Иммунология : учебник. - Хаитов Р. М. - 2009. - 320 с. : ил.
|
|
|
|
ГЛАВА 11 ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ
Первичные иммунодефициты (ПИД) - наследственные заболевания, обусловленные дефектами генов, контролирующих иммунный ответ. Клиническая симптоматика и адекватные лабораторные анализы позволяют дифференцировать патологию на уровне лимфоцитов и патологию на уровне нелимфоцитарных механизмов деструкции и выведения Аг.
• Суммарная частота ПИД составляет 1 случай на 10-100 тыс. живых новорождённых. Селективный дефицит IgA встречается гораздо чаще - 1 на 500-1500 жителей общей популяции. Большинство ПИД манифестирует в раннем детстве. ПИД следует заподозрить, если маленький ребёнок болеет инфекционными заболеваниями более 10 раз в год. У детей с ПИД инфекции могут принять персистирующий характер. Следует обращать внимание на отставание по возрастным показателям развития, рекуррентные синуситы, отиты, пневмонии, диареи, мальабсорбцию, кандидозы.
• Инфекционный синдром. Ведущим в клинической картине ПИД является так называемый инфекционный синдром - повышенная восприимчивость к инфекциям вообще, необычно тяжёлое клиническое течение инфекционных болезней, рекуррентное течение, атипичные возбудители (часто оппортунистические). Инфекционный синдром характерен, но не патогномоничен для истинных иммунодефицитов, т.е. он всегда имеется при иммунодефицитах, но возможен и без иммунодефицита.
• Физикальный осмотр: можно выявить отсутствие лимфатических узлов, миндалин.
• Лабораторные исследования. Если клинические данные заставляют заподозрить ПИД, то выполняют следующие исследования: - анализ на ВИЧ-инфекцию; - определение формулы крови; - определение уровней IgG, IgA и IgM в сыворотке крови; - кожные пробы ГЗТ на Аг (столбнячный, дифтерийный, стрептококковый
Аг, туберкулин, Аг Proteus mirabilis, Trichophyton mentagrophytes, Candida albicans); - подсчёт субпопуляций T- и B-лимфоцитов. по специальным показаниям: - анализ состояния фагоцитов (наиболее простой и информативный анализ - тест на восстановление тетразолиевого синего); - анализ на содержание компонентов комплемента (начинают с С3 и С4); - молекулярно-генетические исследования (если есть перспективы генной терапии).
ДЕФЕКТЫ ИММУНОГЛОБУЛИНОВ
X-сцепленная агаммаглобулинемия Брутона
Болеют мальчики, сыновья (ﭏ, ρ) носительниц дефектного гена btk (OMIM 300300, Xq21.3-q22), кодирующего специфичную для Б- лимфоцитов протеинтирозинкиназу Btk). В результате развиваются нарушения внутриклеточных сигнальных путей, рекомбинации тяжё- лых цепей Ig, дифференцировка пре-Б-клеток в B-лимфоциты.
Лабораторные данные. Отсутствуют периферические Б-лимфоциты. В костном мозге есть пре-Б-клетки с μ-цепью в цитоплазме. Число T-лимфоцитов и функциональные тесты на T-лимфоциты в норме. IgM и IgA в крови не определяются; IgG может быть, но в малых количествах (0,4-1,0 г/л). Нет АТ к Аг групп крови и к вакцинным Аг (столбнячному, дифтерийному токсинам и др.). Гистологическое исследование лимфоидной ткани: нет герминативных центров и плазматических клеток.
Клиническая картина. Если семейный анамнез неизвестен, то диагноз становится очевидным к возрасту в среднем 3,5 года. Для заболевания характерны тяжело протекающие гнойные инфекции, инфекции верхних (синуситы, отиты) и нижних (бронхиты, пневмонии) дыхательных путей, возможны гастроэнтериты, пиодермии, септические артриты (бактериальные или хламидиозные), септицемия, менингиты, энцефалиты. Инфекции дыхательных путей чаще вызваны Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus, диареи - кишечными бактериями или Giardia lamblia. Из вирусов типичны нейротропные вирусы ECHO-19, вызывающие персистирующий менингоэнцефалит. У больных детей при иммунизации живой полиовакциной, как правило, наблюдают продолжительное выделение через слизистые оболочки вируса полиомиелита, причём с восстановленной и нарастающей вирулентностью (т.е. в детском коллективе реальна опасность заражения здоровых
детей полиомиелитом в результате контакта с вакцинированным иммунодефицитным ребёнком).
Осмотр. Обращают внимание на отставание в росте, пальцы в виде барабанных палочек, изменения формы грудной клетки, характерные для заболеваний нижних дыхательных путей, гипоплазию лимфатических узлов и миндалин.
Лечение. - Противомикробная химиотерапия. - Заместительная терапия: внутривенные инфузии донорских препаратов сывороточных Ig каждые 3-4 нед пожизненно. Дозы Ig подбирают так, чтобы создать в сыворотке больного концентрацию Ig, перекрывающую нижнюю границу возрастной нормы. - Обсуждается возможность генной терапии. Ген Btk клонирован, но его гиперэкспрессия ассоциирована со злокачественной трансформацией кроветворной ткани.
Х-сцепленная агаммаглобулинемия с синдромом гипериммуноглобулинемии M
Х-сцепkенный гипер-IgМ-синдром (OMIM 308230, 300386) развивается при дефекте гена, кодирующего CD40LG (CD154, Xq26-q27.2) из семейства TNF - лиганда для CD40. Недостаточность экспрессии CD40L в T-лимфоцитах приводит к невозможности переключения синтеза Ig в Б-лимфоцитах с IgM на другие изотипы и нарушению формирования Б-клеток памяти. Болеют мальчики (К).
Лабораторные данные. IgG, IgA, IgE не определяются или их очень мало. Уровень IgM повышен, зачастую значительно. IgM поликлональны, иногда моноклональны. В лимфоидной ткани отсутствуют герминативные центры, но есть плазматические клетки.
Клиническая картина. Рекуррентные бактериальные и грибковые инфекции, в том числе оппортунистические (Pneumocystis carinii). Могут быть лимфаденопатия и спленомегалия. Похожую клиническую картину описывают у детей, перенесших внутриутробную инфекцию вирусом краснухи.
Лечение: противомикробная химиотерапия и регулярные пожизненные инфузии препаратов донорских сывороточных Ig.
Общий вариабельный иммунодефицит
При общем вариабельном иммунодефиците [OMIM 240500, ген TNFRSF13B (ρ и R) Рц суперсемейства TNF (17p11)] происходит нарушение способности Б-лимфоцитов дифференцироваться в плазматические клетки, развиваются дефекты антителопродукции,
возможна дисфункция T-лимфоцитов, наблюдаются частые рецидивирующие бактериальные инфекции. Синдром может манифестировать в раннем детстве, в подростковом возрасте или у молодых людей.
Лабораторные данные. Существенно снижены уровни IgG и IgA (примерно у 50% больных) и IgM (вплоть до неопределимых количеств). Число Б-лимфоцитов в крови соответствует норме или снижено. Число T-лимфоцитов у большинства больных в норме. Способность вырабатывать специфические АТ в ответ на иммунизацию снижена или отсутствует.
Клиническая картина. Рекуррентные бактериальные инфекции с локализацией преимущественно в дыхательных путях и околоносовых пазухах. К моменту постановки диагноза инфекции дыхательных путей могут прогрессировать до бронхоэктазов и разлитых поражений лёгочной ткани. Возможны инфекции ЖКТ, проявляющиеся диареей, стеатореей и мальабсорбцией (соответственно, потерей массы тела). Часто встречается инфекция, вызванная Giardia lamblia. От агаммаглобулинемии Брутона вариабельный иммунодефицит отличают более поздний возраст манифестации, равное распределение по полу, тенденция к нарушению функций T-лимфоцитов, склонность к аутоиммунным болезням, повышенная частота лимфом и инфекций, вызванных Pneumocystis carinii и вирусами семейства Herpetoviridae.
Лечение - противомикробная химиотерапия. При её неэффективности - заместительная терапия внутривенными препаратами донорских Ig (по мере необходимости).
Дефицит иммуноглобулина A
Селективный дефицит IgA (OMIM 137100) развивается при дефекте гена tnfrsf13b (R или ρ). Дефицит IgA при наличии Ig других классов встречается в общей популяции с высокой частотой (1 случай на 500-1500 жителей, а у аллергических больных ещё чаще). Различают недостаточность IgA селективную, т.е. заключающуюся в дефиците отдельных подклассов (30% случаев), и полную (70% случаев). Дефицит подкласса IgA2 приводит к более выраженной клинической картине, чем дефицит подкласса IgA1. Возможны и сочетания дефицита IgA с другими нарушениями: дефектным биосинтезом IgG и аномалиями T-лимфоцитов.
Клиническая картина. В половине случаев заболевания развиваются рекуррентные инфекции верхних дыхательных путей.
Лечение. Случаи бессимптомного течения не требуют никакого специального лечения; при наличии клинических проявлений проводят противоинфекционную химиотерапию и симптоматическое лечение. Заместительная терапия донорскими Ig не показана ни при селективном, ни при полном дефиците IgA, так как высока вероятность образования у реципиента антиизотипических АТ к IgA и обусловленные этим трансфузионные осложнения.
Транзиторная гипогаммаглобулинемия у детей. У здоровых детей к 3-месячному возрасту уровень IgG в крови снижается до минимума, так как распадается значительная часть материнских АТ, а затем начинается выработка собственных Ig. У некоторых детей, однако, нарастание уровня Ig задерживается. Такие дети могут страдать рекуррентными бактериальными инфекциями. Тем не менее, и в этих случаях следует избегать инфузий донорских Ig, так как даже однократное введение препаратов донорской крови подвергает пациента риску заражения ретровирусными (ВИЧ и др.) и прионными инфекциями.
ДЕФЕКТЫ T-ЛИМФОЦИТОВ
Тяжёлые комбинированные иммунодефициты
Клинический синдром тяжёлой комбинированной иммунной недостаточности (ТКИН) характерен для следующих состояний:
• недостаточность аденозиндезаминазы [OMIM 608958, КФ 3.5.4.4, ген ada (20q12-q13.11, ρ];
• недостаточность пуриннуклеозидфосфорилазы [OMIM 164050, КФ 2.4.2.1, ген pnp (14q11.2), ρ];
• T- и Б-негативная, NK-положительная ТКИН [OMIM 601457, гены RAG1 и RAG2 рекомбинации V(D)J-сегментов Ig и TCR
(11р13), ρ];
• синдром Оменна [OMIM 179615 и 179616, гены RAG1 и/или RAG2 рекомбинации V(D)J (11p13-p12), ρ];
• синдром Оменна с гиперэозинофилией [OMIM 603554, гены RAG1 и RAG2 рекомбинации V(D)J (11p13-p12), ρ];
• ТКИН с повышенной чувствительностью к ионизирующей радиации [OMIM 605988, ген репарации ДНК Artemis (10p), при мутации гена происходит нарушение V(D)J-рекомбинации, ρ];
• недостаточность протеинтирозинкиназы ZAP-70 [OMIM 176947, ген ZAP-70 (2q12, ρ]. При мутации гена страдает фосфорили-
рование доменов ITAM ζ-цепи TCR и ITAM-содержащих Рц NK-клеток;
• недостаточность тирозинкиназы Janus3 [OMIM 600802 и 600173, ген JAK3(19p13.1), ρ]. При дефектах гена нарушаются внутриклеточные сигнальные пути, обеспечивающих пролиферацию, дифференцировку и выживание клеток (в частности, интерлейкины 2, 4, 7, 9, и 15 не индуцируют клеточную активацию);
• ТКИН с дефицитом ИЛ-2 (OMIM 147680, ген IL-2, 4q26-q27);
• X-сцепленная ТКИН с недостаточностью Рц для ИЛ-2 [OMIM
300400, ген IL-2RG (γ-цепь Рц для ИЛ-2), Xq13.1-q21.1, К];
• недостаточность протеинтирозинфосфатазы (CD45, OMIM 151460, ген PTPRC, 1q31-q32). При дефекте гена происходит усиление ингибирующей активности киназы Csk на протеинтирозинкиназу Src с нарушением фосфорилирования ITAM-доменов TCR и BCR. Лабораторные данные. Вариабельная, иногда глубокая лимфопе-
ния; лимфоциты не способны пролиферировать в ответ на специфический Аг; часто выраженное снижение уровня Ig в сыворотке крови. На рентгенограмме грудной клетки отсутствует тень тимуса.
Клиническая картина. Важно как можно раньше распознать ТКИН у новорождённых, так как для них фатальна, например, иммунизация живыми вакцинами. Обычно клинический диагноз становится ясен в первые 6 месяцев жизни. В клинической картине на первый план выходят тяжёлый инфекционный синдром и отставание в развитии. Возбудители инфекций принадлежат к разным таксономическим группам: грибы (Candida, Pneumocystis carinii), бактерии, вирусы. Пневмония часто бывает вызвана P. carinii, диареи - ротавирусами, Campylobacter, Giardia lamblia. Нередко манифестирует вирусный гепатит.
Лечение. Без трансплантации костного мозга дети с ТКИН, как правило, умирают на первом году жизни. Описаны единичные случаи, когда ребёнок в особо санированных условиях доживал до 2-3 лет.
• X-сцепленная тяжёлая комбинированная иммунная недостаточность
[OMIM 300400, ген IL-2RG (γ-цепь Рц для ИЛ-2, Xq13.1-q21.1, К ]. Болеют мальчики.
Лабораторные данные. Снижено (вплоть до полного отсутствия) количество T- и NK-лимфоцитов. Количество Б-лимфоцитов не ниже нормы, но имеется выраженная гипогаммаглобулинемия.
Лечение. Трансплантация HLA-совместимого костного мозга. Здесь отметим, но это будет относиться ко всем вариантам ТКИН: при трансплантации костного мозга больным с ТКИН (кроме подбора
совместимых по HLA донора и реципиента) обязательна процедура удаления перед трансплантацией из донорского костного мозга T- лимфоцитов во избежание такого осложнения, как «реакция трансплантат против хозяина» (РТПХ). Совместимость по HLA необходима не для профилактики отторжения костного мозга реципиентом (ему отторгать нечем), а для достижения цели - создания иммунокомпетентности реципиента: позитивная селекция T-лимфоцитов в тимусе «идёт» на MHC, экспрессированных на эпителиальных клетках тимуса (т.е. на MHC реципиента), а АПК на периферии (ДК, Б-лимфоциты, макрофаги) - костномозгового происхождения (т.е. от донора). Если совпадения по MHC не будет, то T-лимфоциты не будут узнавать Аг и, следовательно, иммунодефицит останется и при наличии прижившихся костномозговых клеток донора.
• Тяжёлый комбинированный иммунный дефицит при недостаточности аденозиндезаминазы [OMIM 608958, КФ 3.5.4.4, ген ada (20q12- q13.11), ρ]. При недостаточности аденозиндезаминазы (АДА) в клетках накапливается токсичный полупродукт метаболизма пуринов - дезоксиаденозин. Больше других от этого страдают лимфоциты.
Клинические признаки ТКИН (рекуррентные респираторные инфекции, диареи, отставание в развитии), как правило, становятся очевидны к 2-3-месячному возрасту.
Лабораторные данные. Отсутствуют T- и Б-лимфоциты (NK сохранны) и иммуноглобулины. Анализ на наличие АДА проводят в эритроцитах или лимфоцитах.
Лечение: - заместительная терапия препаратом АДА крупного рогатого скота, конъюгированной с полиэтиленгликолем (инъекции 3 раза в неделю), может поддерживать жизнеспособность пациента в течение нескольких лет; - трансплантация костного мозга от HLA-сов- местимого донора; - регулярные трансфекции гена ada в лимфоциты периферической крови (есть случаи успеха такой терапии).
• Синдром Оменна. Дефект затрагивает гены RAG1/RAG2. Благодаря невысокой остаточной активности этих генов всё же развивается некоторое количество клонов T-лимфоцитов, аутореактивных к эпителию кожи и ЖКТ, где они и размножаются. Эти клоны продуцируют большие количества ИЛ-4 и ИЛ-5, вызывая гиперэозинофилию и продукцию IgE остаточными Б-лимфоцитами. Клиническая картина. Вскоре после рождения манифестируют
эритродермия (покраснение кожи) и пахидермия (утолщение кожи) с алопецией в области скальпа и бровей, изнуряющая диарея, жиз-
неугрожающий инфекционный синдром. Пальпаторно определяют гепатоспленомегалию и увеличение лимфатических узлов.
Лабораторные данные. В крови повышено содержание эозинофилов и IgE при отсутствии Ig других классов. В биоптатах увеличенных лимфатических узлов: гиперплазия макрофагов и клеток соединительной ткани и почти полное отсутствие лимфоцитов.
• Лечение симптоматическое. Дефект ZAP70. Клиническая картина этого варианта ТКИН такая же, как и при других ТКИН. Лабораторные данные: отсутствие CD8+ T-лимфоцитов, наличие подчас значительных количеств CD4+ T-лимфоцитов (которые, однако, функционально недееспособны). Лечение симптоматическое.
Синдром «голых лимфоцитов»
Так называют патологию, когда в организме не экспрессируются молекулы MHC-I (тип 1, OMIM 170261) или MHC-II (тип 2, OMIM 209920). Когда нет экспрессии молекул MHC-I, полностью отсутствуют CD8+ Tαβ-лимфоциты, MHC-II - нет CD4+ T-лимфоцитов. Охарактеризовано несколько генетических дефектов. Клиническая картина синдрома «голых» лимфоцитов аналогична остальным ТКИН. Лечение симптоматическое.
Синдром Ди Джорджи
При синдроме Ди Джорджи, или синдроме третьего и четвёртого глоточных карманов [OMIM 188400, делеции в 22q 11, в том числе гена TBX1 (22q11.2),R] наблюдается гипоплазия или аплазия тимуса, гипоплазия паращитовидной железы, дефицит T-лимфоцитов, вариабельные количества B-лимфоцитов.
Диагностика для опытного врача не представляет затруднений. Иммунодефицитный компонент представлен гипоплазией или аплазией тимуса и рекуррентными, тяжело протекающими инфекциями. Наблюдают также гипопаратиреоидизм (гипокальциемия и, как следствие - тетания, заметная на 1-2-й дни после рождения); сердечно-сосудистые пороки (правый разворот дуги аорты, стеноз правого желудочка, дефекты в межжелудочковой и межпредсердной перегородках, тетрада Фалло, атрезия или гипоплазия лёгочной артерии); впадина нёба; характерное лицо (увеличенное расстояние между парными органами, челюсти уменьшенного размера, особенно нижняя, низко посаженные ушные раковины с усиленной вывернутостью назад, короткий подносовой желобок).
Лабораторное обследование: количество T- и Б-лимфоцитов у разных больных значительно варьирует. Лечение симптоматическое.
X-сцепленный лимфопролиферативный синдром
Иммунодефицит, инициируемый вирусом Эпштейна-Барр (EBV), или X-сцепленный лимфопролиферативный синдром (обусловлен дефектами локуса в Xq25, ﭏ). Клиническая картина. Болеют мальчики: острая первичная EBV-инфекция прогрессирует с развитием тяжёлого поражения печени. Если мальчик выживает, то в дальнейшем нередко развиваются стойкая гипогаммаглобулинемия, инфильтраты в лимфатических узлах, селёзенке, печени, ЦНС. Среди отсроченных последствий - повышенная частота лимфом. Лечение. Противовирусная химиотерапия (ганцикловир или другие противогерпетические препараты) и симптоматическая терапия.
Атаксия-телеангиэктазия
Синдром Луи-Бар, или атаксия-телеангиэктазия [OMIM 208900 и 607585, КФ 5.99.1.3, дефект гена ДНК-топоизомеразы АТМ (11q22), ρ]. Этот синдром (частота 1:300 тыс. новорождённых) с весьма гетерогенным фенотипом описан бельгийским врачом Луи-Бар. Характерны: гипоплазия тимуса, лимфатических узлов, селезёнки, миндалин, количественная и функциональная недостаточность T-лимфоцитов, снижен уровень IgA, IgE, IgG2; необычайно высока (в 200 раз превосходит среднюю частоту в общей популяции) частота новообразований (преимущественно лимфомы и карциномы), нередко становящихся непосредственной причиной летального исхода к 10-12-летнему возрасту. Клиническая картина. Симптомы атаксии можно обнаружить у ребёнка уже в 2-4-месячном возрасте. Атаксия обусловлена прогрессирующей дегенерацией клеток Пуркинье в мозжечке. Телеангиэктазы на носу, ушах, конъюнктиве появляются несколько позже, к 3-6 годам. Иммунодефицит проявляется в виде снижения выработки иммуноглобулинов изотипов IgA, IgE, IgG2, при этом примерно у 80% больных возникает соответствующая инфекционная клиническая симптоматика. Число T-лимфоцитов у большинства пациентов нормальное. Лечение симптоматическое.
Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича [OMIM 301000, дефект гена WASP (Xp11.23-p11.22), К; также OMIM 277970 (ρ) и OMIM *600903 (R)]. Ген WASP (от Wiskott-Aldrich syndrome) экспрессируется в лимфо-
цитах, ткани селезёнки и в тимоцитах. Мутации этого гена ассоциированы с аномальной экспрессией в нейтрофилах и T-лимфоцитах (CD4 и CD8) молекулы CD43 (лиганд для ICAM-1, выполняет антиадгезивную функцию). Клинически заболевание проявляется триадой признаков: тромбоцитопенией, экземой и рекуррентными инфекциями. Лабораторные показатели. Тромбоцитопения, тромбоциты меньшего размера, чем у здоровых людей. Уровень Ig (особенно IgM) в сыворотке крови снижен в разной степени у различных пациентов, при этом количество IgE повышено. Число T-лимфоцитов варьирует. Лечение симптоматическое.
ДЕФЕКТЫ ФАГОЦИТОЗА
Хроническая гранулематозная болезнь
Хроническая гранулематозная болезнь (ХГБ) - нарушения функциональной активности фагоцитов (образование активных форм кислородных радикалов, внутриклеточный киллинг и фрагментация фагоцитированных патогенов) с постоянными бактериальными и грибковыми инфекциями. Развивается ХГБ у лиц с различными генетическими дефектами [ген gp91-phox (Xp21.1), ﭏ; ген р47-phox (7q11.23), ρ; ген р67-phox (1q25), ρ; ген р22-phox (16q24), ρ], приводящими к нарушениям в системе НАДФ-оксидазы. При гибели короткоживущих (несколько часов) нейтрофилов неубитые бактерии «вытекают» в очаг воспаления. Макрофаги - долгоживущие клетки, и их предшественники (моноциты) стекаются к очагу в повышенных количествах (это и создаёт гранулёмы), фагоцитируют что могут, но убить микроорганизмы они также не в состоянии [дефектный ген(ы) общий у всех клеток организма]. Клинически заболевание манифестирует в детском возрасте инфекционным синдромом (внутри- и внеклеточные инфекции) и образованием гранулём. Лечение. Противомикробная терапия (особенно ко-тримоксазол). Показана эффективность терапии ИФHγ: в группе пациентов с ХГБ, получающих этот препарат, 70% больных не имели тяжёлых проявлений инфекций в течение года, в контрольной группе (плацебо) таких пациентов было 30%. В редких случаях прибегают к трансплантации костного мозга. Прогноз неблагоприятен.
ДЕФЕКТЫ СИСТЕМЫ КОМПЛЕМЕНТА
Болезни с дефицитом компонентов комплемента
Проявления генных дефектов отдельных компонентов системы комплемента приведены в табл. 11.1.
• Наследственный ангионевротический отёк. Болезни, обусловленные дефицитом компонентов комплемента, встречаются редко, поскольку для их манифестации необходимо гомозиготное состояние по аутосомным аллелям. Есть единственное исключение - C1inh (ингибитор C1-эстеразы): мутация гена C1inh, приводящая к дефициту ингибитора, проявляется в гетерозиготном состоянии фенотипом, известном как наследственный ангионевротический отёк.
• Болезни иммунных комплексов. Недостаточность C1-C4 проявляется в болезнях иммунных комплексов - системных васкулитах и повреждениях почек, что обобщённо называют синдромом системной красной волчанки (СКВ).
• Пиогенные инфекции. Дефицит C3 (также факторов H и I) ассоциирован с повышенной восприимчивостью к пиогенным инфекциям. Дефицит компонентов, участвующих в альтернативном пути активации комплемента, а также дефицит компонентов C5-C8 ассоциированы с повышенной восприимчивостью к инфекции, вызываемой Neisseria spp. Дефицит C9 клинически бессимптомен.
Таблица 11.1. Клинические проявления дефектов отдельных компонентов системы комплемента
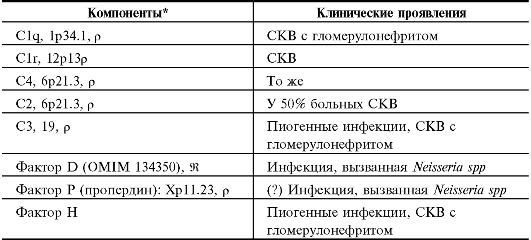 Продолжение табл. 11.1.
Продолжение табл. 11.1.
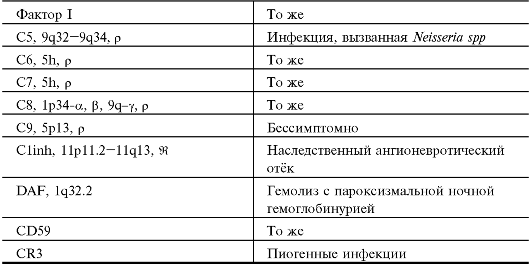 * в том числе ген, наследование
* в том числе ген, наследование
Дефицит связывающего маннозу лектина
Дефицит связывающего маннозу лектина (СМЛ) обусловлен дефектом гена MBL (разные точечные мутации и делеции в гене MBL встречаются у 17% людей европеоидной расы). При дефектах гена нарушается активация протеаз, расщепляющих компоненты комплемента C2 и C4, и активации системы комплемента по лектиновому пути. Клинически эта патология проявляется инфекционным синдромом. Лабораторные данные. Анализ субпопуляций лимфоцитов, лейкоцитов, а также Ig не показывает отклонений, адекватных клиническим симптомам. В сыворотке крови отсутствует СМЛ. Лечение. Ген MBL клонирован. Следовательно, не является проблемой получение рекомбинантного белка, который может быть использован как фармакологический препарат для этиопатогенетической заместительной терапии у пациентов с этим наследственным дефектом. Заместительная терапия рекомбинантным препаратом не несёт и опасности заразить больного кровяными инфекциями (ретровирусными, прионными и др.). При этом заболевании нет настоящего иммунодефицита, следовательно, не показана иммунокоррекция (противопоказана!) иммунотропными средствами.