальных оболочек. Одновременно в органах накапливается ферритин, а в коже и сетчатке глаза — меланин. Классическая триада симптомов первичного гемохроматоза —
бронзовая окраска кожи, сахарный диабет (бронзовый диабет) и пигментный цирроз печени. Нарушение обмена меланина связывают с поражением эндокринных желез, участвующих в процессе синтеза меланина. Особенно резко изменяется печень — она приобретает темно-коричневую окраску, плотная на ощупь, с мелкобугристой поверхностью; микроскопически во всех ее клетках (гепатоцитах, звездчатых ретикулоэндотелиоцитах, гистиоцитах портальных трактов, в эндотелии сосудов и в эпителии желчных капилляров) видно отложение гемосидерина, отмечаются разрастание соединительной ткани и формирование цирроза печени. Иногда гемосидерин откладывается в кардиомиоцитах и развивается "пигментная кардиомиопатия", приводящая к смерти от сердечной недостаточности.
Вторичный гемохроматоз. Развивается в случае приобретенной недостаточности ферментных систем, обеспечивающих всасывание и метаболизм пищевого железа. Подобная ситуация возникает при избыточном поступлении железа с пищей (прием железосодержащих препаратов), алкоголизме, повторных переливаниях крови, после резекции желудка и при гемоглобинопатиях — наследственных заболеваниях, при которых нарушается синтез гема (сидероахрестическая анемия) или глобина (талассемия). В случае вторичного гемохроматоза имеет место двоякий генез нарушения обмена железа: оно накапливается и в сыворотке, и в депо. Типичными являются поражение печени (цирроз), поджелудочной железы (сахарный диабет), сердечной мышцы, что, как правило, оказывается фатальным — больные погибают от сердечной недостаточности.
Желтуха• Желтуха — нарушение обмена билирубина, обусловленное избыточным накоплением его в плазме крови, проявляется желтушным прокрашиванием кожи, склер, слизистых и серозных оболочек и внутренних органов. В зависимости от того, какое звено синтеза пигмента нарушено, различают три вида желтухи: гемолитическую (надпеченочную), печеночную (паренхиматозную), обтурационную (подпеченочную, механическую).
Гемолитическая (надпеченочная) желтуха. Развивается вследствие усиленного внутрисосудистого гемолиза эритроцитов и образования в связи с этим большого количества билирубина. Возникает при интоксикациях (гемолитические яды) и инфекциях (сепсис, малярия, возвратный тиф), переливании несовместимой крови и резус-конфликте, некоторых заболеваниях крови (анемии, гемобластозы). Помимо того, существует группа наследственных болезней, которые проявляются различными дефектами эритроцитов и сопровождаются гемолитической анемией. К ним относят наследственные ферментопатии (микросфероцитоз, овалоцитоз), гемоглобинопатии, или гемоглобинозы (талассемия, или гемоглобиноз F, серповидно-клеточная анемия, или гемоглобиноз S), пароксизмальную ночную гемоглобинурию и др. При всех этих болезнях нарушается первое звено обмена билирубина — захват его гепатоцитами, в крови увеличивается количество не связанного с глюкуроновой кислотой билирубина.
Печеночная (паренхиматозная) желтуха. Возникает при заболеваниях печени (гепатиты острые и хронические, гепатозы, в том числе пигментные, циррозы, поражения печени лекарственные и при аутоинтоксикациях, например при беременности), которые сопровождаются повреждением гепатоцитов, нарушением захвата ими билирубина, конъюгации его и экскреции.
Обтурационная (подпеченочная, механическая) желтуха. Возникает вследствие нарушения оттока желчи по желчным протокам при обтурации их просвета (камень, опухоли) или сдавлении извне (рак головки поджелудочной железы, большого сосочка двенадцатиперстной кишки, метастазы рака в перипортальные лимфатические узлы). В результате нарушается экскреция желчи, и она начинает поступать в кровь через синусоидальный полюс гепатоцита. В последующем нарушается сам процесс синтеза желчи, она становится "белой". Особенно тяжелые изменения при механической желтухе развиваются в печени: в результате холестаза она увеличивается в размерах, приобретает желтовато-зеленый цвет; внутрипеченочные желчные протоки расширены, переполнены желчью. Гистологически желчный пигмент выявляется всюду: в желчных протоках, желчных капиллярах, в печеночных клетках. В условиях холестаза быстро развивается холангит.
