Клиническая фармакология и фармакотерапия: учебник. - 3-е изд., перераб. и доп. / под ред. В. Г. Кукеса, А. К. Стародубцева. - 2012. - 840 с.: ил.
|
|
|
|
Глава 4. КЛИНИЧЕСКАЯ ФАРМАКОКИНЕТИКА
Ответить на вопрос, как ЛС будет действовать на организм человека, невозможно без информации о том, как этот препарат усваивается организмом, распределяется в органах и тканях, а в последующем разрушается и выводится. От каждого из этих процессов зависит выраженность и продолжительность эффекта ЛС, кроме того, его излишнее накопление может быть причиной НЛР.
Существует четкая связь между концентрацией препарата в крови, других тканях организма и его эффектом. Для большинства ЛС определена так называемая терапевтическая концентрация, при которой препарат оказывает оптимальное лечебное действие. В середине ХХ в. появилась возможность измерять концентрации препаратов в крови больного. Это позволяет выбрать оптимальную индивидуальную дозу и избежать нежелательных (токсических) эффектов, связанных с излишним накоплением препарата в организме.
Изучением процессов, которые происходят с препаратом в организме больного, занимается клиническая фармакокинетика (от греч. pharmakon - лекарственное вещество и kinein - движение) - раздел клинической фармакологии, изучающий пути поступления, биотрансформацию, связь с белками плазмы и других тканей организма, распределение и выведение ЛС.
4.1. ОСНОВНЫЕ ФАРМАКОКИНЕТИЧЕСКИЕ ПАРАМЕТРЫ
Измерить концентрацию ЛС непосредственно в ткани органа (например, антиаритмического препарата в сердечной мышце или диуретика в тканях почек) у человека обычно невозможно. Однако, зная концентрацию препарата в крови, можно с высокой точностью предсказать его концентрацию непосредственно в области рецепторов. Именно поэтому клиническая фармакокинетика изучает преимущественно концентрации препаратов в плазме крови, хотя иногда определяют концентрацию ЛС и в других жидкостях орга-
низма, например в моче или мокроте. Определить концентрацию ЛС в плазме крови можно при помощи жидкостной или газожидкостной хроматографии, радиоиммунологического, ферментохимического или спектрофотометрического анализа. Проведя серию измерений концентрации ЛС в плазме крови через определенные промежутки времени, можно построить график «концентрация - время», получивший название фармакокинетической кривой.
ЛС, попадающие в организм человека, подвергаются абсорбции (проникают из просвета ЖКТ в кровь), затем распределяются по организму, попадая в различные органы и ткани, разрушаются под воздействием специализированных ферментов (метаболизма) и выводятся в неизмененном виде или в виде метаболитов (экскреции). На этом основании выделяют фазы абсорбции, распределения и экскреции, хотя обычно эти три процесса протекают практически одновременно: едва поступив в организм, часть препарата сразу же подвергается метаболизму и выводится.
В большинстве случаев скорость всех этих процессов пропорциональна концентрации препарата, например чем больше доза принятого ЛС, тем быстрее нарастает его концентрация в плазме крови (рис. 4-1). Скорость метаболизма и экскреции также зависит от концентрации препарата. Процессы абсорбции, распределения и экскреции подчиняются закону действующих масс, согласно которому скорость химической реакции или процесса пропорциональна массе реагирующих веществ.
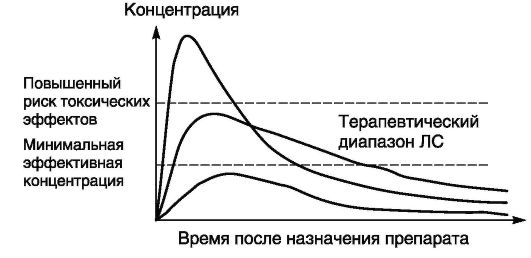
Рис. 4-1. Формы фармакокинетических кривых при приеме препарата внутрь
КЛИНИЧЕСКАЯ ФАРМАКОКИНЕТИКА
Процессы, скорость которых пропорциональна концентрации, получили название процессов первого порядка. При этом скорость элиминации ЛС пропорциональна его концентрации и соответствует кинетике первого порядка. Большинство ЛС подчиняются законам кинетики первого порядка. Скорость процессов (метаболизма или элиминации) при этом непостоянна во времени, но пропорциональна концентрации препарата, а график «концентрация - время» представляет собой кривую: чем выше концентрация ЛС, тем быстрее его метаболизм и выведение из организма (рис. 4-2).
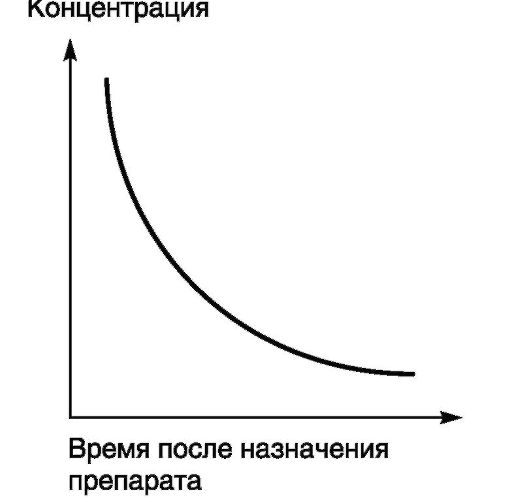
Рис. 4-2. Фармакокинетическая кривая (кинетика первого порядка)
Если ЛС подчиняется законам кинетики первого порядка, при увеличении его дозы (например, в 2 раза) происходит пропорциональное увеличение концентрации препарата в плазме, а период времени, за которое концентрация ЛС снижается наполовину (период полувыведения), - постоянная величина.
Если скорость элиминации не зависит от концентрации препарата (например, скорость метаболизма ЛС ограничена количеством участвующего в этом процессе фермента), то элиминация происходит в соответствии с кинетикой нулевого порядка (кинетика насыщения). При этом скорость выведения препарата постоянна, а график «концентрация - время» представляет собой прямую. Кинетика нулевого порядка характерна для алкоголя, фенитоина и нестероидных противовоспалительных средств (НПВС) в высоких дозах. Так, этанол
(алкоголь) в организме человека трансформируется в ацетальдегид при участии дегидрогеназ. Этот процесс происходит в соответствии с кинетикой первого порядка. Однако если концентрация этанола в крови превышает 100 мг/л, наступает насыщение ферментов и скорость его метаболизма больше не изменяется по мере увеличения концентрации в крови. Таким образом, при высоких концентрациях алкоголя его элиминация подвержена кинетике нулевого порядка.
Порядок кинетики представляет собой взаимосвязь между скоростью элиминации и концентрацией ЛС. При кинетике нулевого порядка за равные промежутки времени из организма выводится одинаковое количество препарата (например, по 20 мг в час), а при кинетике первого порядка - одинаковая доля препарата (например, по 20% каждый час).
После однократного внутривенного введения ЛС его концентрация в крови быстро (в течение нескольких секунд) повышается. Затем концентрация быстро снижается путем перераспределения ЛС в тканях и жидкостях организма (фаза распределения), которое сменяется более медленным снижением концентрации в процессе экскреции препарата (фаза элиминации) (рис. 4-3).
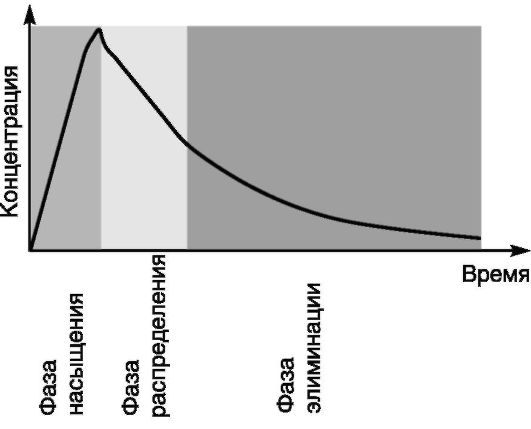
Рис. 4-3. Динамика концентрации препарата в крови после внутривенного введения
Для анализа особенностей фармакокинетики используют условную модель, в которой организм представлен в виде камеры. ЛС поступает в эту камеру (равномерно распределяясь по всему ее объему) и затем постепенно выводится согласно законам кинетики
первого порядка. Понятие камеры условно, так как за ним не стоит какое-либо анатомически ограниченное пространство. В некоторых случаях для фармакокинетических расчетов применяют многокамерные модели. При этом за центральную (обычно меньшую) камеру принимают плазму крови и органы с хорошим кровоснабжением (сердце, легкие, печень, почки, эндокринные железы), а за периферическую - органы и ткани (мышцы, кожу, жировую ткань) с низкой скоростью кровотока.
В однокамерной модели после введения ЛС начинается его элиминация согласно законам кинетики первого порядка. Снижение концентрации препарата на 50% происходит за равные промежутки времени, получившие название периода полуэлиминации ЛС в плазме (Т1/2) (рис. 4-4). Период полуэлиминации ЛС - наиболее важный из математических параметров, с помощью которых описывают фар-макокинетику и рассчитывают концентрацию препарата.
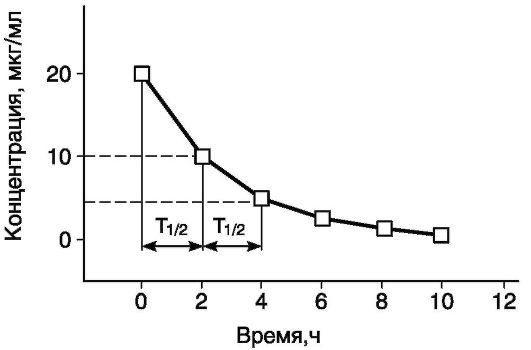
Рис. 4-4. Период полувыведения
Несколько другая картина отмечена при продолжительной внутривенной инфузии или после повторных назначений ЛС (как внутривенно, так и внутрь). В этом случае концентрация препарата повышается линейно при длительной инфузии (рис. 4-5) или скачкообразно при многократных назначениях (рис. 4-6). Концентрация ЛС увеличивается до тех пор, пока не будет достигнуто равновесие между поступлением препарата и скоростью его элиминации. Такое состояние (поступление препарата в организм равно его элиминации) называют равновесным. При назначении препарата в виде отдельных
доз колебания концентрации сохраняются и при равновесном состоянии, но средняя концентрация остается неизменной.
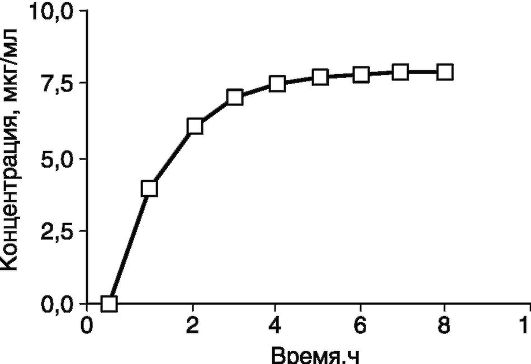
Рис. 4-5. Концентрация препарата в плазме при длительной инфузии

Рис. 4-6. Достижение равновесной концентрации ЛС при многократном приеме
Для достижения равновесной концентрации требуется время, равное примерно пяти периодам полуэлиминации. Время достижения равновесной концентрации зависит только от величины Т1/2 и не зависит ни от дозы ЛС, ни от частоты его назначения. При применении различных доз одного и того же препарата равновесие
наступает в одно и то же время, хотя равновесные концентрации различаются.
Равновесная концентрация ЛС имеет большое практическое значение, она обеспечивает постоянство фармакологического эффекта ЛС. Зная величину Т1/2, можно не только рассчитать время наступления равновесного состояния, но и предсказать снижение концентрации препарата в плазме после прекращения его введения. Препараты с малым Т1 / 2 (несколько минут) имеют высокую управляемость: уже спустя 10 мин после отмены добутамина или лидокаина их концентрация в плазме становится ничтожной, и действие прекращается. Очевидно, что назначать эти ЛС можно только в виде постоянных внутривенных инфузий. Препараты с длительным периодом полуэлиминации (фенобарбитал - 85 ч, дигитоксин - 150 ч, амиода-рон - 700 ч) сохраняют свои эффекты даже через несколько суток после прекращения введения, что следует учитывать при их назначении. В частности, после отмены барбитуратов или бензодиазепинов несколько суток сохраняются снижение внимания и сонливость, соответственно в этот период больные должны отказаться от управления автомобилем и выполнения работы, требующей повышенного внимания.
Если после достижения равновесного состояния потребовалось увеличить или уменьшить дозу ЛС, равновесие нарушается. Концентрация препарата в плазме изменяется (уменьшается или увеличивается) до тех пор, пока равновесие не будет достигнуто вновь, но уже на другом уровне концентрации. Для достижения нового равновесия также требуется время, равное пяти периодам полуэлиминации препарата. Естественно, что быстрая реакция организма больного на увеличение или уменьшение дозы возможна только при коротком периоде полуэлиминации ЛС (хорошо управляемые препараты).
Иногда равновесная концентрация может изменяться, даже если режим дозирования ЛС не был изменен. В частности, при применении аминогликозидных антибактериальных препаратов возможно развитие почечной недостаточности (побочный эффект ЛС этой группы), при этом скорость элиминации ЛС уменьшается, а их концентрация в плазме возрастает (как и токсическое действие). Известны ЛС, которые вызывают индукцию (усиление активности) или ингибирование (подавление активности) ферментов микросомального окисления в печени. Например, на фоне применения циметидина или эритромицина (ингибиторы цитохрома Р-450) концентрация теофиллина в плазме может существенно увеличиваться.
Показатель Т1 / 2 - один из важнейших фармакокинетических параметров. Основываясь на величине Т1 /2, можно рассчитать время наступления равновесного состояния, время полной элиминации препарата или предсказать концентрацию ЛС в любой момент (если препарат обладает кинетикой первого порядка).
Однако существуют и другие фармакокинетические параметры, речь о которых пойдет ниже.
• Максимальная концентрация (Сmах). Применение препарата безопасно только тогда, когда величина Сmах находится в пределах терапевтического диапазона данного ЛС.
• Время наступления максимальной концентрации (Тmах) часто (но не всегда) совпадает с максимумом фармакологического действия ЛС при однократном назначении.
• Площадь под фармакокинетической кривой (AUC) - величина, пропорциональная общему количеству препарата в системном кровотоке.
• Среднее время удержания препарата в организме (МRТ).
• Биодоступность (F) - доля препарата (процент общей дозы), достигшая системного кровотока.
При внутривенном введении весь препарат достигает системного кровотока, о биодоступности говорят лишь тогда, когда препарат назначают каким-либо другим путем (внутрь, внутримышечно, ректально). Эта величина определяется как отношение AUC после внесосудистого введения к AUC после внутривенного введения: F = (AUC внутрь/AUC внутривенно) ? 100%, где F - биодоступность, AUC - площадь под фармакокинетической кривой.
• Общий клиренс (С1) - объем плазмы или крови, который полностью очищается от препарата в единицу времени. Этот параметр отражает элиминацию препарата из организма и выражается в миллилитрах в минуту или в литрах в час.
Клиренс можно выразить так: С1 = D/AUC, где D - доза, AUC - площадь под фармакокинетической кривой.
ЛС выводятся в основном почками и печенью, и общий клиренс представляет собой главным образом сумму почечного и печеночного клиренса (под печеночным клиренсом подразумевается метаболический клиренс в печени и выведение препарата с желчью). Так, почечный клиренс циметидина составляет примерно 600 мл/мин, метаболический - 200 мл/мин, желчный - 10 мл/мин, следовательно, общий клиренс равен 810 мл/мин. Другие пути выведения или внепеченочный метабо-
лизм не имеют существенного практического значения и при расчете общего клиренса их во внимание обычно не принимают.
Величину клиренса в основном определяют функциональное состояние важных систем организма, а также объем и скорость кровотока в органе. Например, клиренс лидокаина, который подвергается интенсивному действию ферментов печени, зависит прежде всего от скорости его доставки к печени (от объема притекающей к печени крови). При снижении печеночного кровотока на фоне сердечной недостаточности клиренс лидокаина снижается. В то же время клиренс других препаратов может зависеть в основном от функционального состояния метаболизирующих ферментов. При поражении печени клиренс многих ЛС резко снижается, а концентрация в крови возрастает.
• Объем распределения (Vd) - это гипотетический объем жидкости организма, необходимый для равномерного распределения всей введенной дозы ЛС в концентрации, аналогичной концентрации в плазме крови.
Таким образом:
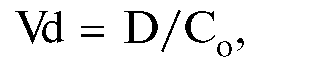
где D - доза, Со - начальная концентрация.
Высокие значения объема распределения свидетельствуют о том, что препарат максимально проникает в биологические жидкости и ткани. Если ЛС активно связывается (например, жировой тканью), его концентрация в крови может быть очень низкой, а объем распределения будет достигать нескольких сотен литров, намного превышая реальный объем организма человека. Из-за этого Vd также называют кажущимся объемом распределения. На основании объема распределения можно рассчитать нагрузочную дозу, необходимую для создания эффективной концентрации ЛС в крови (чем больше Vd, тем большей должна быть нагрузочная доза: D = Vd-C).
Объем распределения зависит от многих факторов (молекулярная масса ЛС, его ионизация и полярность, растворимость в воде и жирах). Возраст, пол, беременность пациентов, общее количество жира в организме также влияют на величину объема распределения. Объем распределения изменяется при некоторых патологических состояниях, особенно при заболеваниях печени, почек и сердечнососудистой системы.
Существует взаимосвязь между периодом полуэлиминации, объемом распределения и общим клиренсом, которая выражается формулой:
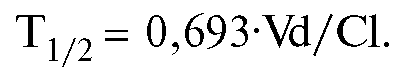
Уровень равновесной концентрации (Css) также можно рассчитать математически. Эта величина прямо пропорциональна дозе ЛС [вернее, произведению дозы на биодоступность (F) - реальному количеству препарата, поступившему в организм], величине T1/2-Css и обратно пропорциональна объему распределения:
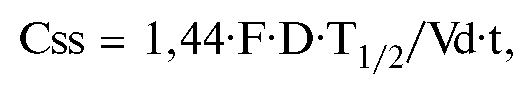
где t - интервал времени.
4.2. КОНТРОЛЬ КОНЦЕНТРАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ В КЛИНИЧЕСКОЙ ПРАКТИКЕ
Представление о фармакокинетических параметрах ЛС позволяет предсказать концентрацию ЛС в плазме в любой момент времени, но в ряде случаев полученные расчеты могут оказаться неточными. Например, больной неаккуратно принимал назначенное ЛС (пропуски приема, ошибки в дозах) или существуют факторы, влияющие на концентрацию ЛС, значение которых не поддается математическому моделированию (одновременный прием нескольких препаратов, различные заболевания, способные изменять показатели фармакокинетики). Из-за этого часто приходится прибегать к экспериментальному исследованию концентрации ЛС в крови.
Необходимость экспериментальных исследований также возникает при внедрении в клиническую практику новых ЛС или их форм, а также при исследовании биоэквивалентности препаратов различных производителей.
В клинической практике к измерению концентрации ЛС прибегают только в некоторых случаях.
• Когда концентрация в плазме четко коррелирует с клиническим эффектом ЛС, но его эффективность трудно оценить клинически. Например, если препарат назначен для профилактики редких проявлений заболевания (эпилептический припадок или пароксизм аритмии). При этом более целесообразно однократно оценить уровень концентрации ЛС, чем ожидать клинического эффекта или неудачи лечения неопределенно
долгое время. Иногда оценка клинического эффекта может быть затруднена из-за неадекватного контакта с больным.
• Когда трудно отличить клиническое и нежелательное действие одного и того же препарата. Например, дигоксин, назначенный для профилактики аритмий, при превышении терапевтической концентрации сам способен вызвать у больного аритмию. В этом случае тактика дальнейшего лечения (отмена дигоксина или увеличение его дозы для достижения большего противоаритмиче-ского эффекта) полностью зависит от концентрации препарата в крови.
• При наличии у препарата потенциально опасных побочных эффектов (аминогликозиды, цитостатики).
• При отравлениях и передозировке ЛС (для оценки тяжести и выбора тактики лечения).
• При нарушениях, связанных с метаболизмом или элиминацией ЛС [печеночная или хроническая почечная недостаточность
(ХПН)].
Необходимость в исследовании концентрации ЛС отсутствует в следующих ситуациях:
• в тех случаях, когда ЛС представляется вполне безопасным и обладает большим терапевтическим диапазоном;
• если эффект ЛС легко поддается клинической оценке;
• если эффект ЛС мало зависит от концентрации и /или продолжается длительное время после того, как препарат полностью выводится из плазмы [гормональные препараты, некоторые средства, используемые для лечения рака, ингибиторы моноаминоксидазы (МАО) и ацетилхолинэстеразы];
• если действие ЛС происходит путем образования активных метаболитов;
• у ЛС, для действия которых более важна их тканевая концентрация (некоторые антибактериальные препараты).
В настоящее время существует возможность оценить эффективность лечения исходя из концентрации ЛС в моче (антибактериальные препараты при мочевой инфекции), мокроте, а также определить концентрацию ЛС непосредственно в тканях и органах человека радионуклидными методами. Однако эти способы исследования фармакокинетики используют только в научных исследованиях и пока не вводят в клиническую практику.
4.3. ФАКТОРЫ, ВЛИЯЮЩИЕ НА ВСАСЫВАНИЕ,
РАСПРЕДЕЛЕНИЕ И ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ
СРЕДСТВ
Общая скорость всасывания зависит от морфологической структуры органа, в который вводят ЛС, и прежде всего от величины абсорбирующей поверхности. Наибольшую абсорбирующую поверхность имеет ЖКТ благодаря ворсинкам (около 120 м2), несколько меньшую - легкие (70-100 м2). Кожа имеет малую абсорбирующую поверхность (в среднем 1,73 м2), кроме того, всасывание ЛС через кожу затруднено из-за особенностей ее анатомического строения.
Для большинства препаратов проникновение в область рецепторов связано с прохождением нескольких барьеров:
• слизистую оболочку кишечника (или полости рта при сублинг-вальном приеме), эпителий кожи (при наружном применении препарата), эпителий бронхов (при ингаляциях);
• стенку капилляров1;
• Специфические капиллярные барьеры2:
- между системным кровотоком и системой кровоснабжения головного мозга (гематоэнцефалический барьер);
- между организмом матери и плода (плацента3).
Некоторые препараты взаимодействуют со своими рецепторами на поверхности клеток, другие должны преодолеть клеточную мембрану (глюкокортикоиды), мембрану ядра (фторхинолоны) или мембраны клеточных органелл (макролиды).
Состояние сердечно-сосудистой системы - определяющий фактор в распределении ЛС. Так, при шоке или сердечной недостаточности кровоснабжение большинства органов уменьшается, что ведет
1 Капилляры - мельчайшие кровеносные сосуды, через которые главным образом и происходят обмен веществ и поступление ЛС в ткани и органы человека. Препараты попадают в системный кровоток через капиллярную сеть кишечника, бронхов (ингаляционный путь введения), полости рта (при сублингвальном применении), кожи (трансдермальный путь введения) и подкожной жировой клетчатки (внутримышечный путь введения). Для достижения органа-мишени ЛС должно вновь преодолеть стенку капилляра.
2 Эти барьеры образованы двойной системой капилляров, например кровь, поступающая в головной мозг, распределяется по капиллярам, из которых кислород и питательные вещества не поступают напрямую к клеткам, а адсорбируются в другую (внутреннюю) капиллярную систему.
3 Со способностью ЛС проникать через плаценту связано, как правило, нежелательное действие препаратов на плод.
к снижению почечного и печеночного клиренса ЛС. В результате концентрация ЛС в плазме крови, особенно после внутривенного введения, будет возрастать.
ЛС способны преодолевать клеточные оболочки, не нарушая их целостности, с помощью ряда механизмов.
• Диффузия - пассивный транспорт ЛС в ткани под воздействием градиента концентраций. Скорость диффузии всегда пропорциональна разнице между концентрациями ЛС снаружи и внутри клетки и подчиняется законам кинетики первого порядка. Процесс диффузии не требует энергетических затрат. Однако преодолеть клеточные оболочки, состоящие из гидрофобных липидов, способны только жирорастворимые ЛС.
• Фильтрация позволяет ЛС поступать в организм через особые водные каналы в эпителиальных оболочках. Путем фильтрации в организм поступают только некоторые водорастворимые ЛС.
• Активный транспорт - перемещение некоторых ЛС в организме независимо от градиента концентраций (при этом используется энергия АТФ). Активный транспорт может происходить быстрее, чем диффузия, но это потенциально насыщаемый механизм: молекулы сходного химического строения конкурируют между собой за ограниченное число молекул-переносчиков. С использованием этого механизма в организм поступают только те ЛС, которые по химическому строению близки к естественным веществам (препараты железа, фторурацил).
Для абсорбции и транспорта ЛС в организме имеют значение растворимость, химическая структура и молекулярная масса ЛС. Переход препарата через клеточную оболочку определяется в первую очередь его растворимостью в липидах. Растворимость в жирах - свойство всей молекулы в целом, хотя ионизация молекулы ЛС способна уменьшать ее липофильность. Растворимость в воде увеличивается при наличии в ЛС спиртовой группы (-ОН), амидной группы (-CO-NH2), карбоксильной группы (-СООН), конъюгатов с глюкуроновым радикалом и конъюгатов с сульфатным радикалом. Растворимость в липидах увеличивается при наличии в молекуле ЛС бензольного кольца, стероидного ядра, галогеновых групп (-Вг, -С1, -F). Способность молекулы к ионизации характеризуется константой ионизации (Ка), которую выражают в виде отрицательного логарифма (рКа). При рН раствора, равном рКа, 50% вещества находится в ионизированном состоянии.
Особенности выведения ЛС также могут быть связаны со степенью ионизации: рН мочи может варьировать в значительных пределах
(от 4,6 до 8,2), обратное всасывание ЛС из первичной мочи1 в значительной степени зависит от ее рН. В частности, ацетилсалициловая кислота становится более ионизированной при щелочном рН мочи и в этом случае почти не подвергается реабсорбции. Это обстоятельство используют при лечении передозировки салицилатами: в этом случае назначают ЛС, увеличивающие рН мочи, что способствует более быстрому выделению салицилатов.
Некоторые ЛС (например, дигоксин и хлорамфеникол) вообще не имеют ионизируемых групп, и их транспорт не зависит от рН среды, другие (гепарин натрия) обладают химической структурой с настолько выраженной ионизацией, что остаются ионизированными практически при любых значениях рН. Некоторые патологические состояния способны изменять внутреннюю среду организма, например среда в полостях абсцессов кислая, что может повлиять на эффективность антибактериальных препаратов с высокой гидро-фильностью.
4.4. ПУТИ ВВЕДЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Стремление влиять на параметры кинетики препаратов отразилось в многообразии путей введения ЛС. Применяя различные пути введения, можно:
- обеспечить разную скорость развития эффекта и его различную продолжительность у одного и того же ЛС;
- значительно увеличить концентрацию ЛС в органе-мишени (например, при применении бронхорасширяющих препаратов в ингаляциях);
- увеличить системную концентрацию ЛС при внутривенном введении или ректальном применении по сравнению с приемом внутрь (для ЛС с эффектом первого прохождения через печень);
- уменьшить выраженность НЛР (наружное применение глю-кокортикоидов, парентеральное введение ЛС, раздражающих слизистую оболочку желудка).
1 В структурной единице почек - нефроне - первоначально образуется большое количество так называемой первичной мочи (до 150 л/сут), состав которой (за исключением белков) близок к составу плазмы крови. Большая часть этой жидкости с растворенными в ней веществами подвергается обратному всасыванию (реабсорбция) в канальцах нефрона.
Энтеральное введение ЛС. К энтеральному пути введения ЛС относится прием препаратов внутрь, буккальный и ректальный путь введения. При этом объем и скорость всасывания ЛС из ЖКТ зависит, с одной стороны, от физико-химических свойств препаратов (водо- и жирорастворимости, константы диссоциации, молекулярной массы), особенностей лекарственной формы (препараты с медленным высвобождением), а с другой - от функционального состояния ЖКТ (рН и присутствия пищеварительных ферментов в просвете кишечника, скорости перемещения пищи, кровотока в стенке кишечника). Кроме того, некоторым ЛС свойствен метаболизм в стенке кишечника или под действием кишечной микрофлоры. Некоторые ЛС при одновременном назначении могут взаимодействовать в ЖКТ между собой (инактивация одного ЛС другим или конкуренция за всасывание).
Прием препаратов внутрь. Преимущества этого пути введения заключаются в простоте и удобстве для пациента. Обычно антибактериальные препараты рекомендуют принимать до еды (абсорбция многих из них зависит от пищи), гипогликемические средства назначают до еды или во время еды, препараты, раздражающие слизистую оболочку желудка (НПВС), - после еды.
Недостатки приема ЛС внутрь:
- абсорбция многих ЛС зависит от приема пищи, функционального состояния ЖКТ и множества других факторов, которые на практике с трудом поддаются учету;
- не все ЛС способны хорошо всасываться в ЖКТ;
- некоторые ЛС (препараты инсулина, антибактериальные препараты пенициллинового ряда) разрушаются в желудке;
- часть ЛС оказывает нежелательные действия на ЖКТ - вызывают изъязвления (НПВС, доксициклин, калия хлорид) или отрицательно влияют на моторику желудка и кишечника (некоторые антациды);
- наконец, ЛС нельзя назначать внутрь больным в бессознательном состоянии и пациентам с нарушением глотания.
На абсорбцию (всасывание) ЛС при приеме внутрь влияют следующие факторы.
Моторика ЖКТ, от которой зависит продолжительность пребывания ЛС в его различных отделах. Так, у пациентов с мигренью моторика желудка замедлена, его опорожнение наступает позже, чем в норме. В результате этого при приеме НПВС у этих больных снижается абсорбция, а эффекты НПВС становятся отсроченными.
Эту проблему можно преодолеть, если одновременно с НПВС назначить средство, повышающее моторику желудка, - метоклопрамид.
Кислотность в желудке способна изменяться в довольно широких пределах, влияя на абсорбцию ЛС. Например, слабые органические основания (эритромицин, хинидин, теофиллин) в кислой среде подвергаются ионизации, препятствующей их всасыванию. Такие ЛС лучше принимать натощак и /или запивать слабощелочными растворами.
У больных с высокой кислотностью желудочного сока замедляется опорожнение желудка, что также влияет на всасывание препаратов. В этом случае перед приемом ЛС можно назначать вещества, нейтрализующие избыточную кислотность (молоко, минеральные воды). При антацидном (сниженная кислотность) состоянии опорожнение желудка наступает быстро и ЛС быстрее поступают в тонкую кишку.
Ферменты в просвете кишечника. В кишечнике находится большое количество ферментов с высокой липолитической и протеолити-ческой активностью. Ряд ЛС белковой и полипептидной природы, гормональные препараты (десмопрессин, кортикотропин, инсулины, прогестерон, тестостерон) в этих условиях почти полностью дезактивируются. Компоненты желчи способствуют растворению липофиль-ных препаратов, а также растворяют оболочки таблеток и капсул с кишечно-растворимым покрытием.
Пища. При одновременном приеме пищи и ЛС адсорбция препаратов может замедляться или ускоряться. Например, яйца уменьшают всасывание железа; молоко, богатое ионами кальция, инактивиру-ет тетрациклин и фторхинолоны, образуя с их молекулами хелат-ные комплексы. Абсорбция изониазида, леводопы и эритромицина уменьшается независимо от характера пищи. При приеме синтетических пенициллинов после еды их всасывание замедляется, а всасывание пропранолола, метопролола и гидралазина, напротив, ускоряется (но абсорбция и биодоступность остаются прежними). Всасывание гризеофульвина увеличивается в несколько раз при приеме жирной пищи.
Некоторые ЛС, особенно при длительном применении, могут нарушать всасывание ряда ингредиентов пищи и в итоге вызывать различные патологические состояния. Так, гормональные оральные контрацептивы нарушают всасывание фолиевой и аскорбиновой кислот, рибофлавина, антикоагулянты непрямого действия подавляют
всасывание витамина К, слабительные средства - всасывание жирорастворимых витаминов и т.д.
Лекарственная форма. Скорость и полнота всасывания ЛС в ЖКТ зависят также от лекарственной формы. Лучше всего всасываются растворы, затем следуют суспензии, капсулы, простые таблетки, таблетки в оболочке и, наконец, лекарственные формы с замедленным высвобождением. ЛС любой формы лучше всасывается, если его принимают через 2-3 ч после еды и запивают 200-250 мл воды.
Иногда внутрь назначают ЛС, которые почти не всасываются в ЖКТ (аминогликозидные антибиотики, противогельминтные ЛС). Это позволяет лечить некоторые заболевания кишечника, избегая нежелательных системных эффектов препаратов.
Буккальное применение ЛС. Слизистая оболочка рта активно кровос-набжается, и при применении препаратов буккально (или сублинг-вально) действие ЛС начинается быстро. При таком пути введения препарат не вступает во взаимодействие с желудочным соком, скорость всасывания не зависит от приема пищи или одновременного назначения других ЛС, кроме того, препараты, всасывающиеся в полости рта, не подвержены пресистемному метаболизму1.
Спектр ЛС, применяемых буккально, невелик и включает в себя нитроглицерин и изосорбида динитрат (при стенокардии), нифеди-пин, каптоприл и клофелин (при гипертоническом кризе) и эрго-тамин (при мигрени). Действие препарата можно прервать в любой момент.
Ректальное назначение ЛС. Кровь от нижних отделов прямой кишки также поступает в системный кровоток, минуя печень. Этот путь введения используют для препаратов с высоким пресистемным метаболизмом. Кроме того, ректально назначают некоторые ЛС, раздражающие слизистую оболочку желудка (НПВС). К ректальному введению препаратов прибегают при рвоте, морской болезни, у детей грудного возраста. Дозы ЛС при ректальном применении, как правило, равны (или незначительно превосходят) дозы для приема внутрь. Ректально также назначают ЛС для местного лечения (при заболеваниях прямой кишки).
1 Кровь, оттекающая от желудка и кишечника (исключая прямую кишку), собирается в воротную вену, в результате чего весь объем ЛС, принятого внутрь, первоначально проходит через печень, где может подвергнуться пресистемному (до поступления в системный кровоток) метаболизму. Из-за этого ЛС с преимущественным метаболизмом в печени не следует назначать внутрь. От слизистой оболочки рта кровь, минуя печень, поступает сразу в системный кровоток (через верхнюю полую вену).
Недостатки этого пути введения заключаются в неприятных для пациента психологических моментах, кроме того, всасывание может замедляться, если прямая кишка содержит каловые массы.
Парентеральное введение ЛС. К парентеральному пути введения ЛС относят внутрисосудистое, внутримышечное, подкожное введение препаратов, кроме того, ингаляционное, эндотрахеальное введение, местное применение ЛС и трансдермальные системы.
Внутрисосудистое (обычно внутривенное) введение ЛС обеспечивает быстрое поступление ЛС в кровь, быстрое создание высокой системной концентрации и возможность управлять ей. Таким путем можно назначать ЛС, разрушающиеся в ЖКТ (пенициллины, инсулины), раздражающие ЖКТ или не всасывающиеся в нем (аминогликозид-ные антибиотики). Внутрисосудисто вводят большинство препаратов для лечения неотложных состояний. К недостаткам этого пути введения относят технические сложности сосудистого доступа, риск развития инфекции в месте инъекций, быстрое нарастание концентрации препарата, тромбозы вен в месте введения ЛС (эритромицин) и болевые ощущения (калия хлорид).
Препараты с длительным периодом элиминации вводят струй-но (болюсно), с коротким периодом полуэлиминации (лидокаин, окситоцин) - в виде длительных инфузий. Некоторые ЛС способны адсорбироваться на стенках систем для переливания (инсулин).
Внутримышечное введение. При внутримышечном введении всасывание препарата в кровь занимает около 10-30 мин. Принципиальных преимуществ этот путь введения ЛС не имеет. Следует помнить о риске развития местных осложнений (абсцессы), особенно при использовании концентрированных растворов препаратов.
Подкожно вводят препараты инсулина и гепарин натрия. После соответствующего обучения больной может делать инъекции самостоятельно. Повторные инъекции инсулинов вызывают атрофию жировой ткани в месте введения, что сказывается на скорости всасывания ЛС.
Ингаляционно назначают препараты для лечения заболеваний легких и бронхов. Ингаляционный путь обеспечивает быстрое начало действия этих ЛС и их высокую концентрацию в области рецепторов. Биодоступность большинства ЛС при этом способе введения не превышает 15-40% (из-за всасывания ЛС в полости рта и со слизистой оболочки крупных бронхов). Это обстоятельство позволяет ослабить нежелательные системные эффекты бронхолитиков и глюкокорти-коидов.
Эндотрахеально ЛС назначают в реанимационной практике. Ряд ЛС (эпинефрин, атропин, налоксон) можно вводить больному в критическом состоянии через интубационную трубку, не дожидаясь создания внутрисосудистого доступа. Эти ЛС хорошо и очень быстро всасываются в трахее, а эндотрахеальное введение не уступает по скорости развития эффекта внутривенному.
Кроме вышеперечисленных способов введения, иногда ЛС назначают местно (при лечении кожных, глазных, гинекологических заболеваний). Некоторые ЛС (нитраты, препараты для лечения морской болезни, половые гормоны) выпускают в виде пластырей с медленным трансдермальным высвобождением действующего вещества.
4.5. РАСПРЕДЕЛЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
В ОРГАНИЗМЕ
ЛС циркулируют в плазме крови частично в свободном виде, а частично в связанном с транспортными белками1. При этом фармакологически активна только фракция, не связанная с белками. Свободная и связанная фракции находятся в состоянии равновесия: молекулы ЛС быстро (Т1 / 2 связи ЛС с молекулой альбумина составляет около 20 мс) переходят из одной фракции в другую.
Основной белок плазмы крови, связывающий ЛС (главным образом со свойствами кислот), - альбумин. Он обладает отрицательным зарядом. Альбумина в плазме настолько много, что полное насыщение каким-либо ЛС всех молекул альбумина происходит очень редко. Например, для насыщения всех белковых связей феноксиметилпе-нициллином этот препарат нужно вводить в чрезвычайно высоких дозах - 50-100 млн ЕД/сут2. Насыщение связи с альбумином может быть актуальным при применении клофибрата® и дизопирамида®.
Помимо альбумина, за связь с ЛС отвечают липопротеины и а1-кис-лый гликопротеин (с этими переносчиками связываются ЛС, имеющие свойства оснований). Концентрация гликопротеина увеличивается при стрессе, ИМ и некоторых других заболеваниях. Некоторые ЛС связываются с поверхностью эритроцитов и других форменных элементов крови (хинидин, аминазин).
1 Транспортные белки плазмы переносят кортизон, дигоксин, железо, медь и многие другие вещества.
2 Стандартная доза феноксиметилпенициллина при лечении тяжелых инфекций не превышает 12 млн ЕД.
Функцию связывающих веществ могут выполнять практически все белки, а также форменные элементы крови. Набор связывающих компонентов в тканях еще больше. ЛС могут связываться с одним или несколькими белками. Например, тетрациклин на 14% связывается с альбуминами, на 38% - с различными липопротеинами и на 8% - с другими белками сыворотки крови. Обычно, когда идет речь о связывании ЛС с белками плазмы, имеется в виду суммарная связь данного вещества с белками и другими фракциями сыворотки.
Ряд тканевых структур также активно связывает определенные химические вещества. Например, ткань щитовидной железы накапливает соединения йода и меди, костная ткань - тетрациклины и т.д.
Чаще всего белок выполняет функцию депо и участвует в регуляции баланса между связанным препаратом и его активной формой. Каждая удаленная из циркуляции (связь с рецептором, выведение из организма) молекула активного препарата возмещается путем диссоциации очередного белкового комплекса. Однако если сродство препарата к белкам и жирам тканей выше, чем к белкам плазмы, то его концентрация в плазме низкая, а в тканях высокая. В частности, некоторые антибактериальные препараты накапливаются в тканях в большей (5-10 раз и более) концентрации, чем в плазме (макролиды, фторхи-нолоны). Многие НПВС (диклофенак, фенилбутазон) имеют высокое сродство к белкам синовиальной жидкости, и уже через 12 ч после введения они практически отсутствуют в плазме крови, а их концентрация в ткани сустава остается на высоком уровне.
Связывание ЛС с белками крови может изменяться при нарушении функций почек, печеночной недостаточности, некоторых формах анемии и при снижении концентрации альбумина в плазме.
4.6. МЕТАБОЛИЗМ ЛЕКАРСТВЕННЫХ СРЕДСТВ
ЛС, как и другие чужеродные вещества, независимо от своей структуры могут подвергаться биотрансформации. Биологическая цель этого процесса заключается в создании субстрата, удобного для последующей утилизации (в качестве энергетического или пластического материала), или в ускорении выведения этих веществ из организма.
Биотрансформация происходит под воздействием нескольких ферментных систем, локализованных как в межклеточном пространстве, так и внутри клеток. Наиболее активно эти процессы проходят
в печени, стенке кишечника, плазме крови и в области рецепторов (например, удаление избытка медиатора из синаптической щели).
Все процессы метаболизма в организме человека подразделяются на две фазы. Реакции I фазы биотрансформации ЛС обычно несинтетические, II фазы - синтетические.
Метаболизм I фазы включает в себя изменение структуры ЛС путем его окисления, восстановления или гидролиза. Метаболизму I фазы подвергается этанол (окисляется до ацетальдегида), лидокаин (гидролизируется до моноэтилглицилксилидида и глицилксилиди-да) и большинство других ЛС. Реакции окисления при метаболизме I фазы подразделяют на реакции, катализируемые ферментами эндо-плазматической сети (микросомальные ферменты), и реакции, катализируемые ферментами, локализованными в других местах (немикро-сомальные).
Метаболизм II фазы включает в себя связывание молекул ЛС - сульфатирование, глюкуронидацию, метилирование или ацетили-рование. Часть ЛС подвергается метаболизму II фазы сразу, другие препараты предварительно проходят через реакции I фазы. Конечные продукты реакций II фазы лучше растворимы в воде и благодаря этому легче выводятся из организма.
Продукты реакций I фазы имеют различную активность: чаще всего метаболиты ЛС не обладают фармакологической активностью или их активность снижена по сравнению с исходным веществом. Однако в некоторых случаях метаболиты могут сохранять активность или даже превосходить по активности исходное ЛС: так, кодеин в организме человека трансформируется до морфина. Процессы биотрансформации могут приводить к образованию токсичных веществ (метаболиты изониазида, лидокаина, метронидазола и нитрофуранов) или метаболитов с противоположными фармакологическими эффектами, например метаболиты неселективных Р2-адреномиметиков обладают свойствами блокаторов этих же рецепторов. В противоположность этому метаболит фенацетина® парацетамол не оказывает присущего фенацетину® токсического действия на почки и постепенно заменил его в клинической практике.
Если ЛС имеет более активные метаболиты, они постепенно вытесняют предыдущие препараты из употребления. Примеры ЛС, первоначально известных в качестве метаболитов других препаратов, - оксазепам, парацетамол, амброксол. Существуют и про-лекарства, которые исходно не дают полезных фармакологических эффектов, но в процессе биотрансформации превращаются в актив-
ные метаболиты. Например, леводопа, проникая через гематоэн-цефалический барьер, превращается в мозгу человека в активный метаболит допамин. Благодаря этому удается избежать нежелательных эффектов допамина, которые наблюдаются при его системном применении. Некоторые пролекарства лучше всасываются в ЖКТ (талампициллин*3).
На биотрансформацию ЛС в организме влияют возраст, пол, характер питания, сопутствующие заболевания, факторы внешней среды. Поскольку метаболизм ЛС происходит преимущественно в печени, любое нарушение ее функционального состояния отражается на фармакокинетике препаратов. При заболеваниях печени клиренс ЛС обычно уменьшается, а период полувыведения возрастает.
Пресистемный метаболизм (или метаболизм первого прохождения). Под этим термином понимают процессы биотрансформации до поступления ЛС в системный кровоток. Реакции пресистемного метаболизма проходят в просвете кишечника. Некоторые ЛС подвергаются действию неспецифических ферментов кишечного сока (феноксиметилпенициллин, аминазин). Биотрансформация мето-трексата, леводопы, допамина в кишечнике обусловлена ферментами, выделяемыми кишечной флорой. В стенке кишечника моноамины (тирамин®) частично метаболизируются моноаминоксидазой, а хлор-промазин сульфатируется в кишечной стенке. Эти реакции проходят также и в легких (при ингаляционном введении), и в печени (при приеме внутрь).
Печень имеет низкую способность к экстракции (метаболизм + выведение с желчью) диазепама, дигитоксина, изониазида, парацетамола, фенобарбитала, фенитоина, прокаинамида, теофиллина, толбу-тамида, варфарина, промежуточную - ацетилсалициловой кислоты, кодеина, хинидина, высокую - пропранолола, морфина, лидокаина, лабеталола®, нитроглицерина, эрготамина. Если в результате активного пресистемного метаболизма образуются вещества с меньшей фармакологической активностью, чем исходное ЛС, предпочтительнее парентеральное введение такого препарата. Пример ЛС с высоким пресистемным метаболизмом - нитроглицерин, который высокоактивен при сублингвальном приеме или внутривенном введении, однако при приеме внутрь полностью утрачивает свое действие. Пропранолол оказывает одинаковое фармакологическое действие при внутривенном введении в дозе 5 мг или при приеме внутрь в дозе около 100 мг. Высокий пресистемный метаболизм полностью исключает прием внутрь гепарина натрия или препаратов инсулина.
Микросомальное окисление. Большое значение в реакциях биотрансформации I фазы имеют два микросомальных фермента: НАДФ-Н-цитохром С-редуктаза и цитохром Р-450. Существует более 50 изоферментов цитохрома Р-450, сходных по физико-химическим и каталитическим свойствам. Большая часть цитохрома Р-450 в организме человека содержится в клетках печени. Различные ЛС подвергаются биотрансформации с участием различных изоферментов цитохрома Р-450 (подробнее см. на компакт-диске в табл. 4-1).
Активность ферментов микросомального окисления может изменяться под воздействием некоторых ЛС - индукторов и ингибиторов микросомального окисления (подробнее см. на компакт-диске). Это обстоятельство следует учитывать при одновременном назначении нескольких ЛС. Иногда происходит полное насыщение определенного изофермента цитохрома Р-450, что влияет на фармакокинетику препарата.
Цитохром Р-450 способен биотрансформировать практически все известные человеку химические соединения и связывать молекулярный кислород. В результате реакций биотрансформации, как правило, образуются неактивные или малоактивные метаболиты, быстро выводящиеся из организма.
Курение способствует индукции ферментов системы цитохро-ма Р-450, в результате чего ускоряется метаболизм ЛС, подвергающихся окислению с участием изофермента CYP1A2 (подробнее см. на компакт-диске). Влияние табачного дыма на активность гепато-цитов сохраняется до 12 мес после прекращения курения. У вегетарианцев биотрансформация ЛС замедлена. У лиц пожилого возраста и детей до 6 мес активность микросомальных ферментов также может быть снижена.
При высоком содержании в пище белков и интенсивной физической нагрузке метаболизм ускоряется.
4.7. ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
ИЗ ОРГАНИЗМА
ЛС выводятся из организма как в неизмененном виде, так и в виде метаболитов. Большинство ЛС выводятся из организма почками, в меньшей степени - легкими, а также с грудным молоком, через потовые железы, печень (с желчью выводятся хлорамфеникол, морфин, рифампицин, тетрациклин) и слюнные железы.
Выведение ЛС почками происходит посредством следующих механизмов.
• Клубочковая фильтрация (в клубочках нефронов1 каждую минуту фильтруется из крови около 120 мл жидкости, содержащей ионы, продукты метаболизма и ЛС). Преимущественно путем клубочко-вой фильтрации из организма удаляются дигоксин, гентамицин, прокаинамид, метотрексат. Скорость клубочковой фильтрации (СКФ) определяют по величине клиренса креатинина. Клиренс препаратов, выводящихся из организма только путем клубочко-вой фильтрации, равен произведению СКФ на долю препарата, которая находится в плазме в несвязанном виде (f): С1 = f-СКФ.
• Пассивная реабсорбция в канальцах. Из клубочков первичная моча попадает в канальцы нефрона, где часть жидкости и растворенных в ней веществ может всасываться обратно в кровь. При этом клиренс ЛС меньше СКФ: С1 < f-СКФ. Процесс реабсорбции зависит от рН первичной мочи и ионизации ЛС. Например, при рН первичной мочи более 7 слабые кислоты (ацетилсалициловая кислота) будут реабсорбироваться хуже, так как в этом случае увеличивается их ионизация. При этих же условиях увеличится реабсорбция слабых оснований (амфетамин).
• Активная секреция в почечных канальцах (например, фенокси-метилпенициллин). При этом клиренс ЛС всегда больше СКФ: С1 >f ? СКФ.
Нефрон - структурная единица почек, в которой происходит образование мочи.