Аналитическая химия. Количественный анализ. Физико-химические методы анализа: учебное пособие / Ю. Я. Харитонов, Д. Н. Джабаров, В. Ю. Григорьева. - 2012. - 368 с.: ил.
|
|
|
|
ТЕМА II. ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Хроматография - область науки, изучающая процессы, основанные на перемещении зоны вещества вдоль слоя сорбента в потоке подвижной фазы и связанные с многократным повторением сорбционных и десорбционных актов.
В любом варианте хроматографических методов используют сочетание неподвижной (стационарной) фазы (НФ) и подвижной фазы (ПФ). Подвижная фаза (газ, жидкость) в процессе хроматографирования непрерывно перемещается вдоль неподвижной фазы (твердое тело, жидкость) так, что частицы хроматографируемых веществ, переносимые вместе с ПФ, могут многократно переходить из ПФ в НФ и обратно. Разделение веществ основано на их различном сродстве к ПФ и НФ. Различие в сродстве приводит к различию в скоростях движения частиц разделяемых веществ с ПФ и в конце концов к их разделению.
Занятие 1. Тонкослойная хроматография
Общая характеристика метода
Одним из наиболее распространенных методов адсорбционной хроматографии является тонкослойная хроматография (ТСХ) - разновидность плоскостной хроматографии, при которой адсорбент используют в виде тонкого слоя на пластинке.
Принцип и основные понятия
На чистую плоскую поверхность (пластинку из стекла, металла, пластмассы) тем или иным способом наносят тонкий слой сорбента, который чаще всего закрепляется на поверхности пластинки. Размеры пластинки могут быть различными (длина и ширина - от 5 до 50 см, хотя это и необязательно). На поверхности пластинки осторожно, чтобы не повредить слой сорбента, намечают (например, карандашом) линию старта (на расстоянии 2-3 см от нижнего края пластинки) и линию финиша растворителя (рис. 3-2).
На линию старта пластинки наносят (микрошприцем, капилляром) пробу - небольшое количество жидкости, содержащей смесь разделяемых веществ, например двух веществ А и В в подходящем растворителе (см. рис. 3-2). Дают возможность испариться растворителю, после чего
пластинку погружают в хроматографической камере в жидкую фазу ПФ, представляющую собой специально подобранный для данного случая растворитель или смесь растворителей. Под действием капиллярных сил ПФ самопроизвольно перемещается вдоль НФ от стартовой линии до линии финиша растворителя, увлекая с собой компоненты А и В пробы, которые перемещаются с различной скоростью. В рассматриваемом случае сродство компонента А к НФ меньше сродства к той же фазе компонента В, поэтому компонент А перемещается быстрее компонента В. После достижения за время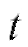 подвижной фазой (растворителем) линии финиша растворителя хроматографирование прерывают, пластинку извлекают из хроматографической камеры, высушивают на воздухе и определяют положение пятен веществ А и В на поверхности пластинки. Пятна (зоны) обычно имеют овальную или круглую форму. В рассматриваемом случае пятно компонента А переместилось от линии старта на расстояние
подвижной фазой (растворителем) линии финиша растворителя хроматографирование прерывают, пластинку извлекают из хроматографической камеры, высушивают на воздухе и определяют положение пятен веществ А и В на поверхности пластинки. Пятна (зоны) обычно имеют овальную или круглую форму. В рассматриваемом случае пятно компонента А переместилось от линии старта на расстояние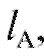 пятно компонента В - на расстояние
пятно компонента В - на расстояние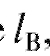 а растворитель прошел расстояние
а растворитель прошел расстояние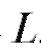 .
.
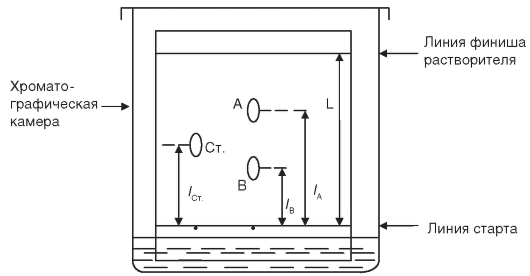
Рис. 3-2. Схема разделения компонентов А и В методом тонкослойной хроматографии
Иногда одновременно с нанесением пробы разделяемых веществ на линию старта наносят небольшое количество вещества-стандарта, а также веществ-«свидетелей» (тех, которые предположительно содержатся в анализируемой пробе).
Для характеристики разделяемых компонентов в системе вводят коэффициент подвижности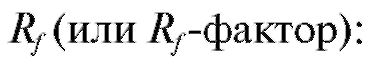
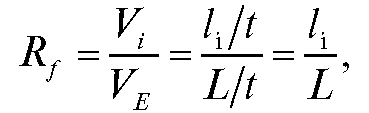
где 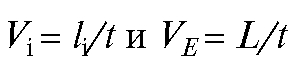 - соответственно скорости перемещения i-го компонента
- соответственно скорости перемещения i-го компонента
и растворителя;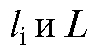 - путь, пройденный i-м компонентом и растворителем соответственно; t - время, необходимое для перемещения растворителя от линии старта до линии фронта растворителя.
- путь, пройденный i-м компонентом и растворителем соответственно; t - время, необходимое для перемещения растворителя от линии старта до линии фронта растворителя.
Расстояния 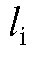 отсчитывают от линии старта до центра пятна соответствующего компонента.
отсчитывают от линии старта до центра пятна соответствующего компонента.
Обычно коэффициент подвижности лежит в пределах: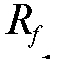 = 0-1. Оптимальное значение составляет 0,3-0,7. Условия хроматографирования подбирают так, чтобы величина
= 0-1. Оптимальное значение составляет 0,3-0,7. Условия хроматографирования подбирают так, чтобы величина 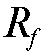 отличалась от нуля и единицы.
отличалась от нуля и единицы.
Коэффициент подвижности является важной характеристикой системы сорбент-сорбат. Для воспроизводимых и строго постоянных условий хроматографирования 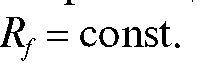
Коэффициент подвижности  зависит от целого ряда факторов: природы и качества растворителя, его чистоты; природы и качества сорбента (тонкого слоя), равномерности его зернения, толщины слоя; активности сорбента (содержания в нем влаги); техники эксперимента (массы образца, длины L пробега растворителя); навыка экспериментатора и т.д. Постоянство воспроизведения всех этих параметров на практике бывает затруднительным. Для нивелирования влияния условий проведения процесса вводят относительный коэффициент подвижности
зависит от целого ряда факторов: природы и качества растворителя, его чистоты; природы и качества сорбента (тонкого слоя), равномерности его зернения, толщины слоя; активности сорбента (содержания в нем влаги); техники эксперимента (массы образца, длины L пробега растворителя); навыка экспериментатора и т.д. Постоянство воспроизведения всех этих параметров на практике бывает затруднительным. Для нивелирования влияния условий проведения процесса вводят относительный коэффициент подвижности  :
:
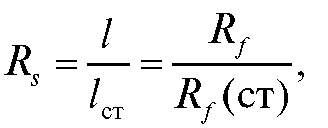
где 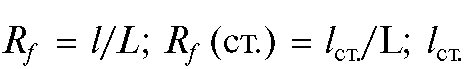 - расстояние от линии старта до центра пятна
- расстояние от линии старта до центра пятна
стандарта (см. рис. 3-2).
Относительный коэффициент подвижности Rs - более объективная характеристика подвижности вещества, чем коэффициент подвижности 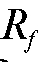 .
.
В качестве стандарта часто выбирают такое вещество, для которого в данных условиях 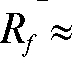 0,5. По химической природе стандарт выбирают близким к разделяемым веществам. С применением стандарта величина
0,5. По химической природе стандарт выбирают близким к разделяемым веществам. С применением стандарта величина 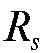 обычно лежит в пределах 0,1-10, оптимальные пределы - около
обычно лежит в пределах 0,1-10, оптимальные пределы - около
0,5-2.
Для более надежной идентификации разделяемых компонентов используют «свидетели» - эталонные вещества, наличие которых предполагается в анализируемой пробе. Если 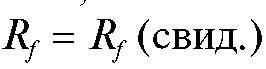 , где
, где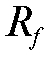 и
и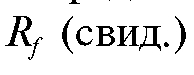 - соответственно коэффициенты подвижности данного
- соответственно коэффициенты подвижности данного
компонента и «свидетеля», то можно с большой вероятностью предположить, что вещество-«свидетель» присутствует в хроматографируемой смеси.
Для характеристики разделения компонентов А и В в данных условиях вводят степень (критерий) разделения  :
:
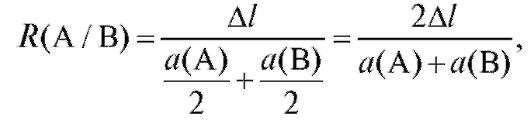
где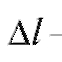 - расстояние между центрами пятен компонентов А и В;
- расстояние между центрами пятен компонентов А и В;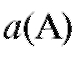 и
и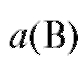 - соответственно диаметры пятен компонентов А и В на хроматограмме (рис. 3-3).
- соответственно диаметры пятен компонентов А и В на хроматограмме (рис. 3-3).
Чем больше величина 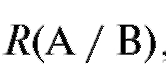 тем четче разделяются пятна компо-
тем четче разделяются пятна компо-
нентов А и В на хроматограмме.
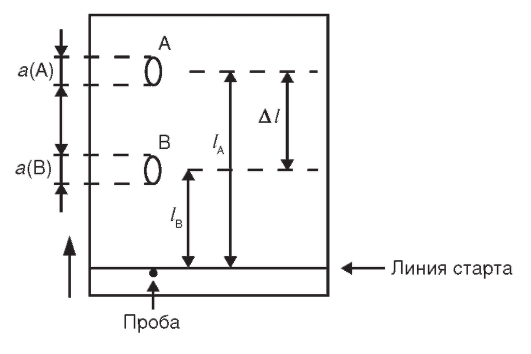
Рис. 3-3. К определению степени разделения компонентов А и В
компонентов А и В
Важная характеристика удерживания вещества - коэффициент емкости: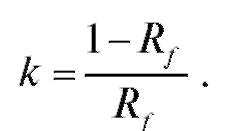
На практике оптимальной считают величину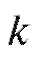 от 1 до 5 (
от 1 до 5 (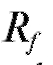 - от 0,5 до ~0,17). При меньших значениях
- от 0,5 до ~0,17). При меньших значениях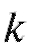 компоненты хроматографируемой смеси плохо разделяются; при больших значениях увеличивается время хроматографирования.
компоненты хроматографируемой смеси плохо разделяются; при больших значениях увеличивается время хроматографирования.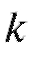
Для оценки селективности разделения веществ А и В используют коэффициент разделения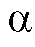 :
: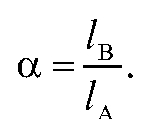
Если 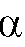 = 1, компоненты А и В не разделяются.
= 1, компоненты А и В не разделяются.
Материалы, применяемые в тонкослойной хроматографии
Сорбенты. Важнейшей характеристикой сорбента считают его активность, т.е. способность сорбировать (удерживать) компоненты разделяемой смеси. Активность сорбента зависит от природы активных центров и их концентрации на поверхности сорбента, от степени дисперсности частиц сорбента, от размеров поверхности сорбента и содержания на нем воды, от природы ПФ, взаимодействующей с сорбентом. Чем больше воды содержит сорбент, тем меньше его активность, поскольку молекулы воды, связываясь с активными центрами сорбента, блокируют их.
В качестве сорбентов чаще всего применяют диоксид кремния - силикагель 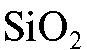 и оксид алюминия
и оксид алюминия 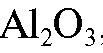 , а также некоторые другие материалы (активированный уголь, сахарозу, карбонат кальция, целлюлозу, тальк, полиамидные смолы и т.д.).
, а также некоторые другие материалы (активированный уголь, сахарозу, карбонат кальция, целлюлозу, тальк, полиамидные смолы и т.д.).
Силикагель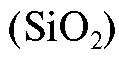 , применяемый в методе ТСХ, обладает довольно
, применяемый в методе ТСХ, обладает довольно
большой удельной поверхностью (до ~500 м /г). Полагают, что активными центрами на поверхности частиц силикагеля являются группы 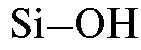 . Молекулы воды могут блокировать эти центры, дезактивируя их, поэтому для дегидратации силикагель активируют нагреванием при ~150-300 °C, но не выше ~400 °C, так как при более высоких температурах активные центры разрушаются.
. Молекулы воды могут блокировать эти центры, дезактивируя их, поэтому для дегидратации силикагель активируют нагреванием при ~150-300 °C, но не выше ~400 °C, так как при более высоких температурах активные центры разрушаются.
Для приготовления хроматографической пластинки с тонким слоем из силикагеля порошкообразный сорбент (с добавкой гипса или крахмала в качестве закрепителей, а также других, например, флуоресцирующих веществ) суспендируют в воде или в водном этаноле, суспензию наносят в виде тонкого слоя (~0,3 мм) на строго горизонтальную поверхность пластинки и высушивают. Обычно такой тонкий слой сорбента хорошо закрепляется на поверхности пластинки.
Оксид алюминия (Al2O3) также находит универсальное применение в качестве сорбента. Его активность сильно зависит от количества поглощенной влаги. На практике обычно применяют оксид алюминия при содержании воды от 6 до 10% (по массе). Оксид алюминия проявляет каталитические свойства в отношении целого ряда реакций, поэтому как сорбент он уступает силикагелю.
Сорбенты, применяемые в ТСХ, должны обладать определенным размером частиц (0,5-20 мкм), большой удельной поверхностью, устойчивостью к действию давления и истирания, однородным составом частиц, химической индифферентностью по отношению к ПФ и анализируемой пробе, не должны растворяться в ПФ.
Тонкий слой сорбента должен быть равномерным и однородным по толщине, обеспечивать воспроизводимость результатов разделения эталонных смесей (стандартов), требуемую скорость перемещения ПФ (часто ~10 см за ~30 мин).
Иногда используют смеси сорбентов различной природы.
Различают пластинки с незакрепленным и закрепленным слоем сорбента.
Для получения пластинок с закрепленным слоем размешивают суспензию порошкообразного сорбента в воде или в органической жидкости, к суспензии добавляют вяжущий (закрепляющий) материал (гипс, крахмал), наносят полученную смесь на строго горизонтальную поверхность пластинки, высушивают на воздухе при комнатной температуре около 20 мин или при нагревании до ~85 °C в течение ~5 мин, затем активируют нагреванием при ~110-120 °C в течение ~30 мин.
На практике используют хроматографические пластинки, полученные в заводских условиях. Такие пластинки можно хранить продолжительное время, они просты в использовании, позволяют получать воспроизводимые результаты. Толщина слоя сорбента на таких пластинках составляет 200-250 мкм, размер зерен сорбента - около 20 мкм. Время хроматографирования обычно равно ~25 мин при длине пути, пройденного растворителем, около 12 см.
Растворители
Выбор растворителя в ТСХ определяется природой сорбента и свойствами анализируемой смеси. Каких-либо строгих правил не существует. Обычно принимают во внимание общие практические рекомендации. Часто готовят смеси нескольких растворителей в качестве ПФ. При выборе растворителей учитывают их элюирующую способность, т.е. способность вытеснять соединения, сорбированные на НФ. Она зависит от сочетания свойств растворителя и НФ. Существуют элюотропные ряды для данного сорбента, облегчающие в какой-то мере выбор растворителя для ТСХ.
В качестве примера приведем элюотропный ряд по Траппе. В этом ряду растворители расположены в порядке увеличения их элюирующей способности, в целом - в порядке возрастания их полярности (диэлектри-
ческой прницаемости): циклогексан, четыреххлористый углерод, трихлорэтилен, толуол, бензол, дихлорэтан, хлороформ, диэтиловый эфир, этилацетат, ацетон, пропанол, этанол, метанол, вода.
Почти таков же ряд растворителей по Шталю: гексан, гептан, циклогексан, четыреххлористый углерод, бензол, хлороформ, диэтиловый эфир, этилацетат, пиридин, ацетон, этанол, метанол, вода.
Систему растворителей, используемую в качестве ПФ, подбирают, смешивая два растворителя из начала и конца элюотропного ряда. Меняя растворители и их количество, часто можно получать ПФ с приблизительно желаемыми свойствами.
Техника эксперимента в тонкослойной хроматографии
• Нанесение пробы. Анализируемую жидкую пробу наносят на линию старта с помощью капилляра, микрошприца, микропипетки, осторожно касаясь слоя сорбента (диаметр пятна на линии старта - обычно от одного до нескольких миллиметров). Если на линию старта наносят несколько проб, то расстояние между пятнами образцов на линии старта не должно быть меньше 2 см. По возможности используют концентрированные растворы. Пятна сушат на воздухе, после чего проводят хроматографирование.
• Развитие хроматограммы (хроматографирование). Процесс проводят в закрытых хроматографических камерах, насыщенных парами растворителя, используемого в качестве ПФ, например, в стеклянном сосуде, покрытом сверху крышкой. В зависимости от направления движения ПФ различают восходящую, нисходящую и горизонтальную хроматографию.
- В варианте восходящей хроматографии ПФ наливают на дно камеры (в качестве последней можно использовать стеклянный химический стакан подходящего размера со стеклянной крышкой), хроматографическую пластинку помещают вертикально или наклонно в камеру так, чтобы слой ПФ на дне камеры смачивал нижнюю часть пластинки (ниже линии старта на ~1,5- 2 см). ПФ перемещается за счет действия капиллярных сил снизу вверх (против силы тяжести) сравнительно медленно.
- В варианте нисходящей хроматографии ПФ подается сверху и перемещается вниз вдоль слоя сорбента пластинки. Сила тяжести ускоряет движение ПФ. Такой вариант реализуют при анализе смесей, содержащих компоненты, медленно перемещающиеся с ПФ.
- В варианте горизонтальной хроматографии пластинку помещают горизонтально. Можно использовать прямоугольные или круглые пластинки. При применении круглых пластинок (круговой вариант горизонтальной хроматографии) стартовую линию обозначают в виде окружности подходящего радиуса (~1,5-2 см), на которую наносят пробы. В центре круглой пластинки вырезают отверстие, в которое вставляют фитиль для подачи ПФ. Последняя перемещается вдоль слоя сорбента от центра круга к его периферии. Хроматографирование проводят в закрытой камере - эксикаторе или в чашке Петри. При круговом варианте можно одновременно анализировать до нескольких десятков проб.
• Расшифровка хроматограмм. Если пятна на хроматограмме окрашены, после высушивания пластинок определяют расстояние от линии старта до центра каждого пятна и вычисляют коэффициенты подвижности. Если же в состав анализируемой пробы входят бесцветные вещества, дающие неокрашенные, т.е. визуально не идентифицируемые пятна на хроматограмме, необходимо провести детектирование этих пятен, для чего хроматограммы проявляют.
Рассмотрим некоторые наиболее распространенные методы детектирования.
• Облучение ультрафиолетовым светом. Используется для обнаружения флуоресцирующих соединений (пятна светятся при облучении пластинки УФ-светом) или нефлуоресцирующих веществ, но с применением сорбента с флуоресцирующим индикатором (сорбент светится, пятна не светятся). Таким образом детектируют, например, алкалоиды, антибиотики, витамины и другие лекарственные вещества.
• Термическая обработка. Высушенную после хроматографирования пластинку осторожно нагревают (до ~200 °C), избегая потемнения слоя самого сорбента (например, тогда, когда тонкий слой сорбента содержит крахмал). При этом пятна проявляются обычно в виде коричневых зон (за счет частичного термолиза органических компонентов).
• Химическая обработка. Хроматограммы проявляют, обрабатывая их реагентами, которые образуют окрашенные соединения с разделяемыми компонентами смесей. Для этих целей применяют различные реагенты: пары йода, аммиака, брома, диоксида серы,
сероводород, специально приготовленные растворы, которыми обрабатывают пластинки. Применяют как универсальные, так и селективные реагенты (понятие «универсальные» достаточно условно).
• Универсальными реагентами могут служить, например, концентрированная серная кислота (при нагревании наблюдается потемнение пятен органических соединений), кислый водный раствор перманганата калия (зоны наблюдаются в виде коричневых пятен на фиолетовом фоне сорбента), раствор фосфорно-молибденовой кислоты при нагревании (появляются синие пятна на желтом фоне) и т.д.
• Селективные реагенты:
- реактив Драгендорфа; -реактив Циммермана;
- водный аммиачный раствор сульфата меди (10% по 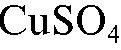 , 2% - по аммиаку);
, 2% - по аммиаку);
- смесь нингидрина 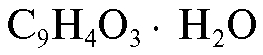 с этанолом и уксусной кислотой.
с этанолом и уксусной кислотой.
Реактив Драгендорфа представляет собой раствор основного нитрата висмута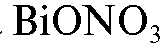 , иодида калия
, иодида калия 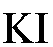 и уксусной кислоты в воде. Использу-
и уксусной кислоты в воде. Использу-
ется для определения аминов, алкалоидов, стероидов.
Реактив Циммермана готовят, обрабатывая раствором щелочи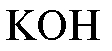 2% этанольный раствор динитробензола с последующим нагреванием смеси при ~70-100 °C. Применяют для обнаружения стероидов.
2% этанольный раствор динитробензола с последующим нагреванием смеси при ~70-100 °C. Применяют для обнаружения стероидов.
С помощью нингидрина детектируют пятна аминов, аминокислот, белков и других соединений.
Применяют и некоторые другие способы детектирования пятен. Например, измеряют их радиоактивность, если некоторые из разделяемых компонентов радиоактивны, либо вводят специально добавки радиоактивных изотопов элементов, входящих в состав разделяемых составляющих смеси.
После детектирования пятен на хроматограмме проводят их идентификацию, т.е. определяют, какому соединению соответствует то или иное пятно. Для этого чаще всего используют эталонные пятна «свидетелей». Иногда пятна идентифицируют по величине коэффициентов подвижности 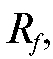 сравнивая их с известными для данных условий величинами
сравнивая их с известными для данных условий величинами 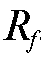 . Однако такая идентификация по величине
. Однако такая идентификация по величине  часто носит предварительный характер.
часто носит предварительный характер.
Окраску флуоресцирующих пятен также используют в целях идентификации, поскольку различные соединения флуоресцируют излучением различной длины волны (разного цвета).
При химическом детектировании пятен селективные реагенты дают окрашенные пятна с соединениями определенной природы, что также используется в целях идентификации.
С помощью ТСХ можно не только открывать, но и количественно определять содержание компонентов в смесях. Для этого либо анализируют сами пятна на хроматограмме, либо извлекают разделенные компоненты из хроматограммы тем или иным способом (экстракцией, элюированием подходящими растворителями).
При анализе пятен предполагают существование определенной связи между площадью пятна и содержанием данного вещества (например, наличие пропорциональной или линейной зависимости), которую устанавливают методом построения градуировочного графика, измеряя площади пятен «свидетелей» - эталона с известным содержанием анализируемого компонента.
Иногда сравнивают интенсивность окраски пятен, полагая, что интенсивность окраски пятна пропорциональна количеству данного окрашенного компонента. Для измерения интенсивности окраски применяют различные приемы.
При извлечении разделенных компонентов из хроматограммы получают раствор, содержащий данный компонент. Последний затем определяют тем или иным аналитическим методом.
Относительная ошибка количественного определения вещества методом ТСХ составляет 5-10%.
ТСХ - фармакопейный метод и широко применяется для анализа и контроля качества разнообразных лекарственных средств.
Высокоэффективная тонкослойная хроматография
В этом современном варианте ТСХ применяют пластинки с толщиной слоя сорбента ~100 мкм и малым размером зерен сорбента - порядка 5 мкм или еще меньше. Благодаря меньшим размерам толщины слоя сорбента и зерен сорбента (по сравнению с обычным вариантом ТСХ) увеличивается эффективность разделения пятен на хроматограмме и уменьшается время хроматографирования (до ~10 мин).
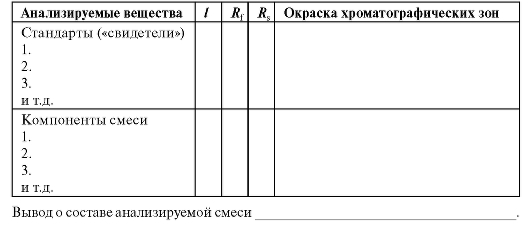
Рекомендуемая форма протокола лабораторной работы
Для получения воспроизводимых результатов разделения необходимо точное фиксирование условий эксперимента.
При ведении протокольных записей рекомендуют использовать следующую форму

Техника подготовки к выполнению лабораторных работ
Приготовление капилляров
Отрезок стеклянной трубки длиной 15-25 см нагревают в пламени горелки с насадкой «ласточкин хвост» до размягчения стекла и вытягивают капилляр диаметром 0,5-0,8 мм. После остывания капилляр разламывают на отрезки длиной около 10 см. (Осторожно! Беречь глаза!) Готовые капилляры хранят в стаканчике.
При использовании пластинок малых размеров наружный диаметр капилляров не должен превышать 0,5-0,8 мм. Для проверки диаметра капилляров измеряют линейкой ширину 10-20 капилляров, сложенных вместе на листе бумаги. Рабочий конец капилляров должен быть достаточно ровно обрезан, чтобы не разрушался слой сорбента. Крупные сколы капилляров осторожно обламывают и зачищают наждачной бумагой. После зачистки капилляр трижды промывают подходящим растворителем, осторожно удаляя его из капилляра кусочком фильтровальной бумаги.
Приобретение навыков работы с капиллярами
Технику нанесения растворов с помощью капилляров на слой сорбента отрабатывают на отдельных пластинках. На пластинках проводят стартовые линии, размечают стартовые точки. Капилляр наполняют раствором красителя, удаляя избыток раствора боковым прикосновением кусочка фильтровальной бумаги. Затем касаются капилляром стартовой точки, держа его вертикально и стараясь не повредить слой сорбента. Диаметр стартового пятна не должен превышать 3 мм.
Подготовка пластинок
В работах можно использовать готовые пластинки, представляющие собой обычно алюминиевую фольгу (подложку) с нанесенным на нее сорбентом - широкопористым силикагелем, закрепленным крахмалом.
Пластинку размером 150 х 150 мм кладут на лист плотной бумаги сорбентом вниз.
ВНИМАНИЕ! К слою сорбента нельзя прикасаться пальцами! Брать пластинку только пинцетом или пальцами за края.
Вырезают скальпелем пластинку нужного размера, например, шириной 5 см и длиной 7,5 см. Продольное направление сорбента различается по царапинам на металле на оборотной стороне пластинки.
Мягким простым карандашом осторожно намечают на слое сорбента линии старта, финиша и, если необходимо, линии «фитиля» (при использовании горизонтального способа хроматографирования)
(рис. 3-4).
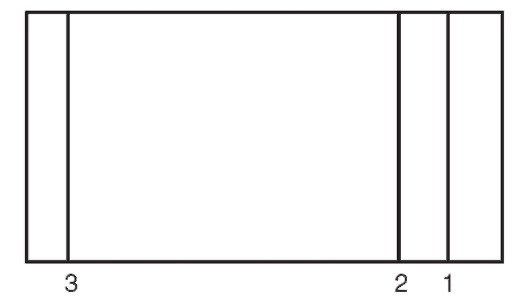
Рис. 3-4. Подготовка пластинки к работе. Линии: 1 - «фитиля»; 2 - старта; 3 - финиша
Линии старта и «фитиля» следует проводить особенно осторожно, не нарушая структуры сорбента.
Прижимая линейкой пластинку на линии «фитиля», загибают ее под углом 90° для образования «фитиля». Затем на стартовой линии размеча-
ют положение стартовых точек - места нанесения всех хроматографируемых компонентов.
Расстояние между точками и от краев пластинки должно быть примерно одинаковым.
Подготовка хроматографической камеры
В качестве хроматографической камеры используют стеклянные сосуды подходящего размера. В работах, не требующих насыщенных камер, используют чашки Петри.
Важным условием успешного хроматографического разделения является строго горизонтальное положение камеры. В качестве подставки для пластинок используют кусочки фольги. Для этого из остатков пластинки вырезают фрагмент размером 5x1 см, удаляют с него сорбент скальпелем, сгибают под углом 60-90° по продольной оси. Установка пластинки при горизонтальном способе подачи подвижной фазы представлена на рис. 3-5.
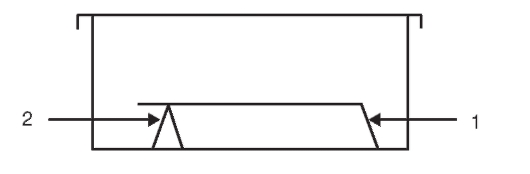
Рис. 3-5. Установка пластинки в камере: 1 - «фитиль»; 2 - подставка
ВНИМАНИЕ! Уровень подвижной фазы должен быть ниже стартовой линии на пластинке, иначе хроматографируемые вещества могут быть смыты в ПФ и разнесены по всему слою.
Лабораторные работы
Тема
«Тонкослойная хроматография в качественном анализе» Цель работ
На основе знаний теоретических основ хроматографического анализа приобрести навыки качественного анализа смесей методом ТСХ.
Целевые задачи
1. Освоение техники нанесения веществ на пластинку с последующим хроматографированием и обнаружением их.
2. Расчет основных хроматографических параметров.
3. Идентификация состава смеси.
Задание для самоподготовки
К занятию необходимо знать
1. Сущность метода тонкослойной хроматографии.
2. Сорбенты, применяемые в ТСХ.
3. Основные этапы получения хроматограммы.
4. Способ идентификации веществ на хроматограмме с применением стандартных веществ («свидетелей»).
К занятию необходимо уметь
1. Готовить ПФ - смесь растворителей.
2. Делать разметку на хроматографических пластинах.
3. Вычислять значения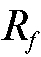 и
и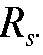
Список литературы
1. Лекции «Хроматографические методы анализа. Тонкослойная хроматография».
2. Учебник. - Книга 1, глава 10. - С. 264-287.
Вопросы для самопроверки
1. Кем и когда предложен хроматографический метод анализа? Назовите ученых, внесших вклад в развитие и применение тонкослойной хроматографии.
2. Что такое хроматография?
3. Какие классификации хроматографических методов применяются?
4. Каковы области применения хроматографических методов анализа? Каковы преимущества ТСХ перед другими методами разделения?
5. Как осуществляется качественный анализ смесей веществ с помощью ТСХ?
6. Каков механизм разделения веществ в ТСХ?
7. Что такое подвижная фаза (элюэнт)? Как осуществляется ее выбор?
8. Что такое неподвижная фаза? Какие вещества чаще всего используются для приготовления закрепленного и незакрепленного слоя?
9. Что такое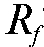 ? От каких факторов зависит его величина?
? От каких факторов зависит его величина?
10. Что такое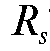 ? Какие факторы влияют на его величину? Как выбирают стандартное вещество?
? Какие факторы влияют на его величину? Как выбирают стандартное вещество?
Материальное обеспечение
Хроматографические пластинки с закрепленным слоем.
Посуда: цилиндры мерные вместимостью 5 и 10 мл; цилиндры мерные с притертой пробкой вместимостью 25 и 50 мл; пипетки мерные на 5 мл.
Прочее: чашки Петри, камеры для хроматографирования с пришлифованной крышкой, предметные стекла, стеклянные капилляры, пульверизаторы, пипетки, бумага наждачная, карандаши, линейки, ножницы, проволока (тонкая).
Лабораторная работа «Анализ смеси аминокислот»
Реактивы
1. Кислота глютаминовая, 0,1% раствор в этаноле.
2. Фенилаланин, 0,1% раствор в этаноле.
3. Нингидрин, 0,2% раствор в н-бутаноле.
4. Н-бутанол.
5. Кислота уксусная концентрированная.
6. Этанол 96%.
1. Подготовка хроматографической пластинки с тонким слоем из силикагеля
Разделение смеси аминокислот проводят на пластинках с тонким слоем из силикагеля размером 30x 50 мм. Мягким карандашом, стараясь не повредить слой сорбента, намечают линии «фитиля», старта и финиша на расстоянии 5, 10 и 45 мм от края пластинки соответственно. Сгибают пластинку по линии «фитиля» под углом 90° (см. рис. 3-4).
2. Приготовление подвижной фазы
В мерном цилиндре смешивают 4 мл этанола, 1 мл концентрированной уксусной кислоты и 1 мл н-бутонола. Смесь перемешивают.
3. Подготовка хроматографической камеры
В качестве камеры для хроматографирования используют чашку Петри, в которую помещают подставку из фольги высотой 5 мм. В камеру помещают подвижную фазу и закрывают крышкой.
4. Нанесение анализируемых веществ
Стеклянными капиллярами, стараясь не повредить слой сорбента, на линию старта наносят на равном расстоянии (7 мм друг от друга и от краев пластинки) растворы глютаминовой кислоты, фенилаланина и контрольной смеси. Диаметр пятен проб не должен превышать 2-3 мм. Удаляют растворитель, подсушивая пластинку на воздухе около 5 мин.
5. Элюирование
Пластинку с пробами веществ помещают в камеру. Верхний край пластинки поддерживается подставкой, нижний - «фитилем». Уровень подвижной фазы в камере - около 2-3 мм. Пластинку вынимают из камеры после достижения ПФ линии финиша и подсушивают на воздухе (под тягой!).
6. Обнаружение хроматографических зон
Проводят, погружая пластинку на 1-2 с в раствор нингидрина (слой сорбента внизу!). Раствор нингидрина предварительно наливают в чистую чашку Петри до уровня не более 5 мм. Пластинку держат за отогнутый уголок пинцетом, после чего ее вновь подсушивают на воздухе, а затем помещают на слой асбеста, осторожно подогревая с помощью горелки (Осторожно! Не обугливать крахмал!) до появления цветных пятен.
7. Обработка хроматограммы
Вычисляют значение 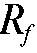 для всех обнаруженных хроматографических зон.
для всех обнаруженных хроматографических зон.
8. Запись результатов анализа
Проводят в соответствии с рекомендациями преподавателя.
9. Вывод
Вывод о составе делают на основе анализа величин 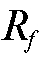 хроматографических зон «свидетелей» и контрольной задачи.
хроматографических зон «свидетелей» и контрольной задачи.
Лабораторная работа «Разделение
и идентификация смеси алкилпроизводных
фенотиазина»
Реактивы
1. Динезина, пропазина, дипразина 0,2% растворы в этаноле.
2. Контрольные задачи, содержащие смеси алкилпроизводных фенотиазина (АФ).
3. Раствор аммиака в воде концентрированный.
4. Этилацетат.
5. Этанол 96%.
6. Йод кристаллический.
1. Подготовка пластинки с закрепленным слоем силикагеля
Хроматографирование проводят на готовых пластинках для ТСХ с тонким слоем силикагеля. Пластинку размером 150x150 мм кладут на лист плотной бумаги сорбентом вниз. Вырезают скальпелем фрагмент размером 75x 75 мм.
Мягким простым карандашом осторожно намечают линию старта (на расстоянии 10 мм от края пластинки), точки нанесения анализируемых растворов (на расстоянии 12-15 мм друг от друга и от края пластинки), линию финиша (на расстоянии 50 мм от линии старта).
2. Приготовление подвижной фазы
Отмеривают мерными цилиндрами 17 мл этилацетата, 2 мл 96% этанола и 0,5 мл концентрированного раствора аммиака в воде, помещают в колбу с притертой пробкой, тщательно перемешивают.
3. Подготовка камеры к хроматографированию
В чистую сухую камеру для хроматографирования диаметром и высотой не менее 10 см помещают часть подготовленной подвижной фазы. Камеру тщательно закрывают и оставляют для насыщения парами подвижной фазы. Время насыщения -  15 мин. Уровень подвижной фазы после насыщения камеры - не более 5-7 мм.
15 мин. Уровень подвижной фазы после насыщения камеры - не более 5-7 мм.
4. Нанесение образцов стандартных растворов и контрольной задачи алкилпроизводных фенотиазина
Осторожно, стараясь не повредить слой сорбента, капиллярами наносят стандартные растворы пропазина, динезина, дипразина и контрольной смеси на заранее намеченные места. Диаметр нанесенных пятен не должен превышать 5 мм, иначе разделение веществ не будет полным.
Пластинку с нанесенными пробами подсушивают на воздухе до полного удаления этанола.
5. Проявление (элюирование)
В работе используют восходящий способ хроматографирования. Пластинку с нанесенными веществами помещают в подготовленную камеру для хроматографирования вертикально. Уровень подвижной фазы должен быть ниже линии старта на пластинке на 4-5 мм.
Как только подвижная фаза достигнет линии финиша, пластинку вынимают из камеры и подсушивают на воздухе в течение 15-20 мин.
Время хроматографирования 10-15 мин.
6. Обнаружение (детектирование) хроматографических зон
А. Высушенную бесцветную хроматограмму помещают на 15 мин в камеру, насыщенную парами йода. Отмечают окраску и положение зон адсорбции на хроматограмме после удаления избытка йода на воздухе.
Б. Возможно термическое обнаружение хроматографических зон на пластинке, для чего пластинку осторожно подогревают на газовой горелке (пластинка не должна обугливаться).
7. Обработка результатов анализа
Линейкой измеряют расстояния между точками нанесения на стартовую линию стандартов («свидетелей») и контрольной задачи и серединой каждой хроматографической зоны. Вычисляют значения 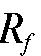 и
и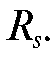
Стандартное вещество - дипразин.
8. Запись результатов анализа
Проводят в соответствии с рекомендациями преподавателя.
9. Вывод
Вывод о составе анализируемой смеси делают на основе анализа величин 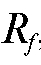 , формы и окраски хроматографических зон стандартных веществ и контрольной задачи.
, формы и окраски хроматографических зон стандартных веществ и контрольной задачи.
Лабораторная работа «Разделение
и идентификация смеси производных фенола»
Реактивы
1. Кислота салициловая 0,5% раствор в 96% этаноле.
2. Кислота аминосалициловая 0,5% раствор в этаноле.
3. Кислота ацетилсалициловая 0,5% раствор в этаноле.
4. Контрольные задачи, содержащие смеси фенолокислот.
5. Хлороформ.
6. Этанол 96% (или метанол).
7. Диоксан.
8. Железоаммониевые квасцы, насыщенный раствор в 70% этаноле.
1. Подготовка пластинки с закрепленным слоем силикагеля
Разделение проводят на готовых пластинках с тонким слоем из силикагеля размером 50x 70 мм. Мягким карандашом осторожно намечают на расстоянии 15 мм от нижнего края пластинки линию старта, линию финиша - 10 мм от верхнего края пластинки и линию сгиба «фитиля» на расстоянии 5 мм от нижнего края пластинки. На линии старта отмечают точки нанесения свидетелей и контрольной задачи - на расстоянии 10 мм от края пластинки и друг от друга.
Сгибают «фитиль» под углом 90° по отношению к основной поверхности пластинки.
2. Приготовление подвижной фазы
Мерными цилиндрами отмеряют 10 мл хлороформа, 2 мл 96% этанола и 1 мл диоксана, помещают в колбу с притертой пробкой и перемешивают.
3. Подготовка камеры для хроматографирования
В качестве камеры для хроматографирования используют чашку Петри, в которую помещают заранее приготовленную из алюминиевой фольги подставку высотой 5 мм и 5-6 мл подвижной фазы.
4. Нанесение стандартных растворов («свидетелей») и контрольной задачи
Капиллярами, стараясь не повредить слой сорбента, наносят на линию старта на расстоянии 10 мм друг от друга контрольную задачу и 3 свидетеля. Диаметр нанесенных пятен - 3-4 мм. Пластинку с нанесенными пробами подсушивают на воздухе до удаления этанола.
5. Проявление (элюирование)
В работе применяют горизонтальный способ подачи подвижной фазы. Уровень подвижной фазы в камере не должен превышать 2-3 мм. Верхний край пластинки поддерживается подставкой, нижний - «фитилем».
Пластинку вынимают из камеры после достижения подвижной фазой линии финиша, подсушивают на воздухе до полного удаления подвижной фазы.
6. Обнаружение (детектирование) хроматографических зон
Высушенную на воздухе пластинку погружают на 1-2 с в насыщенный раствор железоаммониевых квасцов (слой сорбента внизу!). Отмечают окраску и положение зон адсорбции салициловой и аминосалициловой кислоты. Пластинку вновь подсушивают на воздухе до удаления этанола и подогревают на газовой горелке (не обугливать! осторожно!) для обнаружения ацетилсалициловой кислоты.
7. Обработка результатов анализа
Линейкой измеряют расстояния между точками нанесения на стартовую линию свидетелей и контрольной задачи и серединой каждой соответствующей хроматографической зоны. Вычисляют значения 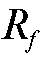 и
и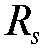 .
.
Стандартное вещество - ацетилсалициловая кислота.
8. Запись результатов анализа
Проводят в соответствии с рекомендациями преподавателя.
9. Вывод
Вывод о составе анализируемой контрольной задачи делают на основе анализа величин 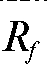 (или
(или 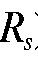 ), формы и окраски хроматографических зон «свидетелей» (стандартных веществ) и контрольной задачи.
), формы и окраски хроматографических зон «свидетелей» (стандартных веществ) и контрольной задачи.
Лабораторная работа «Разделение и идентификация смеси амидопирина, антипирина, хинина»
Реактивы
1. Амидопирин, антипирин, хинин 1% растворы.
2. Контрольные задачи, содержащие двухили трехкомпонентные смеси веществ.
3. Раствор аммиака в воде концентрированный.
4. Железа(III) хлорид 10% раствор.
5. Реактив Драгендорфа.
6. Ацетон.
7. Хлороформ.
8. Диоксан.
9. Пластинки с тонким закрепленным слоем силикагеля.
1. Подготовка пластинки с тонким закрепленным слоем силикагеля
В работе используют пластинки размером 9x 12 см.
На хроматографической пластинке равномерно намечают, слегка касаясь карандашом, 4 точки на расстоянии 1,5 см от нижнего края (на стартовой линии) и 1 см от краев пластинки, распределяя их по стартовой линии. На расстоянии 10 см от линии старта слегка намечают линию финиша.
2. Нанесение стандартных растворов амидопирина, антипирина, хинина и их смеси на пластинку
Проводят с помощью капилляров в намеченные ранее места.
3. Приготовление подвижной фазы
В качестве подвижной фазы используют систему растворителей «ацетон-хлороформ-конц. раствор аммиака в воде-диксан» с объемным соотношением 2:18:1:19. Компоненты смеси отмеривают мерными цилиндрами, помещают в колбу с притертой пробкой, тщательно встряхивают и наливают в камеру.
4. Подготовка хроматографической камеры
В качестве камеры для хроматографирования используют стеклянный сосуд с притертой крышкой высотой 14 см и диаметром не менее 10 см. Подвижную фазу помещают в камеру, закрывают крышкой. Время насыщения камеры - 10 мин. Уровень ПФ в камере не должен превышать 1 см.
5. Элюирование (проявление)
Выполняется методом восходящей хроматографии. Хроматографическую пластинку помещают в камеру вертикально и сразу же закрывают
крышкой. Время проявления хроматограммы - около 80 мин. Как только ПФ достигнет линии финиша, пластинку вынимают и высушивают в вытяжном шкафу для удаления ПФ.
6. Обнаружение хроматографических зон
Хроматографическую зону хинина обнаруживают при освещении пластинки УФ-светом по голубой флуоресценции. Капилляром намечают положение хинина на хроматограмме. Затем хроматограмму обрабатывают 10% раствором железа(III) хлорида. Амидопирин обнаруживают по сине-фиолетовому окрашиванию пятна, антипирин - по оранжево-коричневому.
При последующей обработке пластинки реактивом Драгендорфа хроматографическая зона хинина окрашивается в оранжевый цвет.
7. Обработка результатов анализа
Вычисляют значение Rf и Rs (по антипирину) для всех веществ.
8. Запись результатов анализа
Проводят в соответствии с рекомендациями преподавателя.
9. Вывод
Вывод о составе контрольной задачи делают на основе анализа величин 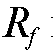 и
и 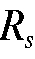 и окраски хроматографических зон стандартных веществ («свидетелей») и контрольной задачи.
и окраски хроматографических зон стандартных веществ («свидетелей») и контрольной задачи.
Лабораторная работа «Идентификация в смеси солей ртути(II), висмута(III), меди(II) и свинца(II)»
Реактивы
1. Ртути(II), висмута(III), меди(II), свинца(II) нитраты 0,2 моль/л растворы.
2. Н-бутанол.
3. Ацетилацетон.
4. Раствор HCl 1,5 моль/л.
5. Натрия сульфид 10% раствор.
6. Хроматографические пластинки с тонким закрепленным слоем силикагеля.
1. Нанесение
Нанесение 0,2 молярных растворов нитратов ртути(II), висмута(III), меди(II) и свинца(II) и контрольной задачи на пластинку размером 9x12 см проводят капиллярами на стартовую линию (в 1,5 см от нижнего края пластинки) на расстоянии 1,5 см друг от друга и от краев пластинки. Карандашом слегка намечают линию финиша на расстоянии 10 см от стартовой линии.
2. Приготовление подвижной фазы
В работе используется система растворителей «н-бутанол-1,5М 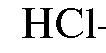 -ацетилацетон» в объемном соотношении 50:10:0,20. Компоненты смеси отмеривают мерными цилиндрами, помещают в колбу с притертой пробкой и перемешивают.
-ацетилацетон» в объемном соотношении 50:10:0,20. Компоненты смеси отмеривают мерными цилиндрами, помещают в колбу с притертой пробкой и перемешивают.
3. Подготовка камеры для хроматографирования
Для хроматографирования используют стеклянный сосуд высотой и диаметром по 14 см с пришлифованной крышкой. Часть подвижной фазы помещают в камеру, закрывают ее крышкой. Время насыщения камеры 10-15 мин.
4. Элюирование (проявление)
Применяют восходящий способ хроматографирования. Подготовленную пластинку погружают в насыщенную камеру почти вертикально и закрывают крышкой. Время элюирования около 40 мин. Как только ПФ достигнет линии финиша, пластинку переносят из камеры под тягу и высушивают на воздухе.
5. Обнаружение хроматографических зон
Проводят опрыскиванием из пульверизатора бесцветной хроматограммы 10% раствором натрия сульфида (работать под тягой!). Отмечают окраску и положение пятен на хроматограмме.
6. Обработка хроматограммы
Вычисляют значение 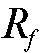 для всех хроматографических зон и
для всех хроматографических зон и 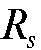 по висмуту(III).
по висмуту(III).
7. Запись результатов анализа
Проводят согласно рекомендациям преподавателя.
8. Вывод
Вывод о составе анализируемой задачи делают на основе сопоставления величин 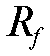 и
и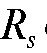 стандартных растворов солей и хроматографических зон задачи.
стандартных растворов солей и хроматографических зон задачи.
Контрольные вопросы
1. Как проводится качественный анализ смесей веществ с помощью ТСХ?
2. Перечислите основные этапы работы при осуществлении методик ТСХ.
3. Каковы особенности нанесения проб анализируемых веществ на пластинку?
4. С какой целью используются в ТСХ стандартные вещества («свидетели»)?
5. Как подготовить камеру для хроматографирования?
6. Что представляют собой хроматографические пластинки?
7. Как получить хроматограмму?
8. Каково значение 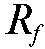 для практически разделенных на пластинке веществ?
для практически разделенных на пластинке веществ?
9. С какой целью в ТСХ вычисляют значения 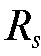 ?
?
10. Что такое подвижная и неподвижная фазы?
11. Каковы возможные способы обнаружения зон адсорбции на хроматограмме?
12. Какие вещества используются для закрепления адсорбента на пластинке?
13. Каковы преимущества ТСХ перед другими видами хроматографии?
Занятие 2. Газожидкостная хроматография
На изучение темы отводится одно занятие, включающее краткое рассмотрение основных теоретических положений, решение задач по теме и экспериментальную часть.
Цель занятия
На основе знания сущности хроматографии, а также основных закономерностей равновесных процессов распределения определяемых веществ между подвижной и неподвижной фазой научиться количественно определять вещества в анализируемых системах с применением хроматографических методов анализа.
Целевые задачи
1. Научиться проводить количественное определение органических веществ с применением метода газожидкостной хроматографии.
2. Научиться проводить количественное определение неорганических веществ с применением метода ионообменной хроматографии.
3. Оформление протокола выполненных лабораторных работ.
Общая характеристика метода
1. Сущность метода
Газожидкостная хроматография (ГЖХ) - метод разделения, качественного и количественного анализа состава многокомпонентной сме-
си, основанный на различиях в равновесном распределении разделяемых компонентов смеси между двумя фазами: подвижной газообразной фазой ПФ и неподвижной фазой НФ (высококипящие жидкости различной химической природы, нанесенные на твердый носитель). Анализ проводят на газожидкостных хроматографах, принципиальная схема которых показана на рис. 3-6.
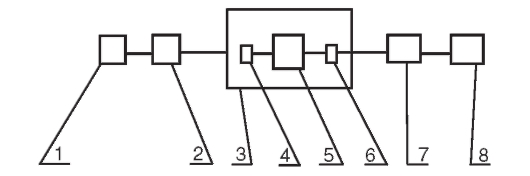
Рис. 3-6. Принципиальная схема газожидкостного хроматографа: 1 - источник газа-носителя; 2 - блок подготовки газа; 3 - блок анализатора; 4 - устройство для ввода пробы в испаритель; 5 - хроматографическая колонка; 6 - детектор; 7 - блок управления; 8 - система усилителя и регистратора
Анализируемую пробу смеси вводят с использованием микрошприца в испаритель (4) анализатора (3), где она переводится в парообразное состояние, смешивается с потоком газа-носителя (1, 2). Под действием газа-носителя разделяемая смесь выносится из испарителя и попадает в начальный участок хроматографической колонки (5). Температура испарителя и хроматографической колонки задается блоком управления (7).
Компоненты подвижной фазы поглощаются (абсорбируются) жидкой НФ. Каждый компонент смеси имеет различное сродство к НФ, различную растворимость в ней и по-разному распределяется между газообразной и неподвижной жидкой фазаой. Равновесное распределение компонента между ПФ и НФ достигается практически мгновенно; участок НФ, на котором устанавливается это равновесие, условно называется теоретической тарелкой.
Основным параметром, характеризующим способность компонента растворяться в неподвижной фазе, является его коэффициент распределения К:
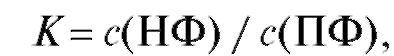
где с(НФ) и с(ПФ) - равновесные концентрации компонента смеси в НФ и ПФ соответственно.
Новая порция газа-носителя переносит десорбируемые из ПФ разделяемые компоненты на новый участок НФ. На новом участке НФ
вновь происходит равновесное распределение компонентов между двумя фазами; этот процесс сорбции-десорбции повторяется многократно по мере продвижения разделяемой смеси вдоль неподвижной фазы под действием газа-носителя. Компонент с большим численным значением коэффициента распределения большую часть времени проводит в НФ и медленнее перемещается в колонке вдоль НФ. Компонент с меньшим численным значением коэффициента распределения двигается по колонке быстрее, первым покидает ее и первым попадает в детектор (6). Таким образом, в процессе продвижения анализируемой смеси по хроматографической колонке происходит ее разделение на компоненты, при этом условием разделения компонентов А и В является их различное сродство к НФ, т.е. различие их коэффициентов распределения.
Каждый компонент, выходящий из колонки, генерирует в детекторе (6) электрический сигнал, который усиливается и фиксируется регистратором (8) в виде соответствующего пика на хроматограмме.
В фармакопейном анализе метод ГЖХ используют при контроле качества субстанций и лекарственных форм для идентификации и количественного определения остаточных летучих растворителей, например ацетона и метанола в пилокарпине, этанола в мелоксикаме, изопропанола в флуконазоле и др.
2. Понятие о теории метода 2.1. Параметры удерживания
Хроматограмма - зарегистрированная во времени последовательность показаний регистрирующего прибора. Общий вид хроматограммы смеси двух компонентов А и В приведен на рис. 3-7.
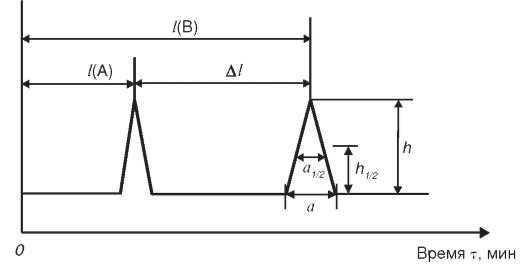
Рис. 3-7. Схематическое изображение хроматограммы двухкомпонентной смеси
Пики на хроматограмме условно изображены в виде равнобедренных треугольников. Положение пика характеризуется временем от момента ввода пробы в испаритель (нулевая точка на оси времени) до момента появления максимума пика компонента на хроматограмме. Это время называется временем удерживания компонента  или
или  . Аналогично положение пика на хроматограмме можно охарактеризовать расстоянием удерживания
. Аналогично положение пика на хроматограмме можно охарактеризовать расстоянием удерживания 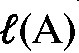 или
или 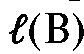 , которое равно расстоянию на хроматограмме от точки ввода пробы в испаритель до максимума пика соответствующего вещества.
, которое равно расстоянию на хроматограмме от точки ввода пробы в испаритель до максимума пика соответствующего вещества.
В общем случае и время удерживания и расстояние удерживания, называемые параметрами удерживания, зависят от природы вещества и условий хроматографирования. При постоянных условиях хроматографирования эти величины являются постоянными для данного вещества и используются для качественной идентификации компонентов анализируемой смеси.
2.2. Степень разделения
Степень разделения двух компонентов смеси 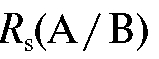 - количествен-
- количествен-
ная характеристика качества разделения двух компонентов А и В анализируемой смеси и определяется по положению соответствующих пиков на хроматограмме (см. рис. 3-7):
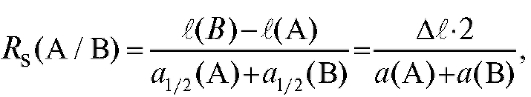
где 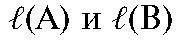 - расстояния удерживания компонентов А и В соответственно, мм;
- расстояния удерживания компонентов А и В соответственно, мм;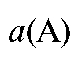
и а(В) - ширина пиков у основания компонентов А и В соответственно, мм;
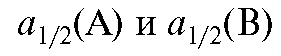 - ширина пиков компонентов А и В на середине их высоты соответственно, мм.
- ширина пиков компонентов А и В на середине их высоты соответственно, мм.
Для равнобедренного треугольника справедливо соотношение:
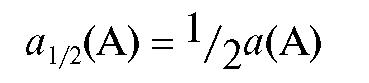
При 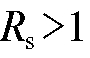 соответствующие пики не перекрываются и разделение веществ хорошее. С ростом величины степени разделения время анализа увеличивается. При значениях
соответствующие пики не перекрываются и разделение веществ хорошее. С ростом величины степени разделения время анализа увеличивается. При значениях 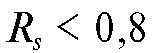 пики перекрываются и разделе-
пики перекрываются и разделе-
ние соответствующих веществ неудовлетворительное. Оптимальным считается разделение веществ при 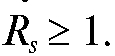
2.3. Эффективность хроматографического процесса
Эффективность хроматографического процесса зависит от условий хроматографирования и качества колонки, которое характеризуется следующими основными параметрами.
2.3.1. Число теоретических тарелок
Теоретической тарелкой (т.т.) называется участок зоны внутри хроматографической колонки, на котором устанавливается динамическое равновесие, характеризующее распределение компонента между ПФ
и НФ.
Число теоретических тарелок n в колонке для каждого компонента рассчитывается по хроматографическим данным по формуле:
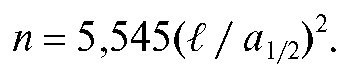
Чем больше число т.т., тем больше число переходов из НФ в ПФ и обратно совершает каждый компонент в процессе прохождения колонки, тем лучше его разделение с другими компонентами и тем выше эффективность хроматографической колонки.
2.3.2. Высота, эквивалентная теоретической тарелке
Высота, эквивалентная теоретической тарелке,- длина участка хроматографической колонки (эквивалентная т.т.), на котором устанавливается динамическое распределение компонента между ПФ и НФ. Высота, эквивалентная теоретической тарелке, зависит от длины хроматографической колонки L и рассчитывается по формуле:
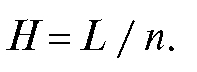
Чем меньше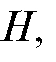 тем больше число т.т. в данной колонке, тем выше эффективность работы такой колонки.
тем больше число т.т. в данной колонке, тем выше эффективность работы такой колонки.
3. Практика метода газожидкостной хроматографии
3.1. Качественный анализ
Качественный анализ состава смеси методом ГЖХ основан на сравнении параметров удерживания стандартных веществ («свидетелей») и компонентов анализируемой смеси. Для этого сначала определяют расстояние (время) удерживания стандартного вещества, присутствие которого предполагается в анализируемой смеси. Затем в тех же условиях хроматографируется анализируемая смесь. Совпадение параметров удерживания стандарта и i-го компонента смеси является основанием для качественной идентификации соответствующего компонента анализируемой смеси.
3.2. Количественный анализ
В основе количественного анализа методом ГЖХ лежит зависимость между площадью пика i-го компонента 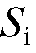 на хроматограмме и массой
на хроматограмме и массой 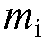 (или массовой долей) этого компонента в смеси:
(или массовой долей) этого компонента в смеси:
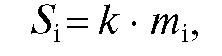
где 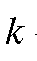 - коэффициент пропорциональности.
- коэффициент пропорциональности.
Площадь пика компонента на хроматограмме часто приближенно (с ошибкой до нескольких процентов) рассчитывается по формуле для равнобедренного треугольника:
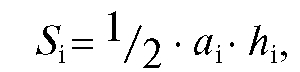
где 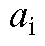 - ширина пика i-го компонента у основания;
- ширина пика i-го компонента у основания; 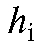 - высота пика i-го компонента.
- высота пика i-го компонента.
Более точно площадь пиков на хроматограмме измеряется интегратором хроматографа.
3.2.1. Метод абсолютной калибровки
Готовят серию эталонных растворов анализируемых веществ с известными массами 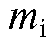 компонентов (или концентрацией), хроматографируют их, рассчитывают площадь пиков
компонентов (или концентрацией), хроматографируют их, рассчитывают площадь пиков 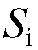 на хроматограммах и строят градуировочный график для этих веществ в координатах
на хроматограммах и строят градуировочный график для этих веществ в координатах 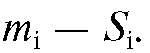 Затем
Затем
в тех же условиях хроматографируют анализируемую смесь, определяют площадь пика, соответствующего i-му компоненту на хроматограмме, и по градуировочному графику определяют массу (или концентрацию) этого вещества в анализируемой смеси.
3.2.2. Метод внутренней нормализации
Хроматографируют анализируемую смесь, рассчитывают площадь всех пиков на хроматограмме и определяют массовую долю каждого i-го компонента смеси по формуле:
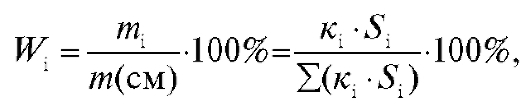
где 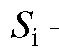 - площадь пика i-го компонента на хроматограмме;
- площадь пика i-го компонента на хроматограмме; 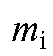 - масса i-го компонента; m (см) - масса смеси;
- масса i-го компонента; m (см) - масса смеси; 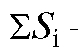 - сумма площадей пиков всех компонентов;
- сумма площадей пиков всех компонентов; 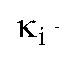 - коэффициент пропорциональности для i-го компонента.
- коэффициент пропорциональности для i-го компонента.
Метод применим для анализа смеси, все компоненты которой элюируются (записываются регистратором хроматографа) в данных условиях. Если коэффициенты пропорциональности к можно принять равными для всех компонентов, то: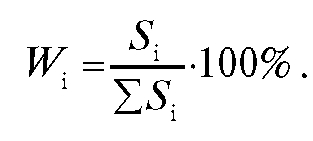
Эту формулу часто используют на практике.
3.2.3. Метод внутреннего стандарта
Готовят несколько (часто - пять) эталонных смесей, каждая из которых включает точно известную массу 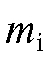 определяемого компонента и массу
определяемого компонента и массу 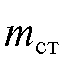 стандарта. В строго одинаковых условиях хроматографируют каждую смесь и на полученных хроматограммах измеряют площади
стандарта. В строго одинаковых условиях хроматографируют каждую смесь и на полученных хроматограммах измеряют площади 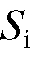 пиков определяемого вещества и площадь
пиков определяемого вещества и площадь 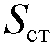 стандарта.
стандарта.
Площадь пика на хроматограмме прямо пропорциональна массе данного вещества:
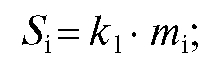
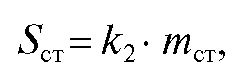
поэтому: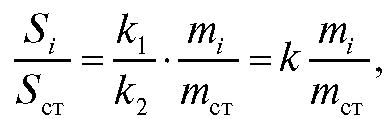
где коэффициент пропорциональности 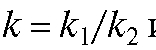 или обратную ему величину
или обратную ему величину 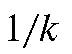
называют поправочным коэффициентом.
Затем к анализируемому раствору, содержащему неизвестную массу mx определяемого вещества, прибавляют точно известную массу стандарта 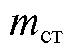 и хроматографируют полученный раствор в тех же условиях, что и эталонные растворы, после чего измеряют площадь
и хроматографируют полученный раствор в тех же условиях, что и эталонные растворы, после чего измеряют площадь 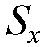 и
и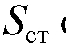 обоих пиков.
обоих пиков.
Иногда, наоборот, к раствору стандарта прибавляют определенное количество определяемого вещества.
По полученным данным вычисляют соотношение 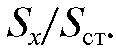 Окончательную обработку результатов можно проводить либо методом градуировочного графика, либо расчетным путем.
Окончательную обработку результатов можно проводить либо методом градуировочного графика, либо расчетным путем.
В первом случае строят градуировочный график в координатах 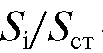 -
- 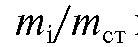 и затем, зная измеренную величину
и затем, зная измеренную величину 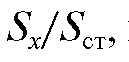 находят по графику со-
находят по графику со-
отношение 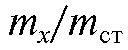 и массу
и массу 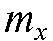 определяемого вещества.
определяемого вещества.
Во втором случае с использованием найденного поправочного коэффициента рассчитывают отношение 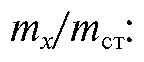
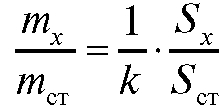
и затем, зная 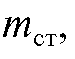 вычисляют массу
вычисляют массу 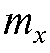 определяемого вещества.
определяемого вещества.
В качестве стандарта используют вещества, родственные определяемому. Чем меньше различаются площади 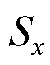 и
и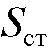 , тем меньше ошибка определения, поэтому анализ обычно проводят в таких условиях, когда
, тем меньше ошибка определения, поэтому анализ обычно проводят в таких условиях, когда
площади 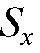 и
и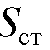 - соизмеримы. Пики стандарта и определяемого вещества не должны перекрываться.
- соизмеримы. Пики стандарта и определяемого вещества не должны перекрываться.
3.2.4. Метод внешнего стандарта (метод стандарта, метод стандартного образца)
Готовят анализируемый раствор, содержащий определяемый компонент, и стандартный раствор, содержащий стандартный образец определяемого вещества в том же растворителе. Концентрацию стандартного раствора стараются подобрать близкой к концентрации определяемого вещества в анализируемом растворе. Последовательно хроматографируют оба раствора в одинаковых условиях и измеряют площади пиков определяемого вещества на обеих хроматограммах.
Пусть S и S (ст) - соответственно площадь пика определяемого вещества на хроматограмме анализируемого и стандартного растворов. Тогда:
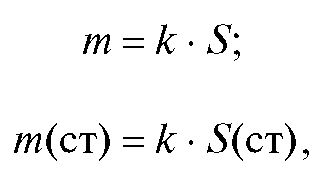
где т и m(ст) - соответственно масса определяемого вещества в анализируемом и стандартном растворах;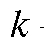 - коэффициент пропорциональности.
- коэффициент пропорциональности.
После простых преобразований вычисляем m:
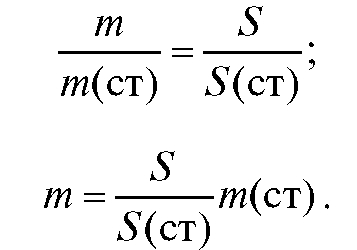
Зная массу т определяемого вещества в объеме пробы, взятой для хроматографирования, можно рассчитать массу, концентрацию, процентное содержание этого вещества во всем объеме анализируемого раствора.
Метод стандартного образца часто используют на практике, однако он требует наличия высокочистого стандартного образца с точно известным содержанием определяемого вещества. Приготовление стандартных образцов - трудоемкий и дорогостоящий процесс.
4. Высокоэффективная жидкостная хроматография
Несмотря на достоинства методов ГЖХ, они неприменимы для разделения и определения веществ с высокой молярной массой (более ~300), нелетучих, термически нестойких, ионогенных соединений.
Эти недостатки отсутствуют в методе высокоэффективной жидкостной хроматографии.
Высокоэффективная жидкостная хроматография, или жидкостная хроматография высокого давления, основана на тех же принципах, что и ГЖХ, только вместо газа-носителя в качестве ПФ применяется поток жидкости, не смешивающейся с жидкой НФ хроматографической колонки. Таким образом, в высокоэффективной жидкостной хроматографии обе контактирующие фазы - НФ и ПФ - жидкости. Разделение компонентов основано на различии их коэффициентов распределения между НФ и ПФ.
Температура хроматографической колонки может быть комнатной, что позволяет хроматографировать белки, аминокислоты и другие термически нестойкие соединения. Молярная масса разделяемых веществ может достигать ~2000.
Расчеты содержания определяемых веществ в анализируемых пробах в методах высокоэффективной жидкостной хроматографии проводятся теми же способами, что и в методах ГЖХ.
Практическое занятие по газожидкостной хроматографии
Цели занятия
Закрепление теоретических основ метода ГЖХ, приобретение навыков работы с газожидкостным хроматографом, проведение качественного и количественного анализа смеси веществ методом ГЖХ.
Целевые задачи
1. Идентификация компонентов анализируемой смеси.
2. Расчет степени разделения компонентов анализируемой смеси.
3. Расчет эффективности газохроматографической колонки.
4. Расчет массовой доли компонентов в анализируемой смеси.
Задание для самоподготовки
К занятию необходимо знать
1. Сущность метода ГЖХ.
2. Принципиальную схему газожидкостного хроматографа.
3. Сущность метода идентификации веществ по временам (расстояниям) удерживания.
4. Принцип метода расчета количественного состава смеси по данным хроматографического анализа.
Уметь
1. Определять времена (расстояния) удерживания компонентов на хроматограмме.
2. Рассчитывать площадь хроматографического пика.
3. Рассчитывать массовые доли компонентов в смеси методом внутренней нормализации.
Список литературы
1. Лекции «Применение хроматографических методов в количественном анализе».
2. Учебник. Книга 2, глава 9, разд. 9.2. - С. 414-435; 438-444.
Вопросы для самопроверки
1. Перечислите варианты газовой хроматографии, отличающиеся механизмом разделения веществ.
2. Какие типы колонок применяются в газовой хроматографии?
3. Укажите типы неподвижных фаз, применяемые в газовой хроматографии.
4. Какие носители применяются в наполненных колонках? Перечислите требования к ним.
5. Как определяются времена (расстояния) удерживания компонентов в газовой хроматографии?
6. Как оценивают эффективность работы хроматографической колонки?
7. Как оценивают качество разделения компонентов в методе
ГЖХ?
8. В каком случае для определения количественного состава смеси можно применять метод внутренней нормализации?
Лабораторная работа «Анализ смеси
«ацетон-гексан-бензол»
методом газожидкостной хроматографии»
Сущность работы
Качественный анализ смеси проводят на основании сравнения времени удерживания компонентов смеси со временем удерживания эталонов ацетона, гексана и бензола. Количественное содержание ацетона, гексана и бензола в анализируемой смеси проводят с применением метода внутренней нормализации.
Хроматографирование анализируемой смеси веществ выполняют на газовом хроматографе того типа, который имеется в данной лаборатории. Целесообразно проводить хроматографирование смеси при давлении азота на входе 200 кПа, температуре испарителя 150 °C и температуре колонки 120 °C.
Современные хроматографы позволяют автоматически регистрировать на хроматограмме площади пиков и времена удерживания компонентов смеси, поэтому практическое определение расстояний удерживания разделяемых веществ и расчет площадей соответствующих пиков носят обучающий характер и способствуют выработке навыков у студентов при работе с хроматограммами.
Реактивы
1. Ацетон (ч.д.а.).
2. Бензол (ч.д.а.).
3. Гексан (ч.д.а.).
4. Растворитель для промывания микрошприца.
Учебная таблица: принципиальная схема газового хроматографа. Прочие принадлежности: ножницы, линейка, клей.
1. Идентификация компонентов анализируемой смеси
Сущность методики
Идентификация веществ в многокомпонентной смеси основана на сравнении расстояний удерживания эталонных веществ с расстояниями удерживания отдельных компонентов анализируемой смеси.
Методика
Сначала хроматографируют эталонные вещества. С использованием хроматографического шприца в испаритель хроматографа вводят 0,5 мкл ацетона и получают хроматограмму, на которой имеется только один пик ацетона. Определяют расстояние удерживания ацетона 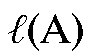 . Аналогично получают хроматограммы двух других эталонов - гексана и бензола и определяют расстояния удерживания этих веществ
. Аналогично получают хроматограммы двух других эталонов - гексана и бензола и определяют расстояния удерживания этих веществ 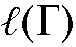 и
и 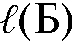 . Полученные данные заносят в таблицу.
. Полученные данные заносят в таблицу.
Затем в тех же условиях хроматографируют полученную задачу, содержащую два или три компонента. Для этого 1 мкл анализируемой смеси вводят в испаритель хроматографа и получают хроматограмму, на которой число пиков равно числу компонентов в смеси. Измеряют расстояния удерживания соответствующих компонентов смеси (1, 2, 3) на хроматограмме 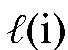 и полученные значения заносят в таблицу. Сравнивают расстояние удерживания пика на хроматограмме смеси и расстояние удерживания эталона; их совпадение является основанием для
и полученные значения заносят в таблицу. Сравнивают расстояние удерживания пика на хроматограмме смеси и расстояние удерживания эталона; их совпадение является основанием для
идентификации сооответствующего компонента смеси. По полученным данным делается вывод о составе анализируемой смеси. Хроматограммы вклеивают в лабораторный журнал.
2. Расчет степени разделения веществ
На полученной хроматограмме анализируемой смеси измеряют расстояние удерживания и ширину пика на середине высоты для каждого компонента. Полученные данные заносят в таблицу.
На основании полученных хроматографических данных рассчитывают степени разделения двух пар веществ: «ацетон-гексан» и «гексан- бензол». Полученные данные заносят в таблицу.
3. Расчет эффективности колонки
Эффективность хроматографической колонки характеризуется числом теоретических тарелок и рассчитывается для каждого компонента смеси по хроматограмме эталона. Затем рассчитывается высота, эквивалентная теоретической тарелке (ВЭТТ), для каждого компонента смеси, и полученные данные заносят в табл. 3-8.
Таблица 3-8. Параметры удерживания веществ и эффективность колонки
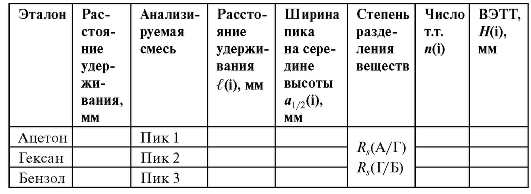
Примечание: т.т.- теоретическая тарелка.
4. Определение количественного состава многокомпонентной смеси
На полученной хроматограмме анализируемой смеси измеряют высоту и ширину пика на середине высоты для каждого компонента смеси и рассчитывают площадь каждого пика. Затем с использованием метода внутренней нормализации рассчитывают массовую долю каждого компонента в анализируемой смеси.
Контрольные вопросы
1. Укажите механизм разделения веществ в ГЖХ.
2. Перечислите основные блоки хроматографа.
3. Объясните принцип действия кондуктометрического и пламенно-ионизационного детекторов.
4. Укажите условие разделения двух веществ в ГЖХ.
5. Для каких целей используются времена (расстояния) удерживания в ГЖХ?
6. Для чего используются величины высоты и ширины пика на середине высоты в ГЖХ?
7. Укажите значения величины степени разделения двух пиков, обеспечивающие удовлетворительное разделение веществ.
8. Что характеризует число т.т, и от чего эта величина зависит?
9. Перечислите способы количественной оценки состава анализируемой смеси, используемые в ГЖХ.
Занятие 3. Ионообменная хроматография Общая характеристика метода
Ионообменная хроматография - метод разделения и количественного анализа катионов и анионов в растворе, основанный на обратимом стехиометрическом ионном обмене между ионитом (неподвижной фазой) и анализируемым раствором (подвижной фазой).
Иониты - твердые, нерастворимые в воде вещества, состоящие из матрицы и ионогенных групп, которые включают прочно связанные с матрицей фиксированные и подвижные ионы, способные обмениваться с одноименными ионами (с тем же знаком заряда) анализируемого раствора. В зависимости от природы ионогенных групп различают катиониты, способные обмениваться с раствором катионами, и аниониты, способные обмениваться с раствором анионами.
Известны также амфотерные иониты (амфолиты), способные обмениваться с раствором как катионами, так и анионами.
Состав сильнокислотного катионита в Н-форме в общем виде можно выразить в виде формулы:
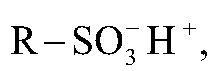
где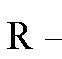 матрица ионита (обычно полимерная синтетическая смола);
матрица ионита (обычно полимерная синтетическая смола); 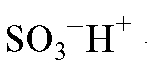 -
-
ионогенная группа; 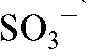 - фиксированный ион, связанный с матрицей проч-
- фиксированный ион, связанный с матрицей проч-
ной ковалентной связью, и поэтому в процессе ионного обмена отщепляться не может; 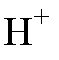 - подвижный ион, который в процессе ионного обмена переходит в раствор (подвижную фазу) и замещается на катион из анализируемого раствора.
- подвижный ион, который в процессе ионного обмена переходит в раствор (подвижную фазу) и замещается на катион из анализируемого раствора.
Аналогично состав основного анионита в ОН-форме можно выразить в виде формулы:
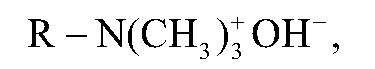
где 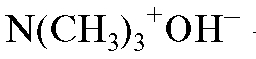 - ионогенная группа;
- ионогенная группа; 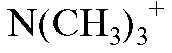 - фиксированный ион, свя-
- фиксированный ион, свя-
занный с матрицей прочной ковалентной связью, и поэтому в процессе ионного обмена отщепляться не может; 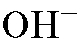 - подвижный ион, который в процессе ионного обмена переходит в раствор (подвижную фазу) и замещается на анион из анализируемого раствора.
- подвижный ион, который в процессе ионного обмена переходит в раствор (подвижную фазу) и замещается на анион из анализируемого раствора.
Заменим фиксированный ион в катионите на 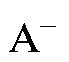 и в анионите на
и в анионите на 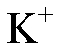 и запишем формулы ионитов в более простой форме
и запишем формулы ионитов в более простой форме 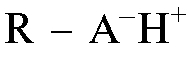 и
и 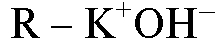 соответственно.
соответственно.
При пропускании раствора с анализируемыми катионами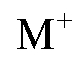 через катионит происходит процесс катионного обмена, который можно записать в виде:
через катионит происходит процесс катионного обмена, который можно записать в виде:
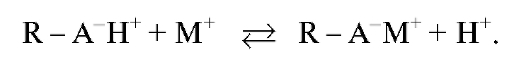
Катион металла вытесняет ион водорода из катионита, сорбируется на нем и удерживается в ионогенной группе. Этот процесс возможен потому, что способность к сорбции на катионите у большинства ионов металлов больше, чем у иона водорода. Ионы водорода выделяются в раствор в количестве, эквивалентном содержанию ионов металла в анализируемом растворе, и уносятся с подвижной фазой.
В результате катионного обмена катионит из Н-формы переходит в солевую форму 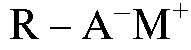 , которая содержит сорбированные из рас-
, которая содержит сорбированные из рас-
твора катионы. Для выделения этих катионов из катионита равновесие ионного обмена необходимо сместить в сторону обратной реакции. Для этого катионит в солевой форме промывают раствором кислоты, и он переходит в Н-форму (происходит регенерация катионита) по схеме:
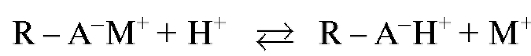
Катионы переходят в подвижную фазу и собираются на выходе из колонки для дальнейшего количественного анализа.
Количественное определение анионов проводится аналогичным образом при пропускании раствора с анализируемыми анионами 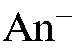 через
через
анионит. В соответствии с сорбционной способностью анионов происходит анионный обмен:
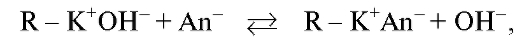
и анионы из раствора сорбируются на анионите, который переходит в солевую форму 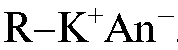 . Затем анионит в солевой форме промывают
. Затем анионит в солевой форме промывают
раствором основания:
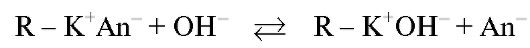
и происходит регенерация анионита. Анионы переходят в подвижную фазу и собираются на выходе из колонки для дальнейшего количественного анализа.
В лабораторных условиях ионообменную хроматографию проводят в хроматографических колонках, представляющих собой обычно стеклянные бюретки, заполненные ионитом, с краном в нижней части для выпуска элюата.
Методы ионообменной хроматографии (ИОХ) применяют в фармакопейном анализе. Например, ионообменная хроматография на катионитах применяется для определения лекарственных веществ, представляющих собой соли органических оснований (гидрохлоридов папаверина, хинина). При определении лекарственных веществ, содержащих ионогенно связанные анионы (атропина сульфат, скополамина гидробромид), применяют ионообменную хроматографию с использованием анионитов.
Цели занятия
Закрепление теоретических основ метода ИОХ, приобретение навыков работы с ионообменными хроматографическими колонками, разделение ионов, содержащихся в анализируемом растворе, и проведение количественного анализа ионов железа(III) и меди(II) с применением метода ИОХ.
Целевые задачи
1. Разделение ионов в анализируемом растворе.
2. Количественное определение катионов в анализируемом растворе.
Задание для самоподготовки
К занятию необходимо знать
1. Сущность метода ИОХ.
2. Классификацию ионитов, их состав и свойства.
3. Сущность процессов ионного обмена на катионитах и анионитах.
4. Возможности количественного определения состава анализируемого раствора по результатам ИОХ.
Уметь
1. Готовить хроматографическую колонку для проведения ионного обмена.
2. Готовить элюирующие растворы (элюенты) для проведения анализа.
3. Собирать необходимые фракции элюата при проведении анализа.
4. Применять титриметрические и физико-химические методы для количественного анализа веществ.
Список литературы
1. Лекции «Применение хроматографических методов в количественном анализе».
2. Учебник. Книга 2, глава 9, разд. 9.1. - С. 402-414; 435-438.
Вопросы для самопроверки
1. Укажите в общем виде состав ионитов и поясните роль матрицы, ионогенных групп и подвижных ионов в ионитах в процессе анализа.
2. Перечислите кислотные катиониты и укажите интервал рН обмена.
3. Перечислите основные аниониты и укажите интервал рН обмена.
4. На чем основан процесс ионного обмена при проведении ИОХ?
5. Что характеризует константа ионного обмена и какие она должна принимать значения?
6. Что такое элюат, элюент?
Лабораторная работа «Разделение ионов железа(III) и меди(II) и их количественное определение в растворе»
Сущность работы
Метод основан на переводе одного из анализируемых катионов (иона железа) в комплексный анион, разделении катионов и анионов в растворе с использованием ионобменной хроматографии на катионите и количественном определении катионов и анионов в раздельно собранных растворах.
Приборы и оборудование
1. Фотоэлектроколориметр (например, типа КФК-2).
2. Хроматографическая колонка, заполненная катионитом (рекомендуется катионит КУ-2).
Реактивы
1. Раствор хлороводородной кислоты с(НС1) = 1 моль/л.
2. Раствор хлороводородной кислоты с(НС1) = 4 моль/л.
3. Раствор сульфосалициловой кислоты (10%).
4. Раствор аммиака (10%).
5. Фильтровальная бумага, пропитанная 
6. Стандартизованный раствор натрия тиосульфата  = = 0,0500 моль/л.
= = 0,0500 моль/л.
1. Подготовка хроматографической колонки к работе
Стеклянную бюретку на 25 мл заполняют катионитом и переводят его в Н-форму. Для этого в верхнюю часть колонки через воронку вносят раствор 1 моль/л хлороводородной кислоты и пропускают 20 мл этого раствора через колонку со скоростью 1-2 мл/мин (что соответствует вытеканию раствора из носика колонки со скоростью 1-2 капли в секунду). После этого через колонку пропускают 20 мл дистиллированной воды с той же скоростью.
ВНИМАНИЕ! При проведении любых операций с хроматографической колонкой над катионитом всегда должен быть столбик жидкости не менее 1-2 см, чтобы в ионит не попали пузырьки воздуха.
2. Разделение катионов железа(III) и меди(II)
Сущность методики
Катионы меди(II) и железа(III), содержащиеся в анализируемом растворе, переводят в комплексные ионы с противоположными зарядами, добавляя в раствор сульфосалициловую кислоту и аммиак. При этом ион меди реагирует с аммиаком с образованием положительно заряженного комплексного катиона синего цвета:
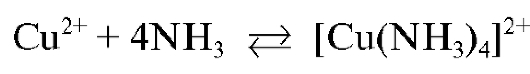
При взаимодействии катиона железа с сульфосалициловой кислотой образуются предположительно при рН ~9,0-15,0 комплексные трисульфосалицилатоферрат(III)-ионы желтого цвета, заряженные отрицательно (комплексы анионного типа) с максимумом поглощения в спектре поглощения около 416 нм.
В результате в анализируемом растворе ионы меди находятся в составе катионного комплекса, а ионы железа - в составе анионного ком-
плекса. При пропускании этого раствора через катионит комплексный катион меди будет сорбироваться на нем в результате протекания реакции ионного обмена:
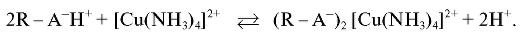
Анионный комплекс железа с катионитом не взаимодействует, уносится с подвижной фазой и собирается в мерной колбе на выходе из колонки для последующего анализа.
Для дальнейшего элюирования сорбированных на катионите комплексных катионов меди колонку промывают раствором хлороводородной кислоты. При этом равновесие сорбции комплексных ионов меди на катионите смещается в сторону обратной реакции; комплексный тетрамминмедь(II)-катион снова переходит в раствор и разрушается под действием хлороводородной кислоты:
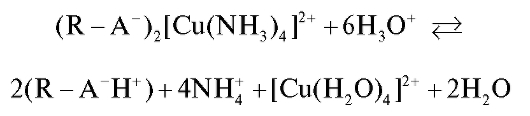
Полученные комплексные ионы меди(II) голубого цвета элюируются из колонки и собираются на выходе из колонки для дальнейшего анализа.
2.1. Выделение из анализируемого раствора ионов железа
Методика
Анализируемый раствор получают в химическом стаканчике (один на двух студентов). К этому раствору добавляют из бюреток 5 мл раствора сульфосалициловой кислоты и затем 10 мл раствора аммиака. Раствор окрашивается в темно-зеленый цвет. Получившийся раствор количественно переносят в хроматографическую колонку, пропуская его со скоростью вытекания 1-2 капли в секунду. Первые 10 мл бесцветного элюата, не содержащего комплексы железа, собирают в градуированную пробирку и отбрасывают. Затем весь элюат, вытекающий из колонки, собирают в мерную колбу на 50 мл. Для количественного перенесения анализируемого раствора в колонку в пустой стаканчик добавляют 2 мл раствора сульфосалициловой кислоты, 2 мл раствора аммиака и пропускают полученный раствор через колонку, собирая элюат в ту же мерную колбу. Эту операцию повторяют дважды.
Затем колонку последовательно промывают элюентами следующих составов:
1) 5 мл раствора сульфосалициловой кислоты и 10 мл раствора аммиака;
2) 3 мл раствора сульфосалициловой кислоты, 3 мл раствора аммиака и 2 мл дистиллированной воды;
3) 1 мл раствора сульфосалициловой кислоты, 1 мл раствора аммиака и 6 мл дистиллированной воды.
Во время промывания колонки поддерживают постоянную скорость вытекания элюата (1-2 капли в секунду). Собранный раствор в мерной колбе доводят дистиллированной водой до метки, перемешивают и оставляют для количественного определения ионов железа(III).
2.2. Выделение ионов меди(II) из анализируемого раствора
Методика
Ионы меди(II) элюируются с катионита промыванием колонки 4 моль/л хлороводородной кислотой. В колонку вносят 10 мл раствора кислоты и выходящий элюат собирают в мерной колбе вместимостью 50 мл. Эту операцию повторяют дважды. Затем колонку промывают дистиллированной водой порциями по 10 мл до полного элюирования меди(II) из колонки, собирая элюат в ту же колбу. Для проверки отсутствия меди(II) в последней порции элюата несколько капель этого элюата помещают на фильтровальную бумагу, предварительно пропитанную калия гексацианоферратом(II). Отсутствие красно-бурого осадка или окрашивания бумаги свидетельствует о полноте элюирования меди(II) из колонки. Собранный раствор в мерной колбе доводят дистиллированной водой до метки, перемешивают и оставляют для количественного определения меди(II).
3. Определение массы ионов меди(II) в растворе
Методика
Содержание меди(II) в растворе определяют йодометрически. В колбу для титрования отбирают с помощью пипетки 2 мл анализируемого раствора, добавляют 2 мл 10% раствора калия йодида, накрывают стеклом и оставляют на 10 мин в темном месте. Выделившийся йод титруют раствором натрия тиосульфата до бледно-желтой окраски, прибавляют 0,5 мл свежеприготовленного раствора крахмала и продолжают титровать до исчезновения синей окраски. Массу меди(II) в анализируемом растворе рассчитывают в граммах с точностью до трех значащих цифр с использованием закона эквивалентов:
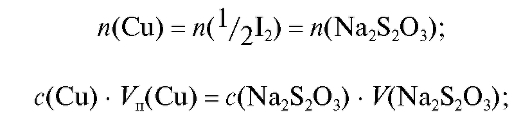
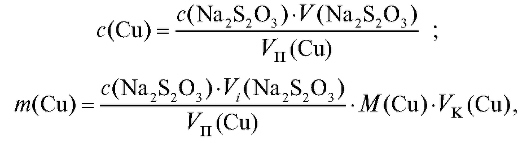
где 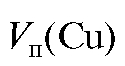 = 0,002 л;
= 0,002 л; 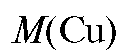 = 63,546 г/моль
= 63,546 г/моль 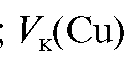 = 0,050 л.
= 0,050 л.
4. Определение массы железа(III) в растворе
Методика
Содержание железа(III) в анализируемом растворе определяют фотометрически методом одного стандарта. Собранный в мерной колбе раствор трисульфосалицилата железа(III) фотометрируют на фотоэлектроколориметре (например, типа КФК-2) при светофильтре 400 нм в кюветах 1 см относительно дистиллированной воды и получают значение оптической плотности A (Х).
Затем готовят эталонный раствор трисульфосалицилата железа(III). Для этого в мерную колбу на 50 мл с помощью пипетки помещают 2 мл эталонного комплекса железа(III) с концентрацией железа(III) 0,10 мг/мл, добавляют 10 мл дистиллированной воды, 5 мл раствора сульфосалициловой кислоты, 5 мл раствора аммиака, доводят объем раствора дистиллированной водой до метки и перемешивают. Полученный раствор фотометрируют в тех же условиях, что и анализируемый раствор, и получают значение оптической плотности А(эт). Массу железа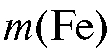 в анализируемом растворе объемом 50 мл рассчитывают в миллиграммах с точностью до двух значащих цифр по формуле:
в анализируемом растворе объемом 50 мл рассчитывают в миллиграммах с точностью до двух значащих цифр по формуле:
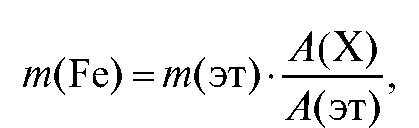
где ,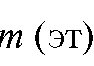 = 0,20 мг - масса железа(III) в эталонном растворе объемом 50 мл.
= 0,20 мг - масса железа(III) в эталонном растворе объемом 50 мл.
По окончании работы в колонку заливают около 10 мл 1 моль/л раствора хлороводородной кислоты и медленно пропускают ее через колонку до достижения уровня раствора над катионитом 2-3 см.
Контрольные вопросы
1. Как проводят подготовку катионита к проведению на нем катионного обмена?
2. Почему анализируемый раствор окрашивается в темно-зеленый цвет при добавлении к нему сульфосалициловой кислоты и аммиака?
3. На чем основано разделение катионов меди(II) и железа(III) при пропускании их через катионит?
4. Укажите элюенты и элюаты на различных стадиях проведения процесса анализа.
5. Сформулируйте условие возможности применения метода одного стандарта для определения железа(III) в данной работе.
6. Укажите метод титрования, использованный для количественного определения меди в растворе.
Лабораторная работа. «Определение массовой доли калия хлорида в образце»
Сущность работы
Метод основан на проведении ионного обмена ионов калия из анализируемого раствора на катионите и на количественном определении ионов водорода методом титриметрического анализа.
При пропускании раствора с анализируемыми катионами калия через катионит в Н-форме происходит процесс ионного обмена, который можно записать в виде:
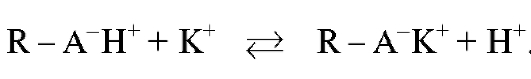
Катион металла вытесняет ион водорода из катионита, сорбируется на нем и удерживается в ионогенной группе. Ионы водорода выделяются в раствор в количестве, эквивалентном содержанию ионов калия в анализируемом растворе, уносятся с подвижной фазой и собираются на выходе из колонки для дальнейшего количественного анализа методом кислотно-основного титрования.
Приборы и оборудование
1. Фотоэлектроколориметр (например, типа КФК-2).
2. Хроматографическая колонка, заполненная катионитом (рекомендуется катионит КУ-2).
Реактивы
1. Раствор хлороводородной кислоты 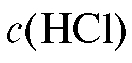 = 1 моль/л.
= 1 моль/л.
2. Универсальная индикаторная бумага.
3. Стандартизованный раствор натрия гидроксида с(NаОН) = = 0,0500 моль/л.
4. Водный раствор индикатора метилового оранжевого 0,1%.
Методика работы
1. Подготовка хроматографической колонки к работе
Стеклянную бюретку на 25 мл заполняют катионитом и переводят его в Н-форму. Для этого в верхнюю часть колонки через воронку вносят раствор 1 моль/л хлороводородной кислоты и пропускают 20 мл этого раствора через колонку со скоростью 1-2 мл/мин (что соответствует вытеканию раствора из носика колонки со скоростью 1-2 капли в секунду). Далее катионит промывают дистиллированной водой с той же скоростью до нейтральной реакции по индикатору метиловому оранжевому.
ВНИМАНИЕ! При проведении любых операций с хроматографической колонкой над катионитом всегда должен быть столбик жидкости не менее 1-2 см, чтобы в ионит не попали пузырьки воздуха.
2. Анализ образца соли
В мерной колбе на 250 мл готовят раствор калия хлорида с молярной концентрацией соли 0,1 моль/л. Теоретическую навеску соли 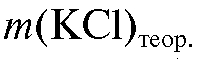 рассчитывают, исходя из содержания калия хлорида в образце 90-95%. Точную навеску образца m (соли) берут на аналитических весах и определяют по разности двух взвешиваний.
рассчитывают, исходя из содержания калия хлорида в образце 90-95%. Точную навеску образца m (соли) берут на аналитических весах и определяют по разности двух взвешиваний.
С помощью мерной пипетки 10,00 мл приготовленного раствора вводят в хроматографическую колонку и элюат, вытекающий из носика колонки, собирают в коническую колбу на 150-250 мл. Затем колонку промывают дистиллированной водой до нейтральной реакции элюата, собирая промывные воды в ту же коническую колбу. Полноту вымывания ионов водорода из колонки проверяют, поместив каплю элюата на универсальную индикаторную бумагу.
Собранный элюат титруют стандартизованным раствором натрия гидроксида в присутствии индикатора метилового оранжевого до перехода окраски титруемого раствора из золотисто-оранжевой в желтую.
Ионный обмен на катионите происходит строго стехиометрически, и закон эквивалентов для этой стадии имеет вид:
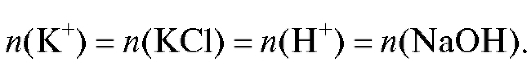
Отсюда массовая доля соли в образце, рассчитанная по результатам прямого титрования, равна:
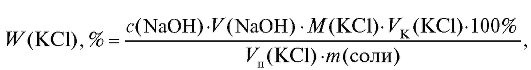
где 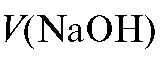 - объем раствора щелочи, израсходованный на титрование.
- объем раствора щелочи, израсходованный на титрование.