Пособие по клинической биохимии / Под ред. Л.В. Акуленко. - 2007. - 256 с.
|
|
|
|
ТЕМА 19 ГЕНЕТИКА И БИОХИМИЯ АПОПТОЗА (ЗАПРОГРАММИРОВАННОЙ КЛЕТОЧНОЙ ГИБЕЛИ)
Поддержание структурно-функционального постоянства тканей и органов биологических систем определяется сбалансированностью процессов отмирания и обновления клеток. В процессе эволюции выработались универсальные механизмы регулирования клеточной гибели и регенерации.
Термин «necrosis» был известен еще со времен Галена. Первое гистологическое описание смерти клеток опубликовано в 1859 г. Вирховым. Он использовал термины «дегенерация», «некроз», «умирание клеток» как синонимы гангрены. В 1950 г. в связи с открытием лизосом Де Дюв выдвинул концепцию клеточного суицида с использованием содержимого лизосом. В 1971 г. I.F.R. Kerr описал особый тип некроза, характеризующийся резким уменьшением объема клетки, как бы ее сжатием, и назвал такой вид гибели «сжатым некрозом». В 1972 г. I.F.R. Kerr с соавт. предложили для этого типа смерти клетки термин «апоптоз», в переводе с греческого означающий «листопад».
В настоящее время принято выделять два основных типа клеточной смерти: апоптоз и некроз.
Сходства некроза и апоптоза:
• изменение содержания кальция в клетках;
• оксидантный стресс;
• активация протеаз.
Морфологическая характеристика клеточной смерти подробно исследована.
В противоположность пассивному некрозу, вовлекающему группы клеток с поврежденными набухшими митохондриями и нарушенным электролитным балансом, апоптоз - активный, тонко регулируемый энергозависимый процесс, находящийся под генетическим контролем.
Апоптоз - двухфазный процесс.
• В первую фазу - латентную - в ответ на разные стимулы, связанные с повреждением ДНК (например, вирусная инфекция, радиация,
• несвойственная экспрессия генов), клетка «приговаривает» себя ко вступлению на путь «самоубийства». Длительность этой фазы варьирует от нескольких часов до нескольких дней. Во второй фазе - активной - клетка подвергается серии морфологических и функциональных изменений, кульминацией которых становится смерть в течение десятков минут. Апоптоз начинается с конденсации ядерного хроматина по периферии ядра, уменьшения последнего (пикноз), его фрагментации (кариорексис). Затем происходят конденсация, уплотнение клеточных органелл, уменьшение объема цитоплазмы. Мембраны приобретают пузырчатый вид. Потом происходит отпочковывание фрагментов клетки с образованием апоптических тел, окруженных мембраной и содержащих уплотненные остатки органелл и ядра. Апоптические тела утилизируются макрофагами или соседними паренхиматозными клетками, причем воспалительная реакция не развивается, что исключает повреждение других клеток.
Таблица 18-1. Морфологические и биохимические характеристики апоптической и некротической гибели клеток
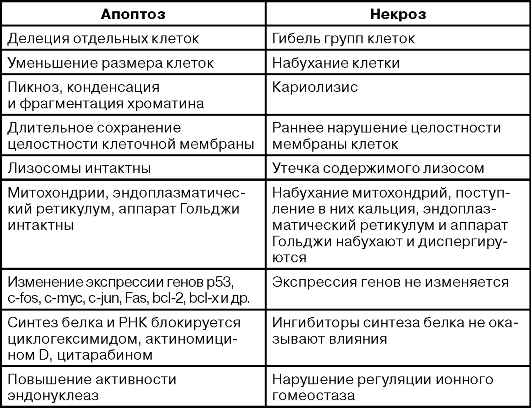 Таблица 18-1. Морфологические и биохимические характеристики апоптической и некротической гибели клеток (окончание)
Таблица 18-1. Морфологические и биохимические характеристики апоптической и некротической гибели клеток (окончание)
 Существует концепция, согласно которой выделяют два понятия:
Существует концепция, согласно которой выделяют два понятия:
• Апоптоз - комплекс морфологических и биохимических особенностей определенного способа клеточной смерти СФОРМИРОВАВШЕЙСЯ ткани ЗРЕЛОГО организма.
• Программированная клеточная гибель (ПКГ) - способ клеточной гибели РАЗВИВАЮЩЕЙСЯ ткани формирующейся биологической системы. Характерны незрелость регулирующих систем и особые тиггерные механизмы клеточного коллапса.
Farber E. (1994) предложил следующую классификацию программированной клеточной гибели:
• Программированная онтогенетически клеточная гибель, происходящая в ходе нормального развития и/или метаморфоза.
• Программированная физиологически гибель клеток взрослых организмов в ходе регрессии гиперплазированных в результате экзо- и эндогенных воздействий органов и тканей. Она проявляется лишь в случае необходимости восстановления клеточного равновесия. Сюда же относят гибель самообновляющихся клеток взрослого организма, например клеток лимфоидной и миелоидной тканей.
• Программированная биохимически клеточная гибель после воздействия патологических агентов различной природы. Этот тип гибели не физиологический, он представляет собой пассивную или активную реакцию на повреждающий стимул.
Выделяют пять типов проявлений апоптоза:
• Смерть клеток в процессе онтогенеза (например, ПКГ ответственна за регрессию личиночных органов в процессе метаморфоза).
• Смерть клеток тканей взрослых особей. Так, in vitro показано, что старение может сопровождаться апоптозом. Важная функция иммунной системы - разрушение лимфоцитов, овариального фолликула, инволюция волосяного фолликула путем апоптоза. Апоптоз уравновешивает результаты митоза в тканях.
• Смерть клеток в процессе патологической атрофии при гиперплазии в эндокринно-зависимых тканях, например, при атрофии простаты после кастрации и коры надпочечников после супрессии секреции АКТГ с помощью глюкокортикоидов и т.п. Патологическое разрастание (гиперплазия) вызывается стимуляцией митозов, а возвращение к нормальным объемам тканей опосредуется апоптозом.
• Альтруистический суицид клеток. Элиминация клеток-мутантов или клеток, пораженных вирусом. Фрагментация ДНК в этом случае предупреждает перенос генетического материала при фагоцитировании апоптозных телец.
• Клеточная смерть, вызванная слабым повреждением. Так, гипертермия клеток в культуре тканей в течение 30 мин при 43-44 °С приводит к апоптозу отдельных клеток, при 46-47 °С - массовый некроз, при 45 °С одновременно наблюдают признаки апоптоза и некроза.
Однако после запуска процесс гибели клетки реализуется с помощью универсальных биохимических механизмов. В основе всех форм ПКГ лежит генетически детерминированная программа клеточного «самоубийства». Об этом свидетельствуют участие ряда генов в реализации этой программы на уровне клетки и наличие специфических генов, контролирующих этот процесс.
Механизм запуска и исполнения программированной клеточной смерти до конца не ясен, но существуют доказательства, что апоптоз регулируется геном p53 и протоонкогенами bcl-2 и bcl-x. Дикий (т.е. нормальный) тип гена р53 (wT p53) индуцирует апоптоз, а протоонкогены bcl-2 и bcl-x его блокируют. В результате мутации дикого типа образуется мутантный тип гена p53 (mt p53) и клетка теряет способность к естественной смерти (апоптозу), вследствие чего развивается опухоль. Ген wt p53 - ген-супрессор, ингибирующий трансформацию клеток. Ген mt p53 - онкоген, наряду с другими онкогенами участвующий в механизмах канцерогенеза. Ген wt p53 не только индуцирует апоптоз, но и блокирует клеточный цикл совместно с геном с-myc на стадии G1 или перехода фазы G1 в S-фазу.
Протоонкоген bcl-2 - результат хромосомной транслокации (из позиции 18q21 в локус Н-цепи Ig в 14q32). Он приводит к гиперэкспрессии
белка bcl-2 в клетках. Белок bcl-2 - интегральный мембранный протеин, локализованный в митохондриальной и околоядерной мембранах. В экспериментальных системах этот белок защищает клетки от программированной клеточной смерти и дает онкогенный эффект, так как снижает апоптоз. Модуляция эндогенного уровня bcl-2 может быть физиологическим механизмом, контролирующим жизнь и смерть лимфоцитов.
Апоптоз и программированная клеточная гибель связаны с экспрессией на клетках белка Fas/АРО-1 (клетки иммунной системы, кишечного эпителия, печени, сердца, легких и яичников). Ген Fas/АРО-! кодирует трансмембранный рецептор Fas/АРО-1 (СД95). Этот ген локализован на участке q23 хромосомы 10 человека (на хромосоме 19 - у мыши). Естественный лиганд рецептора Fas - трансмембранная молекула. Fas вызывает апоптическую гибель клетки. Ген этого лиганда картирован на хромосоме 1 у мыши.
Антиген Fas/АРО^ экспрессирован у человека на кортикальных тимоцитах, активированных Т- и В-лимфоцитах, лимфобластоидных клеточных линиях, гепатоцитах, миелоидных клетках, некоторых типах эпителия и др. Количество этого антигена на лимфоцитах периферической крови зависит от возраста - экспрессия его прогрессивно повышается. Этот антиген представлен на СD8- и СD4-лимфоцитах, несущих СD45RO-антиген, являющийся маркером Т-клеток памяти. Экспрессия этого антигена регулируется экспозицией с интерлейки- ном-2, гамма-интерфероном и на СD34-клетках больных хроническим миелолейкозом в стадии бластного криза.
МЕХАНИЗМЫ, ИНИЦИИРУЮЩИЕ ГИБЕЛЬ КЛЕТКИ
Широкое развитие получили представления о значимости солей кальция (Са) для обеспечения функционирования клеток. Ионы Са и циклические нуклеотиды (цАМФ, цГМФ) - универсальные внутриклеточные посредники. Изменение концентрации цитозольного Са - уникальный внутриклеточный сигнал. В реализации ПКГ Са играет важную роль. Концентрация цитозольного Са составляет 10-7/моль, а во внеклеточной жидкости - 10-3/моль. При значительном повышении содержания Са в клетке за счет поступления по потенциал- и рецептор-зависимым каналам извне, за счет высвобождения ионов из внутриклеточных депо (в мышечных клетках - из саркоплазматического ретикулума, а в остальных - из кальциосом) активируется эндонуклеаза - фермент, способный фрагментировать
ДНК. Возрастание концентрации цитозольного Са сопровождается его переходом в активную форму посредством соединения с внутриклеточным рецептором кальмодулином. Активная форма кальция активирует кальмодулин-зависимые процессы. Ингибиторы кальмодулина блокируют апоптоз, вызванный глюкокортикоидами, без изменения уровня цитозольного кальция. Эти ингибиторы блокируют кальмодулин-опосредованное поступление кальция в ядро, что предполагает наличие Са-зависимых процессов в ядре при активации апоптоза. Увеличение содержания Са приводит к модулирующему воздействию на ядерную эндонуклеазу. При использовании блокаторов синтеза белка и РНК ингибируется увеличение Са.
В тимоцитах происходит увеличение количества белка кальпаина, являющегося Са-зависимой нейтральной протеазой. При ее активации разрушается цитоскелет и формируются поверхностные выступы (пузырьки). В присутствии ингибиторов кальпаина апоптоз блокируется.
Доказана опосредованная роль Са в митохондриальной генерации активных форм кислорода, образующихся при действии цитокинов клеток (фактора некроза опухолей - ФНО). Этот эффект может быть связан с действием Са на активность Са-опосредованной фосфолипазы, способной изменять наружный потенциал мембраны митохондрий и нарушать синтез АТФ. Кроме того, существуют некоторые пути инициации апоптоза, не затрагивающие метаболизм кальция:
• цитотоксическое действие антител к АРО-1;
• Т-клеточная цитотоксичность, связанная с антигеном Fas/АРО-1;
• гибель нейронов при устранении фактора роста нервов (ФРН).
Реализация ПКГ контролируется сложными биохимическими механизмами. В качестве стимула могут выступать различные биомолекулы-сигналы.
Взаимодействие клеточного рецептора с агонистом может опосредованно изменять концентрацию цитозольного кальция. Так, в результате экспрессии генов ПКГ появляются факторы, изменяющие проницаемость потенциал-зависимых Са-каналов, активность систем ионного обмена и вызывающих мобилизацию внутриклеточного кальция из депо.
Все многообразие действия различных активаторов апоптоза можно свести к нескольким системам, передающим сигнал. Активаторы могут влиять на различные звенья этих систем. Пока не найдено апоптоз-специфической системы, передающей сигнал, а перечисленные ниже каскады активируются и при других физиологических процессах клеток.
• Фосфолипаза-Са-зависимый путь передачи.
• Тирозинкиназный сигнальный путь.
• Сфингомиелиновый сигналопередающий путь активации апоптоза.
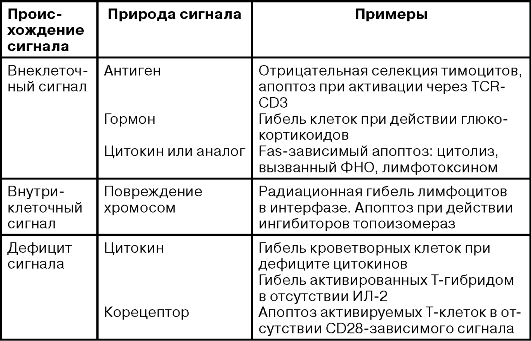
Таблица 18-2. Разновидности апоптоза
 ОКИСЛИТЕЛЬНЫЙ СТРЕСС КАК МЕДИАТОР АПОПТОЗА
ОКИСЛИТЕЛЬНЫЙ СТРЕСС КАК МЕДИАТОР АПОПТОЗА
Многие виды лечения, связанные с индукцией апоптоза, опосредуют свое действие через оксилительный стресс. Генерацию активных форм кислорода (АФК) и апоптоз способны вызывать различные воздействия: ионизирующее и ультрафиолетовое облучение, цитокины (ФНО) и другие прооксиданты. При добавлении антиоксидантов (например, тиоредоксин, N-ацетил-цистеин) происходит ингибирование цитотоксического действия ФНО. Появление избыточного количества АФК приводит к формированию окисленных липидов, полиненасыщенных жирных кислот и холестерина клеточной мембраны, способных индуцировать апоптоз. Увеличение АФК в клетке приводит к изменению фосфорилирования по тирозину многих белков, что может оказывать влияние на различные системы, передающие сигнал, участвующие в активации апоптоза. Не исключено и прямое действие АФК на определенные гены, вызывающие активацию апоптоза.
ВЛИЯНИЕ НА АПОПТОЗ ГЛЮКОКОРТИКОИДОВ И ИММУНОМОДУЛЯТОРОВ
Важное общебиологическое значение имеет возможность воздействия на избирательную регуляцию клеточной смерти. Г.В. Шмарина с соавт (1996) показали усиление апоптоза лимфоцитов периферической крови в присутствии иммуномодулятора синтетического гексапептида иммунофана. Распад ДНК на отдельные олигонуклеосомные фрагменты в лимфоцитах, инкубированных с ионами Са и Mg в присутствии иммунофана и/или циклогексимида (ингибитора синтеза белка), регистрировали на электрофореграмме. Выявленная фрагментация ДНК свидетельствует о высокой активности Са-, Mg-зависимой эндонуклеазы. В ядрах интактных лимфоцитов существенной активации эндонуклеазы в присутствии ионов Са и Mg не происходило. Аналогичное воздействие на индукцию апоптоза через активацию этого фермента выявлено при воздействии на мононуклеары глюкокортикоидных гормонов (дексаметазона). Са- и Mg-зависимая эндонуклеаза не синтезируется de novo под влиянием активаторов апоптоза, а постоянно присутствует в лимфоцитах периферической крови в виде неактивного предшественника в комплексе с белком-ингибитором. В интактных мононуклеарах активность фермента крайне низка и не превышает 30 абс. ед., часовая инкубация с дексаметазоном усиливает активность фермента в 3-5 раз (до 160 абс. ед.). Аналогичная активация отмечена и при обработке лимфоцитов иммунофаном. Снижение жизнеспособности клеток при окраске трипановым синим не выявлено. Однако зафиксированы признаки снижения метаболической активности митохондрий лимфоцитов при одновременной инкубации клеток с иммунофаном и глюкокортикоидами.
Таким образом, при стимуляции дексаметазоном или иммунофаном лимфоциты вступают в латентную фазу программированной клеточной гибели, что сопровождается снижением восстанавливающей функции митохондрий и активацией Са- и Mg-зависимой эндонуклеазы. На этой фазе клетки задерживаются по меньшей мере 24 ч, поэтому в этот период не обнаруживают ни фрагментации хроматина, ни снижения жизнеспособности клеток. Активатор апоптоза иммунофан снижает пролиферативный ответ лимфоцитов на Т-клеточный митоген Кон-А и ФГА. В условиях in vitro он повышает чувствительность клеток к антипролиферативному действию глюкокортикоидов.
ЗНАЧЕНИЕ АПОПТОЗА В ФУНКЦИОНИРОВАНИИ ИММУННОЙ СИСТЕМЫ
Развитие Т-клеток. Ген bcl-2 играет существенную роль в развитии Т-клеток. В вилочковой железе, где происходит созревание Т-клеток, тимоциты подвергаются позитивной и негативной селекции.
• Позитивная селекция состоит в отборе клеток с фенотипом TCR+CD4+CD8+, т.е. клеток, несущих дифференцировочные антигены CD4 и CD8 и Т-клеточный рецептор (TCR), способный связываться с антигенами I и II класса гистосовместимости микроокружения вилочковой железы. Только такие клетки выживают, а остальные погибают путем апоптоза.
• Негативная селекция заключается в элиминации клеток, имеющих ТСR-рецепторы, специфичные по отношению к собственным антигенам организма, представленным в вилочковой железе.
Таким путем создается естественная толерантность к «своему», в ее основе лежит удаление клонов аутореактивных Т-лимфоцитов путем апоптоза. Завершившие дифференцировку Т-лимфоциты TCR+CD4+CD8- и TCR+CD4-CD8+, имеющие TCR, специфичные к чужеродным для организма антигенам, покидают вилочковую железу и расселяются в периферических лимфоидных органах. Экспрессия генов bcl-2 и Fas на лимфоидных клетках имеет фазный характер. Незрелые тимоциты на ранних стадиях развития (TCR-CD4-CD8-) и зрелые Т- клетки (TCR+CD4-CD8+ и TCR+CD4+CD8-), повышенная выживаемость которых должна обеспечить формирование Т-клеточного иммунного ответа и осуществление иммунной функции, обладают наиболее высоким уровнем экспрессии гена bcl-2 и слабой экспрессией гена Fas. Последний ген высоко экспрессирован на тимоцитах TCR+CD4+CD8+, подвергающихся селекции в вилочковой железе.
Развитие В-клеточной системы. Первичные этапы созревания и дифференцировки В-клеток происходят в костном мозге, где их большая часть погибает апоптически в результате селекции. В контроле этого процесса участвует ген bcl-2. На периферии зрелые В-клетки тоже подвергаются селекции. После встречи с антигеном они мигрируют в первичные лимфоидные фолликулы селезенки и лимфатических узлов, пролиферируют и подвергаются селекции. Сохраняются лимфоциты, иммуноглобулиновые рецепторы которых (Ig-рецепторы) имеют высокий аффинитет к антигену. Клетки с низким аффинитетом Ig-рецепторов погибают апоптически. Одновременно формируются долгоживущие В-клетки памяти, отличающиеся повышенной экспрессией bcl-2.
Гибель клеток иммунной системы, обусловленная развитием апоптоза, может быть индуцирована взаимодействием Т-клеточного рецептора (TCR) цитотоксических лимфоцитов с антигенами клетокмишеней. Результатом взаимодействия TCR с антигеном может быть инактивация самих Т-клеток, что происходит с Т-лимфоцитами-хелперами при индукции иммунологической толерантности во взрослом организме. Инактивация В-клеток, если она при этом происходит, осуществляется за счет соединения антигена с поверхностным иммуноглобулиновым рецептором В-лимфоцитов. Основной механизм такой инактивации - физическая элиминация, или делеция, клона антигенспецифических лимфоцитов, характерная для большинства форм искусственно индуцированной иммунологической толерантности (здесь играют роль анергия иммунокомпетентных клеток и активность супрессоров).
Гибель клеток, индуцированная активацией TCR, может быть вызвана не только специфическим антигеном, но и бактериальными токсинами, имеющими сродство с TCR, антителами к СDЗ-антигену, одному из компонентов рецепторного комплекса Т-клеток (TCR/CD3). Каждая активированная через комплекс TCR/CD3 клетка синтезирует все элементы, необходимые для ее деструкции. Гибель может быть предотвращена блокадой системы Fas/Fas-лиганд.
Стимуляция антигенреактивных T-клеток через TCR вызывает не только их пролиферацию и функциональную активацию, но и гибель. Возникает вопрос, каким образом может быть генерирована T-зависимая иммунная реакция. При нормальном иммунном ответе T-лимфоциты становятся чувствительными к опосредованному через Fas апоптозу лишь через несколько дней после TCR-стимуляции, хотя экспрессия Fas происходит в течение 24 ч после стимуляции. По-видимому, эти несколько дней составляют ту фазу, в течение которой активированная клетка осуществляет свои иммунные функции. При иммунной реакции против инфекционных агентов апоптоз (ПКГ) становится механизмом элиминации выполнивших свою роль активированных T-клеток, поскольку они потенциально опасны изза синтеза большого количества цитокинов и могут способствовать гиперплазии лимфоидной ткани, вызванной инфекцией (предшественник злокачественного роста). Каким же образом зрелая клетка, стимулированная связыванием антигена с ее антигенспецифическим рецептором (TCR ил Ig), выбирает между функциональной активацией (иммунным ответом) и апоптозом (иммунологической толерантностью), минуя стадию активации? Большинство исследователей
считают, что стимуляция TCR- или Ig-рецептора ведет к инактивации клетки. Для ее активации же одновременно необходим второй, дополнительный сигнал, поставляемый через другие клеточные рецепторы за счет связывания последних с лигандами соседних клеток или их растворимыми продуктами, в частности цитокинами.
Связывание лигандами TCR активирует через систему тирозинкиназ фосфолипазу С, гидролизующую фосфатидилинозитолдифосфат клеточной мембраны с образованием вторичных мессенджеров - диацилглицерола и инозитолтрифосфата. Последние активируют протеинкиназу С и систему Са/кальмодулин/кальциневрин соответственно. Синергичное действие последних ведет к активации транскрипционных факторов (в частности, NF-АТ, специфичного для Т-клеток). Результатами становятся активация генома и экспрессия ряда генов, в том числе гена ИЛ-2 и его рецептора у Т-клеток. Такая последовательность процессов при активации свойственна и клеткам иммунной системы, и клеткам других типов.
КЛИНИЧЕСКАЯ ЗНАЧИМОСТЬ ЭКСПРЕССИИ BCL-2 И ГЛБ/АРО-1
АЛЛЕРГОЛОГИЯ И КЛИНИЧЕСКАЯ ИММУНОЛОГИЯ
В основе развития аутоиммунных заболеваний лежит нарушение функций Fas/АРО-1-молекулы и ее лиганда. Неспособность к устранению аутореактивных клеток в процессе развития лимфоцитов может привести к аутоиммунным заболеваниям. Одна из критических молекул в регулировании гибели лимфоцитов - поверхностный клеточный рецептор Fas - член семейства рецепторов ФНО. Стимулирование Fas у активированного лимфоцита приводит к индукции апоптоза. Например, при утрате функций этой молекулы у мышей выявлен дефект, сходный с системной красной волчанкой человека и характеризующийся накоплением Т-лимфоцитов в периферических лимфатических узлах и синтезом аутоантител. У больных СКВ растворимый Fas/АРО-1-рецептор утрачивает внутриклеточную и трансмембранную области. В сыворотке крови этих пациентов обнаруживают повышенное содержание растворимой формы Fas. Растворимый белок конкурирует с мембранолокализованным рецептором Fas/АРО^ в связывании лиганда и может ингибировать Fas/АРО-1-опосредован- ный апоптоз in vitro. По этой причине подъем содержания растворимого Fas/АРО-1 может играть роль в патогенезе СКВ и других аутоиммунных
заболеваний. В то же время T-клетки больных СКВ содержат повышенное количество bcl-2, предотвращающего апоптоз.
Активирование клеточной гибели ответственно за изменение негативного отбора самореактивных T-лимфоцитов в вилочковой железе. Нарушение Fas-опосредованного T-клеточного апоптоза в вилочковой железе облегчает персистирование аутореактивных клонов и блокирует образование полного состава апоптозных аутоантигенов, к которым должна развиваться толерантность. При этом действие вызывающего фактора (например, солнечных лучей при СКВ) приводит к тому, что даже низкое содержание антигенов на фоне сенсибилизированной иммунной системы вызывает манифестирование клинических проявлений аутоиммунного заболевания. Существуют данные о роли апоптоза в развитии таких аутоиммунных заболеваний, как ревматоидный артрит, псориаз, аутоиммунный диабет.
ОНКОЛОГИЯ
Повышение в сыворотке крови содержания растворимого Fas/APO-1-рецеп- тора обнаружено и при определенных В- и T-клеточных лейкозах, что указывает на то, что растворимый белок Fas/APO-1 отвечает как за патогенез аутоиммунных нарушений, так и за ускользание от иммунологического надзора и развитие опухолей. Fas/APO-1-антиген широко экспрессирован на опухолевых клетках при заболеваниях как гематологической, так и негематологической природы: T- и В-клеточные лимфомы и лейкозы, хронический миелоидный лейкоз в стадии бластного криза, рабдомиосаркома, плоскоклеточная карцинома, карцинома толстой кишки, меланома, аденокарцинома предстательной железы, поджелудочной железы и эпидермальная карцинома.
Связывание моноклональных антител (МКАT) с антигеном АПО-1 индуцирует апоптоз в АПО-1 - апоптозочувствительных клетках. Однако MКAT не всегда индуцируют ПКГ. При одинаковом количестве Fas/APO-1-антигена на злокачественных клетках MКAT могут индуцировать апоптоз, вызывать стимуляцию пролиферации опухолевых клеток или не оказывать воздействия. Спектр биологических ответов, инициируемых MКAT анти Fas/АРО-1, может быть изменен добавлением к клеткам различных ингибиторов синтеза белка. Tак, комбинация MКAT анти-Fas/APO-1 с циклогексимидом в 90% случаев вызывала ингибицию пролиферации антигенположительных опухолей (Owen-Schaub L.B. и др., 1992). Добавление циклогексимида к MКAT индуцировало апоптоз и в случаях, когда одни MКAT стимулировали пролиферацию опухолевых клеток. Единственная инъекция MКAT
АРО вызывала регрессию В-клеточной лимфомы человека, растущей у голых мышей, однако введение антимышиных Fas/АРО-1-антител вызывало развитие летального молниеносного гепатита.
В настоящее время интенсивно изучают и клиническую значимость экспрессии протоонкогена bcl-2 при злокачественных новообразованиях. Протоонкоген bcl-2 супрессирует апоптоз и защищает от апоптоза, индуцированного многими противораковыми препаратами. Подъем степени экспрессии bcl-2 ассоциирован с резистентностью к различным химиотерапевтическим агентам. Однако при раке молочной железы, при котором экспрессия этого белка достигает 80% случаев, утрата его экспрессии коррелирует с плохим прогнозом течения заболевания. При этой патологии экспрессия bcl-2 коррелирует с экспрессией рецепторов к эстрогену и прогестерону. Лейкозные клетки больных хроническим миелолейкозом резистентны к индукции апоптоза химиопрепаратами вследствие гиперэкспрессии bcl-2, но чувствительны к апоптическому лизису Т-лимфоцитами.
Многие противораковые агенты, включая цитотоксические препараты (ингибиторы изотопоизомераз I и II, ДНК-реактивные препараты, например цисплатин и антиметаболиты), гормональные агенты (антиэстрогены и глюкокортикоиды), действуют, индуцируя программированную клеточную смерть в опухолевых клетках. Апоптоз в опухолевых клетках может быть вызван различными физическими воздействиями: гамма-излучением, ультрафиолетовыми лучами, гипертермией, гипотермией. Фармакологическая регуляция апоптоза может стать новым направлением в лечении онкологических больных.
Блокада механизмов апоптоза способствует развитию рака. Степень устойчивости клеток к апоптозу коррелирует с их устойчивостью к воздействию противоопухолевых препаратов. Возможно, что коррекция блокады апоптоза будет способствовать повышению чувствительности клеток к терапевтическому воздействию. Так, экспрессия bcl-2 может превращать опухолевые клетки в резистентные к ряду физических и химических веществ, в норме индуцирующих апоптоз. Лекарственные препараты, направленные на дезактивацию функций bcl-2, будут повышать чувствительность опухолевых клеток к терапевтическому воздействию, возвращая им способность к апоптозу. Лечение больных хронической миелоидной лейкемией, резистентных к актиномицину D, камптотецину и этопозиду, античувствительными олигонуклеотидами, соответствующими bcr-abl, приводило к снижению содержания химерного протеина Всг-Abl и этим вновь восстанавливало способность к апоптозу (A. McCahon и соавт.)
Эти выводы указывают на перспективность разработки новых методов консервативного лечения рака.
ПОВРЕЖДЕНИЕ МОЗГА
Осенью 1995 г. в США состоялся Международный симпозиум, посвященный механизмам гибели клеток нервной системы. Среди воздействий, индуцирующих апоптоз, особое место занимает оксилительный стресс как один из наиболее важных индукторов ПКГ. Оксилительный стресс можно рассматривать как часть нормального функционирования нейронов, но в случае возрастания его интенсивности он способен стать патогенетическим звеном острых и хронических дегенеративных расстройств. Так, в культуре нервной ткани апоптоз развивается при удалении сыворотки и предотвращается антиоксидантами. Апоптоз инициируется также прямым воздействием оксилительного стресса с помощью фотохимической реакции с образованием синглетного кислорода. Антиоксиданты и в этом случае оказывают нейропротективное действие. Показано, что в патогенезе апоптоза принимают участие протеинкиназа С и фосфатазы. Оксилительный стресс в нейронах может нарушать баланс между активностью киназ и фосфатаз. В работе M.D. Linnik и соавт. показано, что гибель нейронов при ишемическом инсульте развивается по двум механизмам - некроза и апоптоза. Есть сообщения об эффективности ингибитора белкового синтеза циклогексимида, как нейропротективного агента. Обнаружена фрагментация ДНК, характерная для апоптоза при ишемическом и эпилептическом повреждении мозга. Через 6 ч после судорог, вызванных инъекцией каиновой кислоты, часть нейронов погибает, через 18 ч в этих нейронах отмечают явные признаки апоптоза. Применение диазепама предупреждает развитие этих изменений.
Проблеме оксилительного стресса при генетически обусловленных судорожных припадках посвящена работа К.С. Раевского и соавт. Эпилептиформные судороги сопряжены с гиперактивацией глутаматных рецепторов, активацией перекисного окисления липидов и дисбалансом медиаторных аминокислот в ткани мозга. Доказано, что антиконвульсанты (ламотриджин, карбамазепин) способны уменьшать интенсивность оксилительного стресса при судорожных припадках.
Оксилительный стресс играет роль и в тех случаях, когда гибель нейронов обусловлена депривацией фактора роста нервов (NGF). Доказана роль Са при острых (травма, ишемия) и хронических (нейродегенеративные заболевания) состояниях, сопровождающихся
развитием апоптоза. Указывают на два основных механизма нейропротективного эффекта:
• экспрессия антиоксидантных ферментов;
• влияние на экспрессию или функции белков, участвующих в ионном гомеостазе клетки.
Показано, что деполяризующие условия обеспечивают защиту клеток от апоптоза. Активация ионотропных глутаматных рецепторов предупреждает апоптоз, вызываемый недеполяризующими условиями, что связано с деполяризацией нейрональной мембраны.
Tаким образом, во многих случаях нейропротективное действие фармакологических препаратов связано с нейтрализацией цитотоксического эффекта активных форм кислорода, участвующих в активации процесса ПКГ.
МИОКАРДИТЫ
Для миокарда, в физиологическом отношении синтиция, активация апоптоза, сопровождающегося гибелью отдельных кардиомиоцитов, может сказываться на проведении импульсов возбуждения по миокарду и способствовать развитию аритмий. По мнению О. Вing (1994), усиление апоптоза может быть одной из причин перехода от компенсаторной гипертрофии сердца к его дисфункции и сердечной патологии. Tак, некоторые факторы, например кальций, не только причастны к развитию дисфункции миокарда, но и влияют на возникновение апоптоза. Комплекс морфологических изменений у больных с нарушениями ритма в аритмогенных зонах миокарда свидетельствует о кальциевой перегрузке кардиомиоцитов. Аналогичные изменения обнаружены в миокарде и в иных ситуациях, связанных с поступлением кальция внутрь клетки в результате воздействия больших доз катехоламинов или других повреждающих факторов.
Mорфологическое исследование биоптатов субэндокарда из аритмогенных и неаритмогенных зон у больных с различными нарушениями ритма выявило наличие кардиомиоцитов с явлениями апоптоза. В ядре отмечены неравномерное распределение хроматина и накопление его по краям ядра. Набухшие митохондрии с редуцированным количеством крист образовывали скопления среди сокращенных миофибрилл. Фрагменты ядерного вещества вместе с другими разрушенными структурами образовывали апоптические тела. Степень выраженности апоптоза варьировала. Обнаружение апоптических повреждений как на аритмогенных, так и на неаритмогенных участках сердца свидетельствует о генерализованности процесса. Однозначно
ответить на вопрос о первичности апоптоза в развитии нарушений ритма пока не представляется возможным. Однако в эксперименте на ранних стадиях инфаркта миокарда, осложненного фибрилляцией, обнаружены ультраструктурные изменения кардиомиоцитов, свидетельствующие о развитии апоптоза, что подтверждает гипотезу о первичности ПКГ в возникновении нарушений ритма.
ИНФЕКЦИИ И АПОПТОЗ
ВИРУСНЫЕ ИНФЕКЦИИ
При некоторых вирусных заболеваниях анормальный апоптоз становится патогенетическим фактором. К таким заболеваниям относят, в частности, СПИД, ведущую роль в патогенезе которого играет апоптическая гибель активированных Т-клеток TCR+CD4+CD8+. У больных СПИДом лишь небольшое количество Т-лимфоцитов периферической крови (1 клетка из нескольких тысяч) содержит продуктивный вирус. Гибель остальных Т-клеток обусловлена белками вирусов gp120 и Tat, активирующими систему Fas/Fas-лиганд лимфоцитов, что и вызывает их апоптоз. По этой причине одним из путей патогенетической терапии этого заболевания может быть блокирование апоптической гибели.
Биологически важна роль апоптоза в уничтожении клеток, пораженных различными вирусами. Однако некоторые виды вирусов ингибируют программу клеточной гибели, например ДНК-содержащий вирус гепатита В, белок вируса Эпстайна-Барр, гены бакуловируса, аденовируса, вируса герпеса и др.
Предотвращение апоптоза играет важную роль в механизме латентной вирусной инфекции. Вирусный ген LMP-1, синтезируемый при латентности, специфически регулирует содержание bcl-2, обеспечивая выживание латентно инфицированных клеток. Угнетение апоптоза в содержащих вирус клетках объясняет механизм персистенции вирусного заболевания в организме и может стать основой развития онкологического заболевания. Например, гепатит В, при котором наблюдают угнетение апоптоза в гепатоцитах, может переходить в хронические персистирующие формы гепатита, что часто совпадает с последующим развитием гепатомы.
БАКТЕРИАЛЬНЫЕ ИНФЕКЦИИ
При массовом проникновении в организм инфекционных агентов нейтрофилы мигрируют к месту инвазии, активно поглощают
микроорганизмы и выделяют в межклеточное пространство значительное количество факторов, индуцирующих апоптоз. Эти факторы осуществляют деградацию нуклеинового комплекса любых биологических систем и представляют собой опасность для организма, поэтому их действие необходимо заблокировать. Нейтрофилы, «нафаршированные» микроорганизмами, первыми подвергаются апоптозу. Индукторами апоптоза выступают эндотоксины микроорганизмов кишечной группы и экзотоксины (например, столбнячной палочки, стафилококков). Mассовый апоптоз, опосредованный ФНО, развивается при сепсисе.
Tаким образом, в свете изложенного существуют перспективы терапевтического управления механизмами клеточной смерти. Как уже упоминалось, индукцию апоптоза можно использовать как противоопухолевую терапию. Применяемые моноклональные антитела (как конъюгаты с токсинами или лекарствами) могут быть летальны для клеток-мишеней, вызывая в них экспрессию гена анти-Аро-1 и запуская апоптоз. В качестве одного из возможных путей воздействия на апоптоз можно рассматривать изменение свойств цитоплазматических мембран. Tак, перфторан за счет стабилизации клеточных мембран и снижения их проницаемости для ионов Са может стать эффективным средством профилактики реперфузионных повреждений сердца и легких больных, подвергшихся реконструктивной хирургии или трансплантации. Индукция антиоксидантной защиты будет предупреждать развитие апоптоза, а прооксидантное воздействие, стимулирующее образование АФК, - способствовать активации апоптоза. Регулирование процесса апоптоза может приблизить нас к решению вопроса о продолжительности жизни. При разработке тактики терапевтического воздействия с учетом процессов активации или ингибирования ПКГ целесообразно проводить лабораторный контроль экспрессии генов CD95 и bcl-2 на клетках периферической крови или иных заинтересованных тканей, а также содержания растворимого белка Вс1-2 и Fas-лиганда в сыворотке крови.