Лекция 10МОРФОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ НЕДОСТАТОЧНОСТИ• Сердечно-сосудистая недостаточность (ССН) — патологическое состояние, в основе которого лежит сочетание сердечной и сосудистой недостаточности, объединенных общностью этиологии или патогенеза.
Сердечная недостаточность — патологическое состояние, обусловленное неспособностью сердца обеспечить адекватное кровоснабжение органов и тканей.
Сосудистая недостаточность — патологическое состояние, характеризующееся снижением тонуса гладкой мускулатуры сосудистых стенок, что приводит к развитию артериальной гипотензии, нарушению венозного возврата и поступлению крови из депо.
В большинстве случаев развитие сердечно-сосудистой недостаточности обусловлено первичным поражением сердца с развитием его недостаточности, которая неизбежно сопровождается реакцией сосудов. Эта реакция носит компенсаторный характер и при острой сердечной недостаточности проявляется вазоконстрикцией в ответ на прессорные механизмы, что приводит к временному повышению сосудистого сопротивления, некоторому подъему уровня артериального давления и нормализации кровоснабжения жизненно важных органов. При хронической сердечной недостаточности вазоконстрикция сменяется гипертрофией гладких мышечных клеток сосудистой стенки. В случае истощения компенсаторных сосудистых механизмов к сердечной недостаточности присоединяется сосудистая, сопровождающаяся снижением общего периферического сопротивления, резким расширением мелких вен, венул и капилляров -— венозным полнокровием, т.е. развивается сердечно-сосудистая недостаточность. Как синоним сердечно-сосудистой недостаточности часто используется термин
"недостаточность кровообращения".Почти любой процесс, который заставляет сердце усиленно работать в течение длительного времени или вызывает структурные повреждения миокарда, приводит к сердечно-сосудистой недостаточности. Чаще всего она встречается при следующих заболеваниях и состояниях:
▲ ишемической болезни сердца;
▲пороках сердца — врожденных и приобретенных (ревматических, атеросклеротических, после перенесенного бактериального эндокардита и др.);
▲ гипертензивных состояниях;
▲миокардитах;
▲ кардиомиопатиях;
▲ болезнях недостаточного питания, эндокринных и метаболических поражениях, в том числе при тиреотоксикозе, микседеме, бери-бери, карциноидном синдроме, болезнях накопления (жировых, углеводных), амилоидозе и пр. Наиболее частым из перечисленных заболеваний является ишемическая болезнь сердца (ИБС), на которую приходится более 80 % случаев смерти от сердечно-сосудистой недостаточности.
Сердечно-сосудистая недостаточность может развиваться остро или иметь хроническое течение. Наиболее частыми причинами острой сердечно-сосудистой недостаточности являются крупноочаговый инфаркт миокарда, тромбоэмболия крупных ветвей легочной артерии, острые миокардиты, инфекционные заболевания с выраженной интоксикацией, тампонада сердца и пр.
Хроническая сердечно-сосудистая недостаточность возникает при многих заболеваниях сердца — пороках, ишемической болезни сердца, хронических миокардитах, кардиомиопатиях и др.
Сердечная недостаточность может быть левожелудочковой (при ИБС, гипертонической болезни или симптоматических гипертензиях, при ревматических и врожденных пороках сердца, коарктации аорты, кардиомиопатиях, миокардитах, состояниях, сопровождающихся повышением сердечного выброса — токсикозах беременных (гестозы), тяжелых анемиях, гипоксии и гиперкапнии, лихорадочных состояниях, тиреотоксикозе, печеночной недостаточности, бери-бери и др.), правожелудочковой (при легочной гипертензии, эмболии легочной артерии, при некоторых врожденных пороках: дефектах межпредсердной перегородки, стенозе легочной артерии, пороках трехстворчатого клапана, некоторых миокардитах, изредка — при инфаркте миокарда с вовлечением правого желудочка) и тотальной — на поздних стадиях большинства перечисленных заболеваний, а также при тампонаде сердца.
Этиология. Среди многообразия причин, приводящих к сердечно-сосудистой недостаточности, выделяют три основные
группы:
▲ оказывающие прямое повреждающее действие на миокард;
▲ вызывающие функциональную перегрузку миокарда;
▲ нарушающие диастолическое наполнение желудочков. Прямое повреждающее действие на миокард могут оказывать различные факторы: физические (травма, действие электрического тока и др.), химические (высокое содержание некоторых биологически активных веществ: адреналина, тироксина; гипоксия; недостаток витаминов, других субстратов метаболизма; большие дозы некоторых лекарственных препаратов); биологические (инфекционные агенты, токсины, паразиты).
Функциональная перегрузка сердца может быть вызвана следующими факторами:
• чрезмерным увеличением количества притекающей к сердцу крови —
"перегрузка объемом" (при гиперволемии, недостаточности клапанов сердца, наличии артериовенозных вне- и внутри-сердечных шунтов и пр.);
• увеличением сопротивления, которое оказывается при выбросе крови из сердечных полостей —
"перегрузка давлением" (стенозы правого и левого предсердно-желудочкового отверстия, устья аорты и легочной артерии, гипертензия в большом и малом круге кровообращения. Развитию сердечно-сосудистой недостаточности при этом предшествует гипертрофия миокарда (гипертрофируется тот отдел сердца, которому приходится выполнять усиленную работу) и длительный период компенсации с включением как кардиальных, так и сосудистых механизмов, только при срыве которых появляются первые клинические признаки сердечно-сосудистой недостаточности.
Нарушение диастолического наполнения желудочков может быть обусловлено значительным снижением массы циркулирующей крови (при массивных кровопотерях, шоке) или нарушением диастолического расслабления сердца при его сдавлении жидкостью, накапливающейся в полости перикарда (транссудат, кровь, экссудат), при слипчивых перикардитах, рестриктивных кардиомиопатиях и др.
Как правило, сердечно-сосудистая недостаточность является результатом сочетанного действия факторов разных групп, чаще первых двух.
Патогенез. Основным пусковым механизмом сердечно-сосудистой недостаточности является снижение сердечного выброса. Один или оба желудочка теряют способность нормально выбрасывать в кровеносное русло содержащуюся в них кровь. Это ведет, с одной стороны, к увеличению конечного диастолического объема желудочка, повышению давления и объема в предсердии и венозной системе выше него, т.е. развивается венозный застой, который сопровождается повышением системного венозного и капиллярного давления, гипоксией и повышенной транссудацией жидкости в ткани. В случае левожелудочковой недостаточности венозный застой развивается в малом круге кровообращения. Напротив, при правожелудочковой недостаточности венозное полнокровие в основном развивается в большом круге кровообращения. Однако, если сердечно-сосудистая недостаточность сохраняется в течение нескольких месяцев или лет, то венозный застой распространяется на оба круга кровообращения.
С другой стороны, снижение сердечного выброса сопровождается неадекватным поступлением крови в артериальную систему. Для поддержания на нормальном уровне артериального давления при исходно сниженном сердечном выбросе усиливается активность симпатико-адреналовой системы. Гиперкатехоламинемия (в основном за счет содержания адреналина) приводит к сужению артериол и венул и повышению периферического сосудистого сопротивления. Ухудшение кровоснабжения почек вызывает включение почечного звена патогенеза сердечно-сосудистой недостаточности: активируется ренин-ангиотензин-альдостероновая система, что в конечном итоге приводит к задержке в организме натрия и воды, увеличению объема циркулирующей крови и еще большему повышению венозного давления, т.е. возникает порочный круг.
Сердечно-сосудистая недостаточность в результате перегрузки миокарда формируется на фоне более или менее длительной его гиперфункции, что сопровождается гипертрофией, т.е. увеличением мышечной массы сердца за счет увеличения количества и объема внутриклеточных структур кардиомиоцитов. Процесс не сопровождается адекватным энергообеспечением, что в конце концов также приводит к снижению силы и скорости сокращения и расслабления сердца. В обоих случаях — и при перегрузке, и при повреждении сердца снижение его сократительной функции сопровождается включением интракардиальных и сосудистых механизмов компенсации этого сдвига.
Интракардиальные компенсаторные механизмы. Среди них наиболее важными являются: ▲ увеличение развиваемого сердцем напряжения в ответ на растяжение его полостей (механизм Франка — Старлинга); ▲ увеличение силы сокращения в ответ на повышенную нагрузку при неизмененной длине мышечных волокон; ▲ увеличение частоты сердечных сокращений в результате повышения давления в полых венах, правом предсердии и растяжении их (рефлекс Бейнбриджа); ▲ усиление симпатоадреналовых влияний на миокард в связи со снижением сердечного выброса, что увеличивает как силу, так и скорость сердечных сокращений.
Включение названных механизмов обеспечивает экстренную компенсацию снижения сократимости миокарда. Однако это приводит к значительному увеличению интенсивности функционирования сердца, что не сопровождается адекватным энергообеспечением. Следствием этого является структурный полом митохондрий, сопровождающийся нарушением окисления свободных жирных кислот и снижением ресинтеза АТФ. Основным источником АТФ при этом становится гликолитический путь расщепления глюкозы, который в 18 раз менее эффективен, чем аэробный путь, и не может в достаточной мере компенсировать
дефицит макроэргических фосфатов. В кардиомиоцитах при этом возникает
жировая дистрофия — морфологический субстрат сердечной недостаточности. Тоногенная дилатация полостей сердца сменяется миогенной, что приводит к еще большему снижению сократительной функции сердца. Нарушения метаболизма кардиомиоцитов, лежащие в основе сердечной недостаточности, нельзя свести только к снижению продукции АТФ. Они более сложны и не до конца выяснены. По-видимому, играют роль повреждение мембранного аппарата и ферментных систем кардиомиоцитов, а также нарушение сопряжения процессов возбуждения и сокращения, в результате чего снижается доставка ионов кальция к контрактильным элементам. В развитии сердечной декомпенсации большое значение придают истощению симпатоадреналовых механизмов: подавляется биосинтез норадреналина в миокарде, содержание его в ряде случаев составляет лишь 10 % от нормальных значений, снижается количество бета-адренорецепторов. Существует мнение, что на более поздних стадиях сердечной недостаточности, когда содержание норадреналина в миокарде понижено, миокард становится во многом зависимым от внекардиальной адренергической стимуляции, главным образом надпочечниковой.
Сосудистые компенсаторные механизмы. Важным компенсаторным механизмом при снижении кровотока служит перераспределение сердечного выброса: доставка кислорода к жизненно важным органам — мозгу и сердцу, поддерживается на нормальном или субнормальном уровне, в то время как менее важные органы — кожный покров, скелетная мускулатура, органы брюшной полости, снабжаются кровью недостаточно. Основным механизмом перераспределения сердечного выброса является
вазоконстрикция, опосредованная через активацию симпатико-адреналовой системы (в основном за счет адреналина), что приводит к сужению артериол и венул. Этот механизм, с одной стороны, способствует поддержанию артериального давления, а с другой — препятствует распространению венозного застоя на капиллярное русло. Вазоконстрикция в свою очередь служит причиной многих клинических признаков сердечно-сосудистой недостаточности: задержки жидкости вследствие уменьшения почечного кровотока; субфебрильной лихорадки, вызванной снижением кожного кровотока; усталости, обусловленной уменьшением кровоснабжения мышц. Спазм венул и вен при длительно протекающем венозном застое сменяется выраженной гипертрофией мышечной оболочки. Так, в системе верхней полой вены человека при пороках сердца наступает десятикратное увеличение числа мышечных слоев. Гипертрофия мышечной оболочки возникает при забросе крови в обратном направлении (регургитации). Это связано, по-видимому, с сокращением стенок
вен в ответ на растяжение их просвета (реакция Бейлиса — Остроумова). Длительно протекающая регургитация может сопровождаться не только гипертрофией мышечной оболочки, но и врастанием мышечных клеток во внутреннюю оболочку и парадоксальным сужением просвета сосудов.
В стадии сосудистой компенсации сужение мелких вен предохраняет капилляры от полнокровия.
Стадия декомпенсации сосудистой недостаточности возникает при развитии фиброза гипертрофированной мышечной оболочки, сопровождающегося расширением просвета вен и развитием застоя в капиллярах.
Венозное полнокровие не ограничивается перестройкой венозного русла, оно включает веноартериальную реакцию. Последняя заключается в рефлекторном спазме артериол и мелких артерий и сопровождается гипертрофией их стенок. Впервые эта реакция описана в легких при сужении левого предсердно-желудочкового отверстия (митральный стеноз), позже была обнаружена и в других органах. Наиболее интенсивно она выражена в тех органах, где нет других возможностей адаптации — депонирования или коллатерального венозного полнокровия. Сущность этой реакции заключается в предохранении капилляров от полнокровия и предупреждении обратного кровотока из венозной системы в артериальную.
МОРФОГЕНЕЗ И МОРФОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ НЕДОСТАТОЧНОСТИОсновным проявлением сердечно-сосудистой недостаточности является общее венозное полнокровие: острое — при острой и хроническое — при хронической сердечно-сосудистой недостаточности.
Венозное полнокровие служит инициальным моментом для развития всех других изменений в органах при сердечной недостаточности. Ведущим патогенетическим фактором при этом является гипоксия.
Острая сердечно-сосудистая недостаточностьОстрая сердечно-сосудистая недостаточность (схема 12) проявляется острым общим венозным полнокровием, при котором в результате гипоксического повреждения гистогематических барьеров и резкого повышения капиллярной проницаемости, а также увеличения гидростатического давления в капиллярах в тканях наблюдаются плазматическое пропитывание (плазморрагия) и отек, стазы в капиллярах и множественные
диапедезные кровоизлияния; в паренхиматозных органах появляются дистрофические и некротические изменения.
Схема 12. Морфогенез острой сердечно-сосудистой недостаточности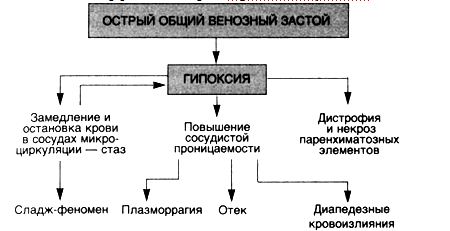
Структурно-функциональные особенности органа, в котором развивается острый венозный застой, определяют преобладание отечно-плазморрагических, геморрагических либо дистрофических и некротических изменений.
В
легких гистофизиологические особенности аэрогематического барьера объясняют развитие при остром венозном полнокровии преимущественно отека и геморрагии. Характерными клиническими проявлениями при этом является пароксизмальная одышка с развитием тяжелого приступа сердечной астмы, сопровождающейся резкой нехваткой воздуха, многочисленными влажными хрипами над всеми легкими, откашливанием кровянистой пенистой жидкости. Острый отек легких — одна из основных причин смерти больных с острой сердечно-сосудистой недостаточностью.
В
почках вследствие особенностей структуры нефрона и кровообращения возникают в основном дистрофические
и некротические изменения эпителия канальцев. Почки при остром венозном полнокровии увеличены в объеме, плотны, их масса достигает 400—500 г. Наиболее полнокровны мозговое вещество и пирамиды; в последних наблюдается радиарная исчерченность, сосочки могут набухать и ущемляться в почечных чашечках. Неравномерность гиперемии объясняется частичным сбросом крови по юкстамедуллярному шунту (по многочисленным анастомозам на границе коркового и мозгового вещества), который возникает при вазоконстрикции артерий и артериол коркового вещества в ответ на снижение сердечного выброса. Тяжесть дистрофических изменений эпителия канальцев нарастает по мере повышения внутрипочечного давления, связанного с отеком паренхимы и нарушением лимфообращения.
В
печени в связи с особенностями архитектоники и кровообращения печеночной дольки при остром полнокровии появляются центролобулярные кровоизлияния и некрозы, которые изредка могут сопровождаться развитием острой печеночной недостаточности.
Селезенка при остром венозном полнокровии увеличена, масса ее достигает 300 г. Капсула селезенки напряжена, с поверхности ее разреза обильно стекает кровь. Микроскопически определяются расширенные синусы, заполненные кровью.
Хроническая сердечно-сосудистая недостаточностьХроническая сердечно-сосудистая недостаточность (схема 13) сопровождается развитием хронического общего венозного полнокровия, при котором гипоксия приобретает хронический характер. Хроническое венозное полнокровие приводит к тяжелым, нередко необратимым изменениям органов и тканей. Длительно поддерживая состояние тканевой гипоксии, оно определяет развитие не только плазморрагии, отека, стаза и кровоизлияний, дистрофии и некроза, но и атрофических, и склеротических процессов. Склеротические изменения, т.е. разрастание соединительной ткани, обусловлены тем, что хроническая гипоксия стимулирует синтез коллагена фибробластами и фибробластоподобными клетками. Соединительная ткань вытесняет паренхиматозные элементы, развивается застойное уплотнение (индурация) органов и тканей. Порочный круг при хроническом венозном полнокровии замыкается развитием капиллярно-паренхиматозного блока в связи с "утолщением" базальных мембран эндотелия и эпителия за счет повышенной продукции коллагена фибробластами, гладкими мышечными клетками и липофибробластами.
Для хронического венозного полнокровия характерны распространенные отеки подкожной основы (жировой клетчатки) — анасарка и скопление жидкости в серозных полостях;
Схема 13. Морфогенез хронической сердечно-сосудистой недостаточности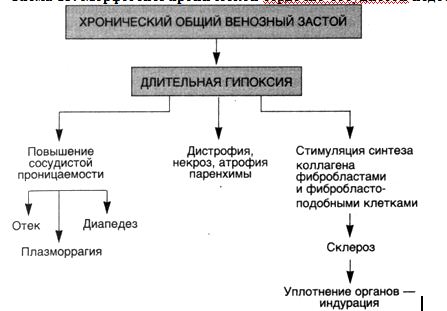
в плевральной — гидроторакс, в полости перикарда — гидроперикард, в брюшной — асцит (обычно при наличии застойного мускатного фиброза печени).
Органы при хроническом венозном полнокровии увеличиваются в объеме, становятся синюшными вследствие повышенного содержания восстановленного гемоглобина, плотными из-за сопутствующего нарушения лимфообращения и отека, а позже в связи с разрастанием соединительной ткани.
Изменения в органах при хроническом венозном полнокровии, несмотря на ряд общих черт (застойная индурация), имеют ряд особенностей.
Кожа, особенно нижних конечностей, становится холодной и приобретает синюшную окраску (цианоз). Вены кожи и подкожной основы расширены, переполнены кровью; также расширены и переполнены лимфой лимфатические сосуды. Выражены отек дермы и подкожной основы, разрастание в коже соединительной ткани. В связи с венозным застоем, отеком и склерозом в коже легко возникают воспалительные процессы и изъязвления, которые длительное время не заживают.
В
слизистых оболочках венозная гиперемия сопровождается цианозом, а также повышенной продукцией слизи
с Развитием катарального воспаления (застойный катар).
Печень при хроническом венозном застое увеличена, плотная, ее края закруглены, поверхность разреза пестрая, серо-желтая с темно-красным крапом и напоминает мускатный орех, поэтому такую печень называют
мускатной. При микроскопическом исследовании видно, что полнокровны лишь центральные отделы долек, где отмечаются кровоизлияния, дискомплексация печеночных балок и гибель гепатоцитов; эти отделы на разрезе печени выглядят темно-красными. На периферии долек гепатоциты находятся в состоянии жировой дистрофии, чем объясняется серо-желтый цвет печеночной ткани.
Хроническое венозное полнокровие печени сопровождается веноартериальной реакцией — гипертрофией мелких ветвей печеночных артерий, имеющих узкий просвет вследствие гипертрофии циркулярного и продольного внутреннего мышечных слоев. В дальнейшем стенки сосудов склерозируются.
Морфогенез изменений печени при длительном венозном застое сложен. Избирательное полнокровие центра долек обусловлено тем, что застой печени охватывает прежде всего печеночные вены, распространяясь на собирательные и центральные вены, а затем и на синусоиды. Последние расширяются, но только в центральных и средних отделах дольки, где встречают сопротивление со стороны впадающих в синусоиды капиллярных разветвлений печеночной артерии, давление в которых выше, чем в синусоидах. По мере нарастания полнокровия в центре долек появляются кровоизлияния, гепатоциты здесь подвергаются дистрофии, некрозу и атрофии. Гепатоциты периферии долек компенсаторно гипертрофируются и приобретают сходство с центролобулярными. Разрастание соединительной ткани в зоне кровоизлияний и гибели гепатоцитов связано с пролиферацией клеток синусоидов — липоцитов, которые могут выступать в роли фибробластов, а вблизи центральных и собирательных вен — с пролиферацией фибробластов адвентиции этих вен. В результате разрастания соединительной ткани в синусоидах появляется непрерывная базальная мембрана (в нормальной печени она отсутствует), т.е. происходит
капилляризация синусоидов, возникает
капиллярно-паренхиматозный блок, который, усугубляя гипоксию, ведет к прогрессированию атрофических и склеротических изменений в печени. Этому способствует также шунтирование крови, развивающееся при склерозе стенок и обтурации просветов многих центральных и собирательных вен, а также нарастающий застой лимфы. В финале развивается
застойный фиброз (склероз) печени, который называют также
мускатным, или
кардиальным, поскольку развивается он при хронической сердечно-сосудистой недостаточности. При прогрессирующем разрастании соединительной ткани в редких случаях возникает перестройка и деформация органа — развивается
застойный (мускатный) цирроз печени, который называют также
сердечным.Почки при хроническом общем венозном застое становятся большими, плотными и цианотичными —
цианотическая индурация почек. Особенно полнокровны вены мозгового вещества и юкстамедуллярной зоны. При хроническом венозном полнокровии почек рано набухает основное вещество мозгового слоя вследствие реакции полисахаридов, содержащихся здесь в большом количестве. Полнокровие почечных клубочков развивается позже, чем наступают изменения в мозговом веществе, так как вступает в силу веноартериальная реакция, сопровождающаяся при длительной гиперемии гипертрофией артерий коркового вещества. Лишь развитие склероза мышечной оболочки этих артерий сопровождается расширением их просвета и капилляров почечных клубочков. На этой стадии возможны огрубение базальной мембраны капилляров и их склероз, приводящие к умеренной протеинурии. На фоне венозного застоя развивается лимфо-стаз. В условиях нарастающей гипоксии возникает дистрофия нефроцитов главных отделов нефрона и склероз стромы, который, однако, не бывает резко выраженным. Снижение сердечного выброса (ударного объема) приводит к уменьшению почечного кровотока, к спазму сосудов коркового вещества, что, с одной стороны, стимулирует ренин-ангиотензиновую систему, а с другой, усиливает ишемические повреждения канальцевого эпителия.
В
селезенке хронический венозный застой также ведет к ее
цианотической индурации. Селезенка увеличена, плотна, темно-вишневого цвета; отмечаются атрофия фолликулов и склероз пульпы. При хроническом венозном полнокровии в условиях портальной гипертензии (при развитии мускатного фиброза печени) масса селезенки может превышать 500 г (спленомегалия). До развития асцита, т.е. в стадии компенсации, строма пульпы аргирофильна, выражена гипертрофия мышечной оболочки селезеночных и трабекулярных артерий. В фазе декомпенсации отмечается фиброз стромы пульпы и стенок указанных сосудов, особенно кисточковых артерий, который следует дифференцировать от возрастного гиалиноза.
В
легких при хроническом венозном полнокровии развиваются два вида изменений — множественные диапедезные кровоизлияния, обусловливающие
гемосидероз легких, и
разрастание соединительной ткани, т.е.
склероз. Легкие становятся большими, бурыми и плотными —
бурая индурация (уплотнение) легких.В морфогенезе бурого уплотнения легких основную роль играют
застойное полнокровие и гипертензия в малом круге кровообращения. Бурая индурация легких развивается лишь в исходе сосудистой декомпенсации нарушенного оттока по легочным венам. Ей предшествует длительная стадия адаптивной перестройки мелких ветвей легочной артерии и вен, выражающаяся в картине посткапиллярной гипертензии в малом круге. При этом развивается резкая гипертрофия мышечной оболочки внутри-дольковых вен, просвет их суживается, что предохраняет капилляры легких от регургитации крови. Гипертрофия мелких ветвей легочной артерии достигает максимума лишь при давлении в системе легочной артерии, в 3 раза превышающем нормальное. В этот период происходит перестройка по типу замыкающих сосудов, что приводит к еще большему сужению просвета. Со временем адаптивные изменения сосудов легких сменяются склеротическими, развивается декомпенсация легочного кровообращения, капилляры межальвеолярных перегородок переполняются кровью. Нарастает гипоксия ткани, в связи с чем повышается сосудистая проницаемость, возникают множественные диапедезные кровоизлияния. В альвеолах, бронхах, межальвеолярных перегородках, лимфатических сосудах и узлах легких появляются скопления нагруженных гемосидерином клеток — сидеробластов и сидерофагов, и свободнолежащего гемосидерина. Возникает
диффузный гемосидероз легких. Гемосидерин и белки плазмы "засоряют" строму и лимфатические дренажи легких, что ведет к резорбционной недостаточности их лимфатической системы, которая сменяется механической. Склероз кровеносных сосудов и недостаточность лимфатической системы усиливают легочную гипоксию, которая становится причиной пролиферации фибробластов, утолщения межальвеолярных перегородок. Возникает
капиллярно-паренхиматозный блок, замыкающий порочный круг в морфогенезе индурации легких, развивается
застойный фиброз легких. Он более значителен в нижних отделах легких, где сильнее выражен венозный застой и больше скоплений кровяных пигментов (гемосидерина), фибрина. Пневмосклероз, как и гемосидероз, при буром уплотнении легких имеет каудоапикальное распространение и зависит от степени и длительности j венозного застоя в легких.
