Иммунология : учебник. - Хаитов Р. М. - 2009. - 320 с. : ил.
|
|
|
|
ГЛАВА 14 АУТОИММУННЫЕ БОЛЕЗНИ
Истинные аутоиммунные болезни - это такие болезни, в патогенезе которых лимфоциты, запускающие механизмы деструкции, распознают именно нативные молекулы мембран собственных клеток или межклеточного вещества и инициируют иммунное воспаление.
• Часто и ошибочно к аутоиммунным относят все патологические процессы, при которых имеется повреждение тканей иммунными механизмами. Это не совсем верно, так как иммунного ответа против чужого патогенного Аг без повреждения собственных тканей вообще не бывает: если патоген проник во внутреннюю среду - он так или иначе вступил в тесную связь с клетками и межклеточным веществом: разрушая патоген, иммунная система разрушает и окружающие собственные ткани, а уж насколько это больно и заметно, зависит от дозы Аг.
• Если вирус заразил клетку, то ЦТЛ или NK, распознавшие вирусные Аг, разрушат всю клетку. Если микробные продукты сорбируются на межклеточном матриксе или стенке сосудов или на эритроцитах, то противомикробные АТ с комплементом вызовут процессы деструкции матрикса, воспаление сосудистых стенок (васкулиты), гемолиз. Но это будут не аутоиммунные процессы, поскольку и ЦТЛ, и АТ направлены против микробных Аг, а разрушение тканей наступает лишь потому, что микробные продукты оказались тесно связанными с этими тканями.
• Ещё один внешний источник повышенной альтерации собственных тканей иммунными механизмами - химические (иногда природные) вещества (ЛС, пищевые добавки, факторы химических производственных процессов и другие вредности подобного рода), которые, проникая во внутреннюю среду организма, в прямом смысле денатурируют молекулы организма, превращая их в раздражающие иммунную систему Аг-мишени. И в этих случаях альтерацию собственных тканей иммунными механизмами тоже
неправильно называть аутоиммунным процессом, поскольку иммунная система борется с внешними повреждениями на поверхности собственных тканей. Патогенетическим в данном случае будет лечение, направленное в первую очередь на элиминацию внешних повреждающих факторов, а не на супрессию собственной иммунной системы, чтобы она «не мешала» присутствию чужеродных факторов на клетках организма. Хотя бывают клинические ситуации, когда процесс иммунного воспаления заходит так далеко, что по жизненным показаниям его приходится подавлять в первую очередь.
ЭТИОЛОГИя И ПАТОГЕНЕЗ
• Аутореактивные клоны лимфоцитов. В норме у каждого здорового организма в периферических лимфоидных тканях есть и T-, и B-лимфоциты с Аг-распознающими Рц для «своего», т.е. манифестация аутоиммунных болезней не является результатом возникновения аномальных аутореактивных клонов лимфоцитов: они есть всегда как нормальное явление. Поэтому манифестация аутоиммунного деструктивного процесса инициируется патогенным внешним фактором. Кстати, конкордантность однояйцовых близнецов по аутоиммунным болезням не превышает их конкордантности по заболеваемости инфекционными болезнями.
• Костимуляторные взаимодействия. Одного факта связывания Рц с Аг недостаточно для запуска иммунного ответа, необходимы ещё своевременные костимуляторные взаимодействия: как минимум «CD40-CD40L» и «B7-CD28» (и для T-, и для В-лимфоцитов). Если связывание с лигандами антигенраспознающего Рц и костимуляторных молекул разобщено во времени [сначала одно, потом (или никогда) другое], то произойдёт не активация лимфоцита к иммунному ответу, а анергия или апоптоз. Это доказано.
• Предшествующий патологический процесс. Таким образом, молекулы собственных клеток и межклеточного вещества - нормальный объект для распознавания по крайней мере T-лимфоцитами, но не для развития иммунного воспаления до тех пор, пока собственные молекулы каким-то образом не попадут во внутриклеточную «машину» профессиональных АПК, активированных к экспрессии костимуляторных молекул. В норме они туда не попадают. Это происходит в результате какого-то предшествующего патологического процесса (всё тех же инфекций или травм), приводящего к
• альтерации тканей и доиммунному воспалению. Иммунный T-лимфоцит, уже прошедший додифференцировку (иммуногенез), отличается от неиммунного T-лимфоцита в двух отношениях: 1) для активации эффекторной функции иммунному лимфоциту достаточно сигнала с TCR и индуцибельных костимуляторных взаимодействий; 2) молекулы адгезии иммунного лимфоцита позволяют ему мигрировать в любые периферические ткани, тогда как неиммунный лимфоцит рециркулирует строго между «своими» зонами в периферических лимфоидных органах и не заходит в иные периферические ткани. Поэтому полагают, что инфекции способны инициировать аутоиммунные процессы по следующим механизмам.
♦ Антигенная мимикрия патогенов (эволюционно достигаемое уподобление микробных продуктов тканевым компонентам макроорганизма) индуцирует иммунный ответ с перекрестной реактивностью со своими Аг. В дальнейшем аутоиммунный процесс не выходит в режим полноценной иммуносупрессии, поскольку свой Аг не может быть элиминирован и продолжает «раздражать» иммунные лимфоциты. В этом отношении особенно «преуспевают» вирусы: размножаясь внутри клеток организма, они время от времени включают в состав своего генома какието из генов этого организма. Это уже не мимикрия, а прямая генетическая «кража» и затем использование «краденого».
♦ Микробные суперантигены вызывают поликлональную активацию. Какие-то из клонов лимфоцитов с реактивностью к своим Аг могут войти в режим эффекторного иммунного ответа.
♦ Деструкция тканей патогеном (цитопатогенное действие вирусов, бактерий и др.) приводит к попаданию тканевых Аг в активированные (тем же патогеном) дендритные клетки, которые транспортируют все Аг в периферические лимфоидные органы, где есть особые условия для индукции продуктивного иммунного ответа.
♦ Провоспалительные цитокины. Индуцированное патогеном локальное доиммунное воспаление сопровождается выработкой провоспалительных цитокинов, которые способны индуцировать экспрессию на клетках тканей (не профессиональных АПК) молекул MHC-II со своими пептидами, что потенциально со- здаёт условия для инициации иммунного ответа на свои Аг.
♦ Два TCR на одном лимфоците. Примерно 30% периферических T-лимфоцитов несут по крайней мере два разных по специ-
фичности TCR (в связи с «плановой» неоднократной перестройкой V-гена α-цепи при уже перестроенной β-цепи). Есть вероятность, что один из TCR может иметь специфичность к патогену, а второй - к аутоантигену. Активация иммуногенеза патогеном приведёт к созданию клона иммунных лимфоцитов, которые будут работать в качестве эффекторов против обоих Аг - чужого и своего. Аутоиммунное воспаление (начавшись так или иначе, но в связи с инфекцией) не может нормальным образом остановиться, потому что аутоантиген не исчезает, пока вся ткань, его экспрессирующая, не будет разрушена и выброшена из организма.
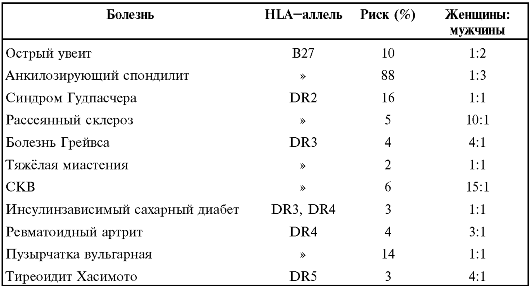
• Ассоциация аутоиммунных болезней с определёнными Аг MHC
(табл. 14.1) может быть понята именно с учётом «инфекционного» компонента патогенеза, так как именно MHC представляют Аг (в том числе и микробные) для распознавания T-лимфоцитам: как представят, такой иммунный ответ и будет.
Таблица 14.1. Ассоциации аутоиммунных болезней с MHC и полом
•  Гиперчувствительность замедленного типа (ГЗТ). Вспомним, каковы эффекторные механизмы нормального иммунного ответа. ♦ Антительный ответ. АТ,
комплемент, фагоцитоз, АЗКЦТ, сосудистые и гладкомышечные реакции,
опосредованные медиаторами тучных клеток и базофилов. Антительный ответ
контролируют и программируют преимущественно либо CD4+ Тh2, либо цитокины из Тh1 и CD8+-ЦТЛ.
Гиперчувствительность замедленного типа (ГЗТ). Вспомним, каковы эффекторные механизмы нормального иммунного ответа. ♦ Антительный ответ. АТ,
комплемент, фагоцитоз, АЗКЦТ, сосудистые и гладкомышечные реакции,
опосредованные медиаторами тучных клеток и базофилов. Антительный ответ
контролируют и программируют преимущественно либо CD4+ Тh2, либо цитокины из Тh1 и CD8+-ЦТЛ.
♦ ГЗТ: клетки-исполнители - активированные макрофаги, клетки-инициаторы и регуляторы - CD4+ Th1.
♦ Деструкция клеток-мишеней CD8+ или CD4+-ЦТЛ.
Те, кто хорошо знают патологию, но меньше - современную иммунологию, всегда рассматривают ГЗТ как патологический (а то и патологический аллергический) процесс. Между тем ГЗТ - нормальный механизм эффекторного этапа иммунного ответа. И воспринимается ГЗТ (как и любой другой эффекторный иммунный процесс) как патология только в случаях, когда доза Аг велика или баланс субпопуляций лимфоцитов ненормален, или нарушены процессы нормальной супрессии иммунного ответа, устанавливаются «порочные круги» и т.п. Запуск деструктивного этапа иммунного ответа, направленного на аутоантигены, - пример тяжёлой патологии, спровоцированной каким-то внешним фактором.
• Причинные антигены. При многих болезнях с явным компонентом иммунного воспаления причинные Аг не известны и задействованы разные эффекторные механизмы иммунного воспаления. Клиницисты такие болезни всё равно часто называют аутоиммунными, хотя этиологический Аг(ы) неизвестен(ы) и, вероятно, большинство болезней - не истинные аутоиммунные.
• Апоптоз. При некоторых заболеваниях этиология иммунного воспаления связана не с конкретными Аг, а например, с нарушениями в нормальных механизмах апоптоза лимфоцитов. При этом органная или тканевая локализация зависит от локализации причинного фактора, действующего на лимфоциты. Например, при ревматоидных артритах иммунное воспаление суставов вызвано тем, что зрелые иммунные T-лимфоциты: в синовиальных полостях своевременно не погибают путём апоптоза, а продолжают продуцировать провоспалительные цитокины, потому что сами получают патологический сигнал на выживание от изменённых (возможно, инфекцией) фибробластов стромы синовиальных хрящей. В синовиальных T-лимфоцитах аномально повышена экспрессия антиапоптозных белков Bcl-2 и Bcl-xL.
• Нозологические единицы. В табл. 14.2. приведены примеры аутоиммунных болезней, а в следующих разделах - краткое описание тех болезней, в течении которых имеет значение не просто иммунное воспаление, а иммунопатогенез ведущих симптомокомплексов. При этом более чем вероятно, что изначально этиологическим фактором в развитии этих болезней явилась та или иная, распознанная или нераспознанная (и скорее всего вирусная) инфекция,
которую иммунная система не сумела санировать, и со временем установились «порочные круги», приводящие к альтерации собственных тканей иммунными механизмами. Мы заведомо не сможем перечислить все подобные болезни, так как их сотни по всем частным медицинским специальностям. В то же время нозологических единиц аутоиммунных болезней выделено не так уж много. Интересно, что у разных пациентов аутоантигены-мишени (в тех случаях, когда они известны) одни и те же в пределах нозологии, а не уникальны для каждого человека.
Таблица 14.2. Примеры аутоиммунных болезней человека
 Продолжение табл. 14.2.
Продолжение табл. 14.2.
 заболевания эндокринных желёз
заболевания эндокринных желёз
Для всех заболеваний, называемых аутоиммунными, характерно длительное, хроническое, прогредиентное течение с периодами ремиссий и обострений, как для хронических инфекционных болезней.
Аутоиммунный гипертиреоидизм
Аутоиммунный гипертиреоидизм (болезнь Грейвса) встречается у 0,5% населения (у женщин в 7 раз чаще) и является наиболее частой причиной гипертиреоидизма. Более чем в 90% случаев удаётся обнаружить АТ к Рц для ТТГ. Эти АТ вместо гормона стимулируют функциональную активность клеток щитовидной железы, что и приводит к развитию симптомокомплекса гипертиреоидизма. Конкордантность по этому заболеванию у гомозиготных близнецов 50%, у дизиготных - 5%. Риск заболевания в 4 раза больше среднего у людей, имеющих аллель HLA-DR3. Лечение. В иммунодепрессантах необходимости нет, кроме тяжёлых случаев экзофтальма (тогда применяют высокие дозы ГКС, циклофосфамид, циклоспорин А). Обыгчно достаточно антитиреоидных блокаторов: тиамазола, пропилтиоурацила. Когда лекарственная терапия перестает поддерживать компенсированное состояние, осуществляют хирургическое удаление части щитовидной железы.
Аутоиммунный зоб
В начальный период болезни Хасимото у 75% пациентов функциональный статус щитовидной железы в пределах нормы, у 20% - гипотиреоидизм, у 5% - гипертиреоидизм. Первые признаки заболевания обнаруживают, как правило, в подростковом возрасте, но клинически значимое состояние развивается в большинстве (90%) случаев после 45 лет. По мере прогрессирования болезни у 50% пациентов развивается гипотиреоидизм. Более чем у 95% больных выявляют АТ к тиреоидной пероксидазе (микросомный Аг), иногда к тиреоглобулину. Лечение. Заместительная терапия тиреоидными гормонами (тироксин и трийодтиронин) под контролем уровня этих гормонов в сыворотке крови.
Сахарный диабет 1-го типа
Инсулинзависимый сахарный диабет (сахарный диабет 1-го типа) развивается в результате селективного разрушения β-клеток островков Лангерганса поджелудочной железы (α- и δ-клетки целы) CD8+ ЦТЛ и CD4+ Th1-опосредованным иммунным воспалением. Этиология болезни предположительна. Конкордантность однояйцовых близнецов не более 40%; следовательно, имеют значение факторы окружающей среды. Прослеживаются клинические ассоциации с рядом вирусных инфекций (краснуха, вирус Коксаки В4, реовирусы 1- го и 3-го типов и др.), а также с интоксикацией такими химическими соединениями, как N-3-пиридил-метил-N-п-нитрофенилмочевина, стрептозотоцин (2-дезоксиметил-нитромочевина-гликопиранозид), мезоксалилмочевина. Поскольку существуют модели на мышах (NOD - Non-Obese Diabetic) и крысах (ВВ - Bio-Breeding diabetesprone), то собственно эффекторный механизм иммунной деструкции β-клеток лимфоцитами CD8+-ЦТЛ и Th1-эффекторами ГЗТ изучен лучше, чем при многих других болезнях. Но незнание инициирующего иммунный ответ Аг(ов) и этиологических кофакторов не позволяет осуществлять ни раннюю доклиническую диагностику, ни профилактику.
Клиническая картина. Болезнь манифестирует симптомами полиурии, полидипсии и быстрого похудания (дни, недели). Но процесс разрушения β-клеток протекает в течение нескольких лет до этого (о чем свидетельствуют результаты патологоанатомических исследований) и при жизни не диагностируется в связи с компенсированностью клинической картины. В преклинической стадии диагностическое значение имеет определение в сыворотке крови АТ к клеточным Аг β-клеток, антиинсулиновых АТ, АТ к глутамат-
декарбоксилазе и АТ к тирозинфосфатазе IA-2, а также тесты на толерантность к глюкозе (внутривенный и пероральный). Лечение. Заместительная терапия инсулином.
Недостаточность надпочечников
Болезнь Аддисона встречается с частотой 6 на 100 тыс. населения, у женщин в 2 раза чаще. Поражается кора, но не мозговое вещество надпочечников. У 80% больных обнаруживают АТ к 17а-гидроксилазе. Лечение. Заместительная терапия гормонами коры надпочечников.
Болезнь Кушинга
Болезнь Кушинга - микронодулярная адренокортикальная гиперплазия. Вызвана АТ к Рц для АКТГ, которые стимулируют надпочечники вместо АКТГ. Как правило, у 50% больных выявляют множественные эндокринопатии и АТ к другим тканевым Аг: у 20% - патология щитовидной железы, у 15% - сахарный диабет, у 8% - патология яичников, у 4% - гипопаратиреоидизм. Лечение симптоматическое.
заболевания желудочно-кишечного тракта
Гастрит и пернициозная анемия
Гастрит и пернициозная анемия встречаются с частотой 0,1% в западных странах. Характеризуются атрофией слизистой оболочки желудка с потерей париетальных и главных клеток. У 90% больных обнаруживают АТ к мембранному белку париетальных клеток (Н+/ К+-зависимой АТФазе) и у 70% - к фактору Касла для витамина B12. Слизистая оболочка инфильтрирована лимфоцитами (CD4+, CD8+, В). Лечение симптоматическое.
Целиакия
Целиакия - хроническое воспалительное заболевание тонкой кишки с атрофией ворсинок, гиперплазией крипт, лимфоцитарным инфильтратом эпителия и собственного слоя слизистой оболочки, мальабсорбцией.
• Заболевание развивается при поступлении в организм с пищей глютена - компонента пшеницы. В 50% случаев выявляется в раннем детском возрасте, в 50% - у взрослых. Распространено
с частотой 1:300 в Ирландии, 1:2000 в Великобритании, 1:6000 в Швеции, крайне редко встречается в странах Азии и Африки. Болезнь ассоциирована с HLA-DQ2: этот аллель экспрессирован более чем у 95% больных целиакией, у остальных 5% экспрессированы HLA-DR4 или HLA-DQ8. Но целиакия развивается далеко не у всех людей, имеющих аллель HLA-DQ2, а лишь у малого процента из них.
• Кроме пшеницы, перекрёстную токсичность проявляют рожь, ячмень и овёс (Аг являются спирторастворимые проламины эндосперма этих злаковых - глиадин, секалин, хордеин, авенин).
• В слизистой оболочке тонкой кишки повышено содержание внутриэпителиальных лимфоцитов - CD7+, Tγδ, CD44+ Th1, a также плазмоцитов, продуцирующих преимущественно IgG и IgM (в норме в слизистой оболочке ЖКТ преобладает продукция IgA), эозинофилов и тучных клеток. Иммунопатогенез тем не менее неизвестен. Не исключён фактор инфекции каким-то энтеровирусом. Гистологическая картина слизистой оболочки даёт основания признать наличие ГЗТ [сначала гипертрофия, затем атрофия ворсин; гиперплазия крипт; увеличенное число внутриэпителиальных лимфоцитов; признаки активации T-лимфоцитов и макрофагов (DR+, ИЛ-2R+, повышенная продукция ИФНу, дегрануляция тучных клеток в слизистой оболочке). В l. propria много T-лимфоцитов, реагирующих на глиадин, представленный в комплексе с HLA-DQ2.
Клиническая картина. У детей отставание в росте, диарея, обильный стул, увеличение живота, анорексия, общая слабость. В подростковом возрасте возможно спонтанное улучшение. У взрослых диарея, потеря массы тела, анемия, герпетиформный дерматит, у 50% - гипоспленизм.
Диагноз ставят по данным лабораторного обследования: определяют в сыворотке крови АТ к глиадину, выполняют гистологический анализ биоптата слизистой оболочки тонкой кишки. Анализ на антиглиадиновые IgG более чувствителен (95%), - более специфичен (нормальные показатели зависят от возраста и наличия в диете продуктов, содержащих глиадин). В связи с повышенной проницаемостью воспалённой слизистой оболочки в сыворотке крови могут быть АТ к другим пищевым Аг - β-лактоглобулину, казеину, овальбумину). Роль этих АТ в патогенезе, однако, непонятна.
Лечение. Пожизненное исключение из диеты всех продуктов, содержащих глиадин и перекрестно реагирующие проламины. В тяжёлых
случаях назначают ГКС и другие иммунодепрессанты до улучшения клинического состояния и нормализации гистологической картины биоптата тонкой кишки. Некупированная целиакия часто осложняется ассоциированной с энтеропатией T-клеточной лимфомой (из CD7+ внутриэпителиальных T-лимфоцитов).
Воспалительные заболевания кишечника
Этим «сборным» термином обозначают идиопатический язвенный колит и нечётко отличающуюся от него болезнь Крона. Встречаются с частотой 10-15 случаев на 100 тыс. населения. Как правило, манифестируют в возрасте 20-40 лет. Этиология неизвестна, но подозревают вирусную инфекцию, особенно в случае болезни Крона. Гистологическая картина: трансмуральное воспаление с лимфоидными агрегатами и гранулемами из синцития и эпителиоподобных клеток, характерна гипертрофия нервов и подслизистого нервного сплетения. Инфильтрат состоит из нейтрофилов, макрофагов, плазмоцитов и лимфоцитов.
При болезни Крона симптомы зависят от конкретной локализации и размеров воспаления: лихорадка, боли в животе, диарея, потеря массы тела. Возможны симптомы обструкции, фистулы, абсцессы.
В крови больных определяют АТ к пищевым Аг и кишечным бактериям-комменсалам, но это рассматривают как эпифеномен (результат патологически повышенной проницаемости воспалённого кишечника). Определяют также АТ, связывающиеся с поверхностными структурами эпителиальных клеток толстой кишки. Достоверно показано, что эти АТ перекрёстно реагируют с микробными продуктами Escherichia coli. Однако, поскольку болезнь Крона и язвенный колит редко сочетаются с другими аутоиммунными расстройствами, более вероятны инфекционная этиология и индуцированное инфекцией хроническое иммунное воспаление.
Лечение. Сульфасалазин, месалазин; в тяжёлых случаях ГКС. В 80% случаев болезни Крона возникает необходимость в хирургическом вмешательстве.
Гепатиты
• Вирусные гепатиты. Хотя этиология их известна как определённо вирусная, в патогенезе существен компонент иммунного воспаления. Описаны 7 вирусов гепатита, точнее 6 идентифицированных и 1 неопределённый: А, В, С, D, Е, G, не -ABCDEG. Вирус гепатита A (HAV) - РНК-содержащий пикорнавирус, реп-
• лицирующийся в клетках кишечника и печени. Заражение - пероральное, возможно парентеральное. Инкубационный период составляет 15-45 дней. Болеют чаще дети или молодые люди. Молекулярный и клеточный патогенез гепатита неизвестен. Клиническая картина гепатита у большинства больных не слишком тяжёлая. Экстрапечёночной симптоматики мало или нет. Диагноз ставят по идентификации вируса в посевах из фекалий и/или по нарастающим титрам противовирусных АТ (IgM) в крови. Выздоровление в большинстве случаев полное. Хронизация гепатита и цирроз - крайне редкие исходы. Вирус гепатита В
Вирус гепатита В (HBV) - ДНК-содержащий гепатотропный вирус - самый распространённый этиологический агент заболеваний печени. Всего инфицированы не менее 500 млн человек, отмечают 50 млн новых случаев заражения в год. Заражение происходит через кровь или слизистые оболочки, включая таковые при стоматологических процедурах и подобных медицинских манипуляциях. Вирус могут переносить постельные клопы. Вирус проникает в плод трансплацентарно и с высокой вероятностью. Подавляющее большинство бессимптомных или субклинических вирусоносителей получили вирус во внутриутробном или раннем постнатальном периоде.
Именно у почти бессимптомных вирусоносителей чаще, чем у остро переболевших людей, в исходе болезни может развиться гепатокарцинома (первичный рак печени). В Англии, например, 0,3% населения - вирусоносители HBV, в странах тропической Африки, Китае и странах Юго-Восточной Азии - 8-35%. Инкубационный период составляет 50-180 дней. Течение заболевания у большинства больных тяжёлое. Выход в хронический гепатит типичен, причём возможны как хроническое вирусоносительство, так и активный хронический гепатит.
Лабораторные маркёры инфекции HBV, которые могут быть обнаружены в крови инфицированных людей: HbsAg - поверхностный Аг (белок) вируса, HbcAg - сердцевинный Аг (белок нуклеоида), HbeAg - внутринуклеоидный Аг вируса. Аг HbsAg можно обнаружить в крови за 2 нед или более до манифестации клинических симптомов и/или повышения содержания в крови печёночных ферментов. Обычно лабораторная диагностика гепатита В состоит в определении в острый период в крови HbsAg. Этот Аг обнаруживают в крови до периода конвалесценции (т.е. до 4-5 мес после заражения, в течение примерно 2 мес после манифестации клини-
ческих симптомов). Если HbsAg персистирует в крови более 6 мес, то пациента следует рассматривать как вирусоносителя. Если при этом у данного человека можно выявить ещё и HbeAg, то существует значимый (20%) риск заразности для других людей. Если HbeAg не определим, то риск заразности данного носителя в контактах рассматривают как не слишком большой (5%).
Кроме того, диагностическое значение имеет лабораторное определение нарастающих во времени титров АТ к HbcAg. Если АТ к данному Аг - IgM, то это является признаком недавней свежей инфекции, если АТ принадлежат преимущественно к классу IgG, то это признак уже инфекции «со стажем» (месяцы или годы). Например, по законодательству Великобритании медицинский персонал с позитивными результатами анализа на HbsAg/HbeAg не допускают к контактному обслуживанию пациентов.
Вирус гепатита В не цитопатогенен, т.е. он не убивает инфицированные им клетки. Гибель инфицированных гепатоцитов и, следовательно, патологическое воспаление печени и клинические симптомы гепатита вызывают ЦТЛ, иммунные к вирусным Аг. Сверхострое (фульминантное) клиническое течение вирусного гепатита бывает при интенсивном развитии (быстром и сильном количественно) иммунного ответа на вирус. Напротив, у иммунодефицитных людей вирусный гепатит течёт субклинически, но с развитием персистирующего варианта инфекции, т.е. без санации.
Выделяют три варианта хронического вирусоносительства: хронический активный гепатит, хроническая персистенция вируса с субклиническим гепатитом, вирусоносительство при относительно нормальной гистологии печени (по данным прижизненной биопсии).
Вирусоносительство существует почти у всех (более 90%) детей, рождённых инфицированными HBV (HbeAg-позитивными) матерями и у 10% переболевших взрослых. Большинство из названных групп при биопсии печени дают картину хронического активного гепатита.
У носителей, которые только HbsAg-позитивны и не имеют в анамнезе явной клиники гепатита, в биоптатах печени выявляют либо нормальную гистологическую картину, либо сглаженные симптомы хронического гепатита. Такие носители заразились или внутриутробно, или в раннем постнатальном периоде, что явилось предпосылкой для формирования у них иммунологической толерантности к вирусу (отсутствию иммунного ответа).
Внепечёночные симптомы при вирусном гепатите обусловлены «выпадением в осадок» иммунных комплексов АТ с вирусными Аг в почках (особенно при избытке Аг) с развитием гломерулонефрита. Иммунные комплексы при избытке АТ крупнее, вызывают системные васкулиты разной степени поражения и могут выглядеть как узелковый полиартериит (polyarteritis nodosa) или криоглобулинемия.
Гепатома (первичная гепатоцеллюлярная карцинома) - часто встречающаяся опухоль у населения тех регионов, где высока частота инфицирования HBV. Риск развития данной опухоли у вирусоносителей в 300 раз выше, чем у невирусоносителей. У 30-95% пациентов с гепатомой достоверно повышен в крови уровень α-фетопротеина (АФП), поэтому АФП является полезным диагностическим маркёром для анализа состояния печени в отношении развития гепатомы.
• Лечение гепатитов В. Примерно в 50% случаев эффективны продолжительные курсы инъекций ИФНа (3 мес и дольше). Наступает продолжительная ремиссия и со временем у многих больных исчезают лабораторные маркёры наличия вируса в организме. В случае исхода в цирроз может быть принято решение о трансплантации печени. Но опыт показывает, что практически в 100% случаев трансплантат быстро инфицируется вирусами гепатита. Существуют рекомбинантные вакцины - HbsAg в дрожжевом векторе. Эффективность защиты не более 80%, продолжительность действия - не более 2 лет. Вирус гепатита С
В 70-х годах XX в. значительное число случаев посттрансфузионных гепатитов в лабораторной диагностике документировали как «ни А, ни В». Позже идентифицировали новый вирус и обозначили его «С» (HCV). Это РНК-содержащий вирус весьма вариабельный. К настоящему времени идентифицировано 6 генетических типов и множество подтипов данного вируса. Для этого вируса пока не подобраны условия культивирования in vitro, но его геном тем не менее клонирован.
Основная заражённая популяция - больные наркоманией (более 70% таких больных в Англии инфицированы HCV). При сексуальных и трансплацентарных контактах риск заражения HCV оценивают в 5%.
Клинический инкубационный период составляет 15-150 дней. Острая первичная инфекция в большинстве случаев мягкотекущая
или бессимптомная. Желтуха проявляется только у 10% больных. Фульминантное течение регистрируют крайне редко. Естественная история развития этой инфекции мало изучена. Вероятно, у 80% инфицированных развивается хронический гепатит С, который через 20-30 лет имеет исход в цирроз печени, ещё через 10 лет - в гепатокарциному.
Лабораторная диагностика состоит в определении противовирусных АТ (против HCV) и применении в дополнение ПЦР на вирусный геном. Вирусные Аг в крови выявить не удаётся.
Лечение (не считая симптоматического). Курс ИФНа в течение 6 мес у 50% инфицированных нормализует активность печёночных ферментов (аминотрансфераз) и улучшает гистологическую картину при биопсии печени. В 50% случаев «улучшения», однако, наступает релапс. В целом лишь 15-20% пациентов хорошо поддаются лечению ИФНа по состоянию на 6-12-й месяц после прекращения курса такого лечения. Ориентировочные факторы, предрасполагающие к хорошим эффектам от интерферонотерапии: возраст до 40 лет; срок инфицирования менее 10 лет; масса тела не выше 70 кг; отсутствие явлений цирроза; низкий уровень виремии (т.е. небольшое количество вируса в организме); генотипы HCV 2 или 3. В комбинации с ИФНа некоторый положительный результат может дать рибавирин, но не в режиме монотерапии.
• Вирус гепатита дельта (HDV) - неполноценный РНК-содержащий вирус, который способен к репликации только совместно с вирусом гепатита В. Такая коинфекция неблагоприятна для организма больного. Парное инфицирование обусловливает более тяжёлое течение острого гепатита, чем при «чистой» инфекции HBV, и более высокую смертность. В исходе - чаще хронический активный гепатит и цирроз печени.
• Вирус гепатита E - энтеральный РНК-содержащий вирус. Распро- странён в Азии (особенно в Китае и Индии, на Среднем Востоке), Африке и в Центральной Америке. У большинства заболевших наступает самоизлечение, но у беременных женщин, особенно заражённых в III триместре, высока (25% и более) летальность. Плод, однако, чаще не инфицирован. Хронические формы не выявлены.
• Вирус гепатита G - парентерально передающийся агент, который сам по себе клинической картины не вызывает. Его обнаруживают у 2% «здоровых» доноров крови и у 20% пациентов, получающих по тем или иным показаниям препараты из крови человека.
• Вирус не-ABCDEG. В 10% случаев острых гепатитов в лабораторных исследованиях не выявляют ни один из известных вирусов. Как правило, у этих пациентов сильная желтуха, высокие уровни печёночных ферментов в крови, но наступает полное выздоровление. Пути трансмиссии не известны.
Аутоиммунные заболевания печени
В настоящее время как аутоиммунные заболевания печени рассматривают три нозологические единицы: аутоиммунный гепатит, первичный склерозирующий холангит, первичный билиарный цирроз. Как и при других аутоиммунных заболеваниях, исходный этиологический агент ни в одном случае доподлинно неизвестен, подозревают инфекционную этиологию, инициировавшую многие месяцы или годы назад аутоиммунное деструктивное воспаление.
Гистологическим признаком, на основании которого можно рассматривать процесс как аутоиммунный, являются лимфоцитарные инфильтраты в очагах поражения печени. Причём при всех трёх указанных нозологиях в лимфоцитарных инфильтратах больной печени (в отличие от здоровой) соотношение Т4-лимфоцитов к Т8-лимфоцитам возрастает в сторону преобладания Т4 (в здоровой печени преобладают Т8-клетки).
Аутоиммунный гепатит
Если по клиническому состоянию напрашивается диагноз хронического гепатита, а лабораторными анализами не определяется ни один из известных маркёров вирусных гепатитов, то эта ситуация часто побуждает выставлять диагноз аутоиммунного гепатита.
• Диагноз хронического гепатита ставят по признаку продолжительности воспалительного процесса в печени более 6 мес. По гистологической картине при биопсии печени выделяют две категории хронического гепатита: хронический персистирующий гепатит (незначительные признаки воспаления) и хронический активный гепатит (выраженные признаки воспаления).
• При хроническом персистирующем гепатите признаки воспалительного процесса есть только в портальных зонах. Это можно наблюдать в исходе вирусных гепатитов В и С, при алкогольном повреждении печени, гиперреактивности организма на лекарства, хронических воспалительных заболеваниях кишечника. Необходимости в применении ГКС нет. Прогноз благоприятен. Прогрессия в цирроз редка.
• При хроническом активном гепатите при гистологическом исследовании биопсийного материала инфильтраты мононуклеарами
обнаруживают как в портальных зонах, так и в паренхиме. Виден некроз отдельных перипортальных гепатоцитов. По мере прогрессирования процесса очаги некроза распространяются от портального тракта в сторону центральной вены - это и есть патоморфология развития цирроза печени.
У многих пациентов с аутоиммунным гепатитом обнаруживают АТ и клоны T-лимфоцитов, реагирующие с внутриклеточными Аг микросом, а именно с Аг цитохрома Р450. Выявлен даже характерный сегмент ДНК V-области β-цепи TCR, экспрессированный в аутореактивных клонах, - νβ3/Jβ1.2. Иммунные T-лимфоциты из очагов воспаления в печени при заболевании продуцируют преимущественно ИЛ-4.
Аутоиммунный гепатит ассоциирован с аллелем HLA-DR4, болеют гораздо чаще женщины, чем мужчины, возраст манифестации - молодой и средний. Не менее чем в 60% случаев у тех же пациентов имеются другие аутоиммунные заболевания: инсулинзависимый диабет, тиреоидит, гломерулонефрит.
Лечение аутоиммунного гепатита. В острый период, как правило, быстрое и заметное улучшение клинического состояния удаётся достичь назначением ГКС (преднизолон 30 мг в день). На диагностической сравнительной биопсии печени можно видеть, что на фоне терапии ГКС существенно уменьшаются и гистологические признаки воспаления, но картина цирроза постоянна. При достижении клинической ремиссии дозу гормонов снижают насколько это возможно. Минимизировать дозу, иногда даже отменить препараты позволяет назначение в дополнение к ГКС иммунодепрессантов (азатиоприна).
Диагностическая биопсия печени показана для взвешенного прогноза у каждого пациента. В случае отсутствия гистологических признаков цирроза и при правильной терапии 10-летнее выживание вероятно более чем у 95% пациентов. При наличии признаков цирроза на биопсии вероятность 10-летнего выживания составляет около 60%. В случае витального цирроза показана трансплантация печени: 5-летнее выживание имеет эмпирическую вероятность около 80%.
Первичный склерозирующий холангит
Этиология первичного склерозирующего холангита, как и при других аутоиммунных процессах, неизвестна. Патологическая морфология: облитерирующий фиброз желчных протоков с интенсивной лимфоцитарной инфильтрацией очагов и деструкцией эпителия
желчных протоков. Среди лимфоцитов печени возрастает доля CD4+ T-клеток и Tγδ (до 10% при 3% в здоровой печени).
Примерно у 80% пациентов первичный склерозирующий холангит ассоциирован с воспалительными заболеваниями кишечника, которые также относят к аутоиммунной патологии. Первичный склерозирующий холангит развивается примерно у 5% пациентов с язвенным колитом (особенно с панколитом). Прогрессирование процесса приводит к билиарному циррозу. Специального лечения не подобрано. При декомпенсации цирроза назначают трансплантацию печени.
Первичный билиарный цирроз
Патоморфологический процесс при первичном билиарном циррозе - гранулёматозное воспаление портальных зон и прогрессивная деструкция мелких и средних желчных протоков. Лимфоцитарный инфильтрат состоит преимущественно из CD4+ T-лимфоцитов и лишь по периферии гранулем присутствуют CD8+ T-лимфоциты. При биохимическом анализе биопсийного материала в ткани печени обнаруживают заметное возрастание содержания меди. Этот факт используют для подтверждения диагноза заболевания на поздних стадиях.
При первичном билиарном циррозе известен нормальный компонент собственных тканей, ставший объектом атаки иммунных лимфоцитов, - это белок митохондрий, а именно Е2-компонент митохондриальной пируватдегидрогеназы, в норме локализованный на внутренней стороне мембраны митохондрий. Известен даже эпитоп для аутоиммунной атаки - домен E2L2, содержащий липоевую кислоту. Есть данные, что T-лимфоциты человека с Рц для данного домена (последовательность АК в эпитопе AVDKA) перекрестно распознают липоилсодержащий эпитоп пируватдегидрогеназного комплекса Escherichia coli с последовательностью АК EGDKA, т.е. не исключено, что в этиологии аутоиммунного первичного билиарного цирроза существенна (если не решающая) молекулярная мимикрия микробных продуктов и нормальных тканей человека. В патогенезе же, очевидно, значим ещё такой компонент, как аберрантная экспрессия молекул MHC-II на эпителиальных клетках желчных протоков (в норме этого нет).
От 20 до 50% пациентов с первичным билиарным циррозом оказываются под наблюдением ещё в клинически бессимптомный период. Их выявляют случайно по данным лабораторных анализов (повышены количество билирубина у 75%, активность щелочной фосфатазы в сыворотке крови у 97% и содержание IgM у 80%;
присутствуют антимитохондриальные АТ у 90% больных). При правильно начатом лечении среднее время выживания таких пациентов около 10 лет. Если больной попал под наблюдение в стадии манифестировавших клинических симптомов (желтуха, зуд кожи, слабость, висцеральные геморрагии, боли в животе, гепатомегалия, спленомегалия, отёки, асцит, усиление пигментации кожи), то прогноз выживания около 5-7 лет.
Лечение. Помимо симптоматического лечения, применяют иммунодепрессанты. При декомпенсированном циррозе - только трансплантация печени с прогнозом на выживание 80% больных в течение 5 лет.
заболевания крови
Аутоиммунная гемолитическая анемия
Различают по крайней мере две формы: тепловую и холодовую.
• При тепловой гемолитической анемии (протекающей при нормальной температуре внутренней среды: организма 36,8-37 °C) эритроциты человека аномально покрыты АТ преимущественно класса IgG и компонентами комплемента C3 и C4. В таком виде эритроциты подвергаются повышенной экстраваскулярной деструкции макрофагами печени и селезёнки. Этиология неизвестна. Бывают идиопатические случаи, встречаются так называемые вторичные варианты, т.е. ассоциированные с другими заболеваниями, в первую очередь лимфопролиферативными, а также с коллагеновыми болезнями, инфекциями, синдромами иммунодефицитов. Лечение. В первую очередь лечат основную болезнь. При тяжёлой анемии показаны гемотрансфузии; иногда улучшение состояния достигают применением ГКС, иммуносупрессивных препаратов, спленэктомии.
• Холодовую гемолитическую анемию вызывают АТ класса IgM, обычно направленные против таких Аг эритроцитов, как 001, i, Pr. Эта анемия бывает также идиопатической или вторичной. Вторичная ассоциирована с заболеваниями, вызываемыми Mycoplasma pneumoniae, и при инфекционном мононуклеозе, как правило, в подростковом и молодом возрасте; саморазрешается при выздоровлении от основной болезни. У пожилых пациентов холодовая гемолитическая анемия чаще всего является осложнением лимфопролиферативных процессов и имеет хроническое длительное течение. Антиэритроцитарные АТ присоединяются к эритроцитам
только в периферических сосудах, где температура крови ниже 32 °C, затем в глубоких сосудах комплекс «эритроцит-АТ» фиксирует комплемент и развивается внутрисосудистый гемолиз. Гемолиз происходит эпизодами в связи с переохлаждением и ознобами, может сопровождаться (или нет) желтухой и гемоглобинурией. Лечение. Избегание переохлаждений - это главное. ГКС и спленэктомия улучшения не приносят. Остановимся ещё на 3 видах разрушения эритроцитов, опосредованного иммунными факторами, но не являющихся аутоиммунными процессами, это гемолиз, связанный с введением в организм ЛС; гемолитическая болезнь новорождённых и гемотрансфузионные осложнения.
Лекарственный гемолиз
Отдельные ЛС у некоторых людей могут сорбироваться на поверхности эритроцитов или образовывать комплексы с белками крови, в том числе и Ig. В обеих ситуациях эритроциты оказываются под угрозой опосредованного комплементом лизиса и/или повышенного фагоцитоза макрофагами печени и селезёнки. В клинической практике явления такого рода замечены при применении пенициллинов, метилдопа, гидрохлоротиазида. Учитывая количество фармакохимических препаратов, следует иметь в виду возможность подобных осложнений в связи с другими ЛС. Прогноз зависит от величины гемолиза. Лечение сводится к коррекции осложнений, из которых наиболее опасна почечная недостаточность.
Гемолитическая болезнь новорождённых
Гемолитическая болезнь новорождённых - экстраваскулярное разрушение эритроцитов плода антиэритроцитарными АТ матери, проникшими через плаценту (IgG). Чаще всего такая ситуация возникает при несовместимости по АВ0 и группе крови 0 у матери. Степень выраженности патологического процесса у новорождённого сильно варьирует, поскольку Аг АВ0 к моменту рождения ещё недостаточно экспрессированы на эритроцитах, а АТ к Аг А и В принадлежат преимущественно к подклассу IgG2, плохо фиксирующему комплемент и слабо опсонизирующему к фагоцитозу. Вопрос об обменном переливании крови или плазмы решают в зависимости от степени анемии, наличия желтухи и общего состояния новорождённого.
Как правило, гораздо сильнее проявляется так называемый резусконфликт в случае Rh-отрицательной матери и Rh-положительного
плода. Аг Rh(D) хорошо экспрессированы на эритроцитах ещё в пренатальном периоде, а АТ к Rh(D) принадлежат к подклассам IgG1 и IgG3, которые хорошо активируют комплемент и опсонизируют к фагоцитозу. При сильно выраженном процессе плод может погибнуть ещё в матке с явлениями водянки. При рождении живого ребёнка часто необходимо обменное переливание крови или плазмы в зависимости от выраженности анемии. Иногда это делают ещё in utero. Если у Rh(D)-отрицательной матери при предыдущих беременностях были травмы, геморрагии плаценты, эктопическая беременность, акушерские процедуры, при которых клетки плода могли бы попасть в организм матери, то для профилактики резуc-конфликта при текущей беременности женщине вводят анти-Rh(D)-сыворотку [что через FcγRIIB на B-лимфоцитах ингибирует продукцию АТ B-лимфоцитами именно клона aнти-Rh(D)].
Гемолиз при трансфузионных осложнениях
Международным обществом переливания крови в 2004 г. признаны следующие группы: крови: ABO [в русскоязычной литературе - AB0 (цифра «0»)], Cartwright, Chido/Rodgers, Colton, Cost, Cromer, Diego, Dombrock, Duffy, Er, Gerbich, GIL, GLOB (Globoside), Hh, Ii, Indian, JMH (John Milton Hagen), Kell, Kidd, Knops, Kx, Landsteiner-Wiener, Lewis, Lutheran, MNS, OK, P, Raph, Rh, Scianna, Wright, Xg, Yt с присвоением им номеров в соответствии с порядком открытия систем (группа АВ0 - 001, Rh - 004). В практике переливания крови (гемотрансфузия) и её компонентов обязательна проверка на совместимость по Аг систем AB0 (4 группы) и Rh (2 группы). Остальные системы могут учитываться в особых случаях тестирования на совместимость и при определении возможности развития гемолитической болезни новорождённого.
Агглютиногены. Эритроцитарные Аг системы AB0 - A, B и 0 - относятся к классу гликофоринов. Их полисахаридные цепи содержат Аг-детерминанты - агглютиногены А и В. Формирование агглютиногенов А и В происходит под влиянием гликозилтрансфераз, кодируемых аллелями гена АВ0. Этот ген кодирует три полипептида (А, В, 0), два из них (гликозилтрансферазы А и В) модифицируют полисахаридные цепи гликофоринов, полипептид 0 функционально не активен. Так, гликозилтрансфераза А катализирует присоединение N-ацетилгалактозамина, что означает экспрессию Аг Α, а гликозилтрансфераза В - присоединение галактозы и экспрессию Аг B. В результате поверхность эритроцитов разных лиц может содержать
либо агглютиноген А, либо агглютиноген В, либо оба агглютиногена (А и В), либо не содержать ни агглютиногена А, ни агглютиногена В. В соответствии с типом экспрессии на поверхности эритроцитов агглютиногенов А и В в системе AB0 выделено 4 группы крови, обозначаемых римскими цифрами I, II, III и IV. Эритроциты группы крови I не содержат ни агглютиногена А, ни агглютиногена В, её сокращённое наименование - 0(I). Эритроциты группы крови IV содержат оба агглютиногена - AB(IV), группы II - A(II), группы III - B(III). Первые три группы крови обнаружил в 1900 г. Карл Ландштейнер, а четвёртую группу немного позже Декастрелло и Штурли.
Агглютинины. В плазме крови к агглютиногенам А и В могут содержаться АТ (соответственно α- и β-агглютинины). Плазма крови группы 0(I) содержит α- и β-агглютинины; группы A(II) - β-агглютинины, B(III) - α-агглютинины, плазма крови группы AB(IV) агглютининов не содержит.
Таблица 14-3. Содержание в крови разных групп (система AB0) агглютиногенов (Аг) и агглютининов (АТ)
 Таким
образом, в крови конкретного человека АТ к эритроцитарным Аг системы
AB0 одновременно не присутствуют (табл. 14-3), но при переливании крови
от донора с одной группой к реципиенту с другой группой может возникнуть
ситуация, когда в крови реципиента одновременно будут находиться и Аг и
АТ именно к этому Аг, т.е. возникнет ситуация несовместимости. Кроме
того, такая несовместимость может возникнуть и по другим системам групп
крови. Именно поэтому стало правилом, что переливать можно только
одногруппную кровь. Если точнее, то переливают не цельную кровь, а
компоненты, так как «показаний к переливанию цельной консервированной
донорской крови нет, за исключением случаев острых массивных
кровопотерь, когда отсутствуют кровезаменители или свежезамороженная
плазма, эритроцитарная масса или их взвесь» (из приказа МЗ РФ). И именно
поэтому теоретическое
Таким
образом, в крови конкретного человека АТ к эритроцитарным Аг системы
AB0 одновременно не присутствуют (табл. 14-3), но при переливании крови
от донора с одной группой к реципиенту с другой группой может возникнуть
ситуация, когда в крови реципиента одновременно будут находиться и Аг и
АТ именно к этому Аг, т.е. возникнет ситуация несовместимости. Кроме
того, такая несовместимость может возникнуть и по другим системам групп
крови. Именно поэтому стало правилом, что переливать можно только
одногруппную кровь. Если точнее, то переливают не цельную кровь, а
компоненты, так как «показаний к переливанию цельной консервированной
донорской крови нет, за исключением случаев острых массивных
кровопотерь, когда отсутствуют кровезаменители или свежезамороженная
плазма, эритроцитарная масса или их взвесь» (из приказа МЗ РФ). И именно
поэтому теоретическое
представление об «универсальном доноре» с кровью группы 0(I) на практике оставлено. Rh-система
Rh-система открыта в 1939 г. (Левин и Стетсон) и 1940 г. (Ландштейнер и Винер) при изучении образования у грызунов АТ к эритроцитам макак-резусов.
Антигены. 6 аллелей 3-х генов системы Rh кодируют Аг: c, C, d, D, e, E. С учётом крайне редко встречающихся Аг системы Rh возможны 47 фенотипов этой системы.
Антитела системы Rh относятся к классу IgG (не обнаружены АТ только к Аг d).
Наследование. Индивидуальные комбинации Аг (фенотипы) определяются гаплотипами системы Rh (c/C, d/D, e/E) каждого родителя.
Rh-положительные и Rh-отрицательные лица. Если генотип конкретного человека кодирует хотя бы один из Аг C, D и E, то такие лица резус-положительны (на практике резус-положительными считают лиц, имеющих на поверхности эритроцитов Аг D - сильный иммуноген). Таким образом, АТ образуются не только против «сильного» Аг D, но могут образоваться и против «слабых» Аг - c, C, e и E. Резус-отрицательны только лица фенотипа cde/cde (rr).
Резус-конфликт (несовместимость) возникает при переливании Rh-положительной крови донора Rh-отрицательному реципиенту либо у плода при повторной беременности Rh-отрицательной матери Rh-положительным плодом (первая беременность и/или роды Rh-положительным плодом). В этом случае развивается гемолитическая болезнь новорождённого.
При гемотрансфузиях активируются процессы внутрисосудистого комплементзависимого гемолиза, внесосудистого разрушения эритроцитов, а также анафилатоксины из системы комплемента - C5a и C3a, система коагуляции крови и кининовая система. Клинически значимые симптомы могут появиться от вливания всего 30 мл чужой крови. Основные клинические симптомы гемотрансфузионных осложнений: озноб, лихорадка, чувство жжения в месте инфузии, боли в грудной клетке и спине, диспноэ, нервное возбуждение, чувство психического дискомфорта (ощущение обречённости); затем гипотензия, олигурия, гемоглобинурия, анурия, шок, кровоточивость.
Лечение. Интенсивная противошоковая терапия и лечение синдрома диссеминированного внутрисосудистого свёртывания.
Идиопатическая аутоиммунная тромбоцитопеническая пурпура
Заболевание развивается у взрослых. У 10% больных наступает спонтанное излечение. АТ направлены против интегринов тромбоцитов GpIIb/IIIa. С этим заболеванием люди живут долго, если не появляются усугубляющие его обстоятельства. Симптомы: петехии, склонность к синякам, рекуррентные носовые кровотечения, меноррагии у женщин. Болезнь необходимо дифференцировать с постинфекционной тромбоцитопенией (подобной той, которая бывает у детей после вирусных инфекций), вторичной тромбоцитопенией ВИЧ-инфицированных, больных хроническими лимфопролиферативными заболеваниями, болезнью Ходжкина, СКВ, ревматоидным артритом, с тромбоцитопенией, индуцированной ЛС (хинином, хинидином, солями золота, ацетилсалициловой кислотой, гепарином). Лечение. ГКС, в тяжёлых случаях спленэктомия.
Аналогичные аутоиммунные и не «ауто», но иммуноопосредованные патологические процессы известны в отношении всех других клеток крови.
заболевания нервной системы с компонентом иммунного воспаления
Перечислим заболевания нервной системы, в патогенезе которых прослеживаются процессы иммунного воспаления. В периферической нервной системе это полинейропатии (синдром Гийена-Бар- ре; хроническая демиелинизирующая полирадикулонейропатия; мультифокальная моторная нейропатия; плексопатии; парапротеинемическая нейропатия). Нервно-мышечные нарушения: тяжёлая псевдопаралитическая миастения (myasthenia gravis), миастенический синдром Лэмберта-Итона. Нарушения спинного мозга: тропический спастический парапарез. В ЦНС это рассеянный склероз, острый диссеминированный геморрагический энцефаломиелит, подострый склерозирующий панэнцефалит, паранеопластический синдром (дегенерация мозжечка, энцефаломиелит). Кратко опишем только рассеянный склероз и myasthenia gravis.
Рассеянный склероз
Заболевание описано в 1868 г. врачом Шарко. Иммунное воспаление в патогенезе заболевания заподозрено в работах патофизиологов 50-х годов XX в. Заболевание в западных странах встречается
с частотой 1:1000 населения (т.е. весьма распространено). При этом заболевании происходит диссеминированная демиелинизация аксонов мозга преимущественно в перивентрикулярных областях и в мозолистом теле. Бляшки демиелинизации бывают размером от менее чем 1 мм до нескольких сантиметров. Олигодендроциты разрушаются, астроциты избыточно пролиферируют, в области бляшек развивается ацеллюлярный фиброз. В периваскулярных областях - лимфоцитарная инфильтрация.
Моделью этого заболевания считают экспериментальный аутоиммунный энцефаломиелит у мышей и крыс. Аутоантиген - ОБМ - основный белок миелина. Иммунопатогенез состоит в повреждении миелина по механизму ГЗТ, т.е. опосредован ТЫ. Мыши, трансгенные по TCR для ОБМ, у которых все T-лимфоциты в организме специфичны к ОБМ, здоровы. Но если таких мышей искусственно иммунизировать ОБМ с полным адъювантом Фрейнда (содержащим микробные продукты), у них быстро разовьётся клиническая картина энцефаломиелита. Спровоцировать клиническую манифестацию можно иначе - инфицировать мышей нейротропным изолятом вируса гепатита мышей, без иммунизации ОБМ. Тем не менее этиологические факторы рассеянного склероза у человека не идентифицированы.
Клиническая картина. Характерны (в соответствии с локализацией бляшек демиелинизации) симптомы неврита зрительного нерва, офтальмоплегия (нистагм при отведении и невозможность полного приведения глазного яблока), диплопия при попытке пристального взгляда, головокружения, гемистезии, гемипарезы, нарушения координации, спинальные синдромы у 30% больных. При прогрессировании - спутанное сознание, депрессия, деменция. Симптомы неврита зрительного нерва практически патогномоничны для рассеянного склероза: у 75% женщин, обратившихся впервые с жалобами на симптомы неврита зрительного нерва, в дальнейшем оказывается рассеянный склероз.
Диагноз ставят только по клинической картине. Адекватных лабораторных методов дифференциальной диагностики нет.
Лечение. Адекватного лечения нет. У некоторых пациентов отмечают временный эффект от больших доз метилпреднизолона (1 г внутривенно в течение 3). Иммунодепрессанты (азатиоприн, циклоспорин, циклофосфамид), как правило, неэффективны. Клинические испытания ИФНγ показали, что он усугубляет течение болезни (что и следовало ожидать). Клинические испытания
ИФНβ (рекомбинантного, негликозилированного), возможно, более обнадёживающи, но ещё не подтверждены. В последнее время для лечения рассеянного склероза предложен селективный иммуномодулятор Натализумаб (рекомбинатные моноклональные АТ против интегринов). Эти АТ блокируют адгезию T-лимфоцитов к эндотелию и тем самым уменьшают выраженность иммунного воспаления.
Прогноз. Через 15 лет от момента манифестации 10% пациентов не могут обходиться без инвалидного кресла, 50% вынуждены пользоваться палкой или посторонней помощью при ходьбе.
Тяжёлая псевдопаралитическая миастения
Заболевание впервые описали сэр Томас Уиллис в 1672 г., Вильгельм Эрб (1879) и Фридрих Йолли (1895). Тимэктомия в лечебных целях впервые была сделана в 1911 г. Заболевание встречается в Европе с частотой 2-4 на 100 тыс. населения, чаще у женщин. Пик манифестации - в возрасте 20-30 лет.
Этиология неясна.
Патогенез обусловлен блокирующими АТ к никотиновым Рц для ацетилхолина, обеспечивающего в нервно-мышечном синапсе передачу возбуждения с двигательных нервов на поперечнополосатые мышцы. У большинства пациентов с myasthenia gravis есть аномалии в тимусе: в 70% случаев они могут быть выявлены только микроскопически - лимфоидная фолликулярная гиперплазия. В 10% случаев макроскопически обнаруживают доброкачественную тимому. При этом опухолевые клетки тимуса имеют морфологические и биохимические признаки подобия клеткам поперечнополосатых мышц. Степень повреждения постсинаптических Рц к ацетилхолину колеблется от 30 до 50%. Соответственно различается и степень выраженности клинических симптомов: от средней степени птоза до тяжёлой мышечной дебильности и дыхательной недостаточности при вовлечении в процесс мышц, участвующих в респираторных движениях. При генерализованной миастении АТ к ацетилхолиновому Рц обнаруживают у 75% пациентов с myasthenia gravis, при только окулярной форме - у 50-60%. У больных с тимомами обнаруживают АТ не только к Рц для ацетилхолина, но также АТ к актину, а-актинину, миозину, Рц для рианодина (кальциевый канал в саркоплазматическом ретикулуме поперечнополосатых мышц). Обнаружение АТ к Рц для рианодина коррелирует с наиболее тяжёлыми клиническими формами myasthenia gravis.
Миастеническая диагностическая проба - у больных с myasthenia gravis после подкожного введения неостигмина метилсульфата (0,5-
1,0 мл 0,05% р-ра) через 20-30 мин мышечная слабость временно уменьшается, затем поражённые мышцы вновь слабеют.
Лечение: ингибиторы ацетилхолинэстеразы, тимэктомия, ГКС, иммунодепрессанты. При применении ингибиторов ацетилхолинэстеразы (неостигмина метилсульфат, пиридостигмина бромид) требуется подбор доз в стационаре, поскольку побочные эффекты излишней стимуляции мускариновых Рц представляют проблему и нуждаются в тщательной коррекции (тошнота, спазмы в животе, диарея, излишнее слюно- и потоотделение). ГКС, а также иммунодепрессанты (азатиоприн, циклоспорин, циклофосфамид) у некоторых пациентов дают улучшение.
Прогноз. Ещё 30 лет назад от myasthenia gravis умирал каждый 4-й больной. Риск летального исхода максимален в течение первого года после постановки диагноза. Если человек переживает этот срок без признаков быстрой прогрессии, то прогноз благоприятен. Если в первые 2 года после манифестации произведена тимэктомия, то у некоторых пациентов достигается перманентная ремиссия. Если в течение 7 лет заболевание при любом способе лечения не выходит в «режим» быстрой прогрессии, то риск тяжёлого релапса невелик.
ПЕРВИЧНЫЕ СИСТЕМНЫЕ ВАСКУЛИТЫ
Первичными системными васкулитами называют заболевания, при которых развиваются (по неизвестным причинам) воспаление и некроз стенок сосудов, а патологические процессы в окружающих тканях вторичны по отношению к поражению сосудов. В связи с неясностью этиологии васкулиты до настоящего времени классифицируют по морфологическим признакам, а именно по калибру поражаемых сосудов и наличию или отсутствию гранулём вокруг поражённых участков сосудов (табл. 14.4).
Таблица 14.4. Классификация нозологических форм первичных системных васкулитов
 Если
этиология ни одного из этих васкулитов неизвестна и можно лишь
предполагать инфекционное начало, то механизмы патогенеза, по крайней
мере гранулёматоза Вегенера (в остальных случаях можно предполагать по
аналогии), в какой-то мере изучены. У 85% больных гранулёматозом
Вегенера обнаруживают АТ к сериновой протеиназе-3 нейтрофилов (Pr3). Эти
АТ обозначают как ANCA (Anti-Neutrophil Cytoplasm Antigens). Фермент
локализован в гранулах нейтрофилов, но при активации клеток
экспрессируется на мембране. ANCA, связываясь с нейтрофилами, индуцируют
выброс из клеток агрессивных метаболитов (активных радикалов кислорода и
пероксида водорода, ферментов), которые разрушают стенки сосудов.
Провоспалительный цитокин TNFα индуцирует экспрессию Pr3 на клетках
эндотелия, и они становятся прямой мишенью иммунной атаки и,
следовательно, деструкции. Кроме ANCA, у больных васкулитом обнаруживают
АТ к другим ферментам нейтрофилов - миелопероксидазе, эластазе,
лизоциму, лактоферрину, катепсину G.
Если
этиология ни одного из этих васкулитов неизвестна и можно лишь
предполагать инфекционное начало, то механизмы патогенеза, по крайней
мере гранулёматоза Вегенера (в остальных случаях можно предполагать по
аналогии), в какой-то мере изучены. У 85% больных гранулёматозом
Вегенера обнаруживают АТ к сериновой протеиназе-3 нейтрофилов (Pr3). Эти
АТ обозначают как ANCA (Anti-Neutrophil Cytoplasm Antigens). Фермент
локализован в гранулах нейтрофилов, но при активации клеток
экспрессируется на мембране. ANCA, связываясь с нейтрофилами, индуцируют
выброс из клеток агрессивных метаболитов (активных радикалов кислорода и
пероксида водорода, ферментов), которые разрушают стенки сосудов.
Провоспалительный цитокин TNFα индуцирует экспрессию Pr3 на клетках
эндотелия, и они становятся прямой мишенью иммунной атаки и,
следовательно, деструкции. Кроме ANCA, у больных васкулитом обнаруживают
АТ к другим ферментам нейтрофилов - миелопероксидазе, эластазе,
лизоциму, лактоферрину, катепсину G.
Отдельные васкулиты имеют общие признаки: цикличность чередования обострений и ремиссий, сезонность обострений (в холодное время года) и общность так называемых конституциональных неспецифических симптомов (общая слабость, лихорадка, потеря массы тела, ночные поты, повышение СОЭ и содержания в сыворотке C-реактивного белка - СРБ).
Лечение. При системных васкулитах оно одинаково, независимо от нозологии. Поскольку неизвестна конкретная этиология отдельных форм, применяют индуктивную терапию на высоте атаки - иммунодепрессанты (циклофосфамид или по подбору на эффективность) и высокие дозы ГКС; поддерживающую терапию - ГКС плюс симптоматические средства в зависимости от локализации процесса и присоединившихся патологий. В периоды ремиссий - отдых от ЛС. Поскольку при всех васкулитах рано или поздно повреждаются почки, в тяжёлых случаях единственным методом спасения жизни становится трансплантация донорской почки.
Гранулёматоз Вегенера
Заболевание встречается в любом возрасте, одинаково распределено по полу.
Клиническая картина. Некротизирующий васкулит мелких сосудов развивается в первую очередь в верхних и нижних дыхательных путях, поражает почки и в разной степени у разных пациентов мо-
жет затронуть другие органы. У некоторых пациентов заболевание развивается бессимптомно в течение многих месяцев и даже лет, симптомы нарастают постепенно. У некоторых больных наблюдают молниеносное развитие симптоматики (за несколько недель) с жизнеугрожающим поражением органов.
Верхние дыхательные пути: выделения из носа с примесью крови, боли в околоносовых пазухах, изъязвление слизистой оболочки носа, перфорация перегородки и деформация седла носа. Возможно развитие хронического гнойного среднего отита в связи с блокадой слуховой (евстахиевой) трубы, боли по ветвям VIII нерва. Возможны осиплость голоса и стридор, стеноз трахеи. В синусах могут образовываться фистулы с бактериальными суперинфекциями.
Нижние дыхательные пути: гранулёмы в паренхиме лёгких вызывают симптомы непродуктивного кашля, диспноэ, плеврита, кровохарканья. Возможны окклюзии бронхов и сегментарные коллапсы лёгких. Васкулит в почках начинает проявляться как клинически бессимптомная протеинурия и гематурия. Скорость развития поражения почек непредсказуема. При биопсии почечной ткани наблюдают картину гранулёматозного тубулоинтерстициального нефрита. Около 50% больных гранулёматозом Вегенера имеют поражение глаз (вплоть до потери зрения): конъюнктивит, увеит, склерит, ишемия зрительного нерва.
Поражения других органов (миалгии, артралгии, инфаркты ногтевых валиков, пурпура на коже, мононевриты, менингеальный синдром, инсульты мозга, поражение коронарных артерий, перикардиты, аритмии и т.д.), как правило, развиваются позже и по-разному у разных пациентов.
Диагноз ставят по клиническим симптомам и данным биопсии. Лабораторным подтверждением является обнаружение АТ ANCA. Из неспецифических лабораторных данных характерны нейтрофильный лейкоцитоз, нормохромная нормоцитная анемия, тромбоцитоз, повышение СОЭ и СРБ, у 80% больных увеличено содержание Ig в сыворотке крови, у 50% определяется ревматоидный фактор.
Микроскопический полиангиит
Микроскопический полиангиит (васкулит гиперчувствительности) - воспаление мелких сосудов, которое может затронуть любой орган или несколько органов, но чаще поражаются почки, кишечник и кожа. У многих пациентов обнаруживают АТ к компонентам нейтрофилов, особенно к миелопероксидазе.
Клиническая картина. Глубокая общая слабость, лихорадка, потеря массы тела, ночные поты. Если затронуты почки, наблюдают протеинурию, гематурию и быстро прогрессирующий нефрит. Лёгкие: кашель с кровохарканьем, диспноэ, симптомы плеврита. Бывают профузные кровотечения. На коже пурпурная сыпь, геморрагии в местах мелких травм (типа занозы). ЖКТ: боли в животе, диареи, гастроинтестинальные геморрагии.
Диагноз ставят по клиническим признакам и данным биопсии почки. Лабораторные данные, характерные для всех васкулитов: нормохромная нормоцитная анемия, нейтрофилёз, повышение СОЭ и СРБ, тромбоцитемия, гиперглобулинемия.
Узелковый полиартериит
Узелковый полиартериит (polyarteritis nodosa) - васкулит артерий среднего калибра. Встречается в любом возрасте, у мужчин в 2 раза чаще. Характерно частое образование аневризм в поражённых участках сосудов. Клиническая манифестация зависит от локализации и размеров поражённых сосудов: инфаркты органов (кишечника, почек, мозга, сердца), типична гипертензия средней или высокой тяжести. У некоторых пациентов polyarteritis nodosa сочетается с наличием микроскопического полиангиита. АТ ANCA обнаруживают редко (только если есть ещё микроскопический полиангиит). Часто выявляют вирус гепатита В. Поражение почек у многих является непосредственной причиной смерти. У некоторых пациентов polyarteritis nodosa поражает преимущественно один внутренний орган и манифестирует под видом острого холецистита, панкреатита или аппендицита, иногда некроза печени.
Клиническая картина. На коже polyarteritis nodosa проявляется в виде пурпуры и крапивницы, а в более тяжёлых случаях в виде подкожных геморрагий, вплоть до гангрены. Пальпируемые узелки по ходу поверхностных артерий являются диагностическим признаком этого заболевания.
Диагноз ставят по клиническим симптомам, подтверждают биопсией и ангиографией. Специфических лабораторных тестов нет. Неспецифические лабораторные данные - те же, что при всех васкулитах: нормохромная нормоцитная анемия, нейтрофилия, повышение СОЭ и уровня СРБ.
Синдром Чёрджа-Стросс
Этот васкулит поражает преимущественно лёгкие и характеризу-
ется эозинофилией (<20х109/л). Проявляется в виде бронхиальной астмы и признаков системного васкулита. Астма чаще имеет тяжёлую форму. Со стороны ЖКТ: симптомы абдоминальных болей, при биопсии - эозинофильный инфильтрат, широко распространённый по разным отделам ЖКТ.
Диагноз ставят по клиническим признакам, подтверждают биопсией. Характерны эозинофилия в крови и эозинофильный инфильтрат при биопсии. Дифференцировать с гиперэозинофильным синдромом (эозинофильная эндомиокардиальная болезнь, или болезнь Лёф- флера) позволяют лабораторные данные: при гиперэозинофильном синдроме число эозинофилов в крови превышает 20х109/л, при микроскопическом исследовании эозинофилы имеют аномальную морфологию (потеря гранул, вакуолизация цитоплазмы).
Пурпура Шёнляйна-Геноха
Это детский некротизирующий васкулит, поражающий кожу, ЖКТ и почки. Заболевают в возрасте от 2 до 10 лет, редко - взрослые (тогда средний возраст манифестации 43 года). При этом заболевании есть какая-то патология IgA, потому что в поражённых капиллярах кожи, мезангии клубочков почек и иных местах откладываются депозиты IgA. Часто уровень IgA повышен и в сыворотке крови.
Клиническая картина. Клиническая триада симптомов у детей: сыпь на коже, абдоминальные и почечные симптомы в сочетании с лихорадкой и артралгиями (что указывает на системный васкулит). Сыпь на коже может начинаться как крапивница, но со временем приобретает типичный для васкулитов вид слегка бугристой пурпурной сыпи на разгибательных поверхностях конечностей и на ягодицах. Боли в животе могут быть весьма сильными, обычно после приёма пищи, возможна мелена. Боли могут выглядеть как «острый живот». Почки поражаются у 40% пациентов, у большинства наступает спонтанное излечение и лишь у 10% нефрит прогрессирует.
Диагноз ставят по клиническим признакам и результатам биопсии, при которой можно обнаружить (методами иммуноцитохимии) депозиты IgA, C3 и фибриногена, преимущественно в посткапиллярных венулах. Специфических лабораторных тестов нет. В сыворотке крови могут определяться повышенные количества свободного IgA, IgA в составе иммунных комплексов, а также ревматоидного фактора класса IgA (АТ к IgG). Дифференциальный диагноз поставить трудно: приходится дифференцировать с другими формами васкулитов.
Гигантоклеточный артериит
Гигантоклеточный артериит - генерализованный артериит крупных артерий. Чаще первым становится очевидным поражение экстракраниальных ветвей сонной артерии. Процесс редко «спускается» ниже шеи. Таких больных учтено около 16 млн, возраст большинства - более 50 лет. Женщины болеют в 3-4 раза чаще. Патогистологически выявляются сегментарные поражения стенки сосуда с инфильтратом преимущественно из CD4+ T-лимфоцитов и гигантскими клетками-синцитием; гладкомышечные клетки частично подвергаются некрозу, часть их пролиферирует, что может прогрессировать вплоть до окклюзии сосуда.
Клиническая картина. Головная боль, боль в плечах, болезненность участков головы (скальпа), тошнота, лихорадка, потеря массы тела. Возможна болезненность в челюстях при жевании и разговоре. У 30% больных в патологический процесс вовлечена артерия сетчатки, что без лечения чревато необратимой потерей зрения.
Диагноз ставят по клиническим данным, подтверждают биопсией сонной артерии. СОЭ обычно повышена до 100 мм/ч. Как всегда, увеличен уровень СРБ и есть нормохромная анемия - около 11-12 г/л.
Лечение. Атаку лечат сначала большими дозами ГКС (40-60 мг/сут), через 4-6 нед дозу плавно снижают до 5-10 мг/сут. ГКС обычно принимают около 2 лет, после чего возможна ремиссия в течение нескольких лет. Только в случае неэффективности ГКС назначают азатиоприн или циклофосфамид или подбирают иммунодепрессанты ех juvantibus.
Артериит Такаясу
Артериит Такаясу встречается у молодых людей в 15-20-летнем возрасте, у женщин на порядок чаще, преобладает в странах Азии (особенно в Японии).
Клиническая картина. Выделяют 3 клинические стадии. Первая - преходящая, саморазрешается за несколько недель. Симптомы гриппоподобны: лихорадка, слабость, тошнота, головная боль, ночные поты, потеря массы тела, миалгии, артралгии. Вторая атака имеет признаки васкулита крупных артерий (подключичных, сонных, почечных, нисходящей дуги аорты, мезентериальных, лё- гочных и подвздошных артерий), но также саморазрешается, после чего наступает бессимптомный период на 5-10 лет. Третья атака у большинства больных заканчивается вазоокклюзией.
Диагноз ставят по клиническим данным, подтверждают на третьей стадии ангиографией.
Лечение. Сначала применяют ГКС, при их неэффективности - иммунодепрессанты.
Болезнь Кавасаки
Болезнь Кавасаки чаще встречается у детей с частотой 60-200 случаев на 100 тыс. населения. Весьма вероятна инфекционная природа заболевания.
Клиническая картина. Развивается как острое фебрильное мультисистемное заболевание: в первые 10 дней высокая температура тела и шейная лимфаденопатия. Через 2-4 нед присоединяются симптомы системного васкулита и миокардита (похоже на polyarteritis nodosa). С 4-й по 6-ю неделю симптомы васкулита стихают, но появляются симптомы фиброза. В течение нескольких лет происходят процессы образования аневризм, рубцов и организации тромбов. Примерно у 25% пациентов развиваются аневризмы коронарных артерий. Летальный исход возможен на любой стадии заболевания.
Лечение: эффективны препараты донорских Ig. Механизм их действия точно неизвестен.
Болезнь Бехчета
Болезнь Бехчета - мультисистемный васкулит, для которого характерны рекуррентные язвенные процессы в полости рта и в половых органах, также возможны кожный васкулит, воспаление синовиальных оболочек, увеит, менингоэнцефалит. Заболевание ассоциировано с HLA-B51. Диагноз можно поставить только по клиническим признакам. Специфических лабораторных тестов нет. Лечение нестандартное: колхицин (0,6 мг 2-3 раза в день), дапсон и нестероидные противовоспалительные средства. На ГКС и иммунодепрессанты ответ не всегда есть, при необходимости иммунодепрессанты подбирают.