Лекция 3
МОРФОЛОГИЯ ПОВРЕЖДЕНИЯПовреждение органов начинается на молекулярном или клеточном уровне, поэтому изучение патологии начинается с познания причин и молекулярных механизмов структурных изменений, возникающих в клетках при их повреждении.
Структура нормальной клетки генетически направлена на осуществление определенного метаболизма, дифференцировку и специализацию. В ответ на воздействие различных факторов в клетках развивается процесс адаптации. В результате этого процесса клетки могут достигать нового устойчивого состояния, позволяющего им приспособиться к подобным воздействиям. Если лимиты адаптивного ответа клетки исчерпаны, а адаптация невозможна, то возникает повреждение клетки, до определенного предела обратимое. Однако, если неблагоприятный фактор действует постоянно или его интенсивность очень велика, развивается необратимое повреждение, или смерть, клетки.
Смерть клетки — конечный результат ее повреждения, главное следствие ишемии, инфекции, интоксикации, иммунных реакций. Кроме того, это естественное событие в процессе нормального эмбриогенеза, развития лимфоидной ткани, инволюции органа под действием гормонов, а также желаемый результат радио- и химиотерапии при раке.
Существует два типа клеточной смерти — некроз и апоптоз.
Некроз — наиболее распространенный тип смерти клетки. Он проявляется ее резким набуханием и разрывом клеточной мембраны, денатурацией и коагуляцией цитоплазматических белков, разрушением клеточных органелл. Апоптоз необходим для нормальной элиминации ненужных клеточных популяций в процессе эмбриогенеза и при различных физиологических процессах. Апоптоз встречается и при патологических процессах; в этом случае он сопровождается некрозом.
ПРИЧИНЫ ПОВРЕЖДЕНИЯ КЛЕТОКРазличают следующие причины повреждения клеток.
1. Гипоксия. Она является исключительно важной и распространенной причиной повреждения и смерти клеток. Уменьшение кровотока (ишемия), возникающее при появлении препятствий в артериях, обычно при атеросклерозе или тромбозе, является основной причиной гипоксии. Другой причиной может быть неадекватная оксигенация крови при сердечно-сосудистой недостаточности. Снижение способности крови к транспортировке кислорода, например при анемии и отравлении СО2 — третья и наиболее редкая причина гипоксии. В зависимости от тяжести гипоксии клетки могут адаптироваться к ней, повреждаться или погибать.
2. Физические агенты. К ним относят механическую травму, чрезмерное снижение или повышение температуры окружающей среды, внезапные колебания атмосферного давления, радиацию и электрический шок.
3. Химические агенты и лекарства. Даже простые химические соединения, такие как глюкоза и поваренная соль, в повышенных концентрациях могут вызвать повреждение клеток непосредственно или путем нарушения их электролитного гомеостаза. Кислород в высоких концентрациях очень токсичен.
Следовые количества веществ, известных как яды (мышьяк, цианиды, соли ртути), могут разрушить достаточно большое количество клеток в течение минут и часов.
Разрушительным действием обладают также многие факторы окружающей среды: пыль, инсектициды и гербициды; промышленные и природные факторы, например уголь и асбест; социальные факторы: алкоголь, курение и наркотики; высокие дозы лекарств.
4. Инфекционные агенты. Они включают как субмикроскопические вирусы, так и ленточных червей. К инфекционным агентам относятся риккетсии, бактерии, грибы, а также более высоко организованные формы паразитов.
5. Иммунные реакции. Могут защищать организм, но могут вызвать и его смерть. Хотя иммунная система защищает организм от воздействия биологических агентов, тем не менее иммунные реакции могут привести к повреждению клеток. Развитие некоторых иммунных реакций лежит в основе аутоиммунных болезней.
6. Генетические нарушения. Многие врожденные нарушения метаболизма связаны с энзимопатиями, чаще отсутствием фермента.
7. Дисбаланс питания. Нередко является основной причиной повреждения клеток. Дефицит белковой пищи и витаминов остается распространенным явлением.
МЕХАНИЗМЫ ПОВРЕЖДЕНИЯ КЛЕТОКМолекулярные механизмы повреждения клеток, приводящие к их смерти, очень сложны. Существуют четыре наиболее чувствительные внутриклеточные системы:
▲ поддержание целости клеточных мембран, от которой зависит ионный и осмотический гомеостаз клетки и ее органелл;
▲ аэробное дыхание, связанное с окислительным фосфорилированием и образованием аденозинтрифосфата (АТФ);
▲ синтез ферментов и структурных белков;
▲ сохранение единства генетического аппарата клетки. Структурные и биохимические элементы клетки тесно взаимосвязаны. Например, нарушение аэробного дыхания повреждает натриевый насос мембраны, который поддерживает ионно-жидкостный баланс клетки, что приводит к нарушению внутриклеточного содержания ионов и воды.
Морфологические изменения становятся очевидными только после того, как нарушения биологической системы клетки проходят некий критический уровень, причем развитие морфологических признаков смертельного повреждения клетки занимает больше времени, чем появление обратимых изменений. Например, набухание клетки обратимо и может развиться в течение нескольких минут, а достоверные светооптические признаки смерти клетки в миокарде выявляются лишь спустя 10—12 ч после тотальной ишемии, хотя известно, что необратимые повреждения наступают уже через 20—60 мин. Естественно, ультраструктурные повреждения будут видны раньше, чем светооптические.
Реакция клеток на повреждающие воздействия зависит от типа, продолжительности действия и тяжести повреждающего фактора. Например, малые дозы токсинов или непродолжительная ишемия могут вызвать обратимые изменения, тогда как большие дозы того же токсина и продолжительная ишемия способны привести к немедленной гибели клетки или медленному необратимому повреждению, вызывающему клеточную смерть. Тип, состояние и приспособляемость клетки также определяют последствия ее повреждения. Для ответа клетки на повреждение важны ее гормональный статус, характер питания и метаболические потребности.
Механизмы действия многих повреждающих агентов хорошо известны. Так, многие токсины вызывают повреждение клеток, воздействуя на эндогенные субстраты или ферменты. Особенно чувствительны к действию токсинов гликолиз, цикл лимонной кислоты и окислительное фосфорилирование на внутренних мембранах митохондрий. Например, цианид инактивирует цитохромоксидазу, а флуороацетат препятствует реализации цикла лимонной кислоты, что в результате приводит к недостаточности АТФ. Некоторые анаэробные бактерии, например Clostridium perfringens, высвобождают фосфолипиды, которые атакуют фосфолипиды клеточных мембран, повреждая их.
Наиболее важными для развития повреждения и смерти клетки считают следующие четыре механизма.1. При недостаточном поступлении кислорода в ткани образуются его свободные радикалы, вызывающие свободнорадикальное пероксидное окисление липидов (СПОЛ), что оказывает разрушительное действие на клетки.
2. Особую роль в повреждении клетки играет нарушение гомеостаза кальция. Свободный кальций в цитозоле присутствует в исключительно низких концентрациях по сравнению с таковым вне клетки. Это состояние поддерживается связанными с клеточной мембраной энергозависимыми Са2+, Mg2+-АТФазами. Ишемия и некоторые токсины вызывают увеличение концентрации кальция в цитозоле путем его избыточного поступления через плазматическую мембрану и высвобождения из митохондрий и эндоплазматической сети. Повышенное содержание
кальция в клетке ведет к активации ряда ферментов, повреждающих клетку: фосфолипаз (повреждение клеточной мембраны), протеаз (разрушение мембраны и белков цитоскелета), АТФаз (истощение запасов АТФ) и эндонуклеаз (фрагментация хроматина).
3. Потеря митохондриями пиридиннуклеотидов и последующая недостаточность АТФ, а также снижение синтеза АТФ являются характерными как для ишемического, так и для токсического повреждения клеток. Высокоэнергетические фосфаты в форме АТФ требуются для многих процессов синтеза и расщепления, происходящих в клетках. К этим процессам относятся мембранный транспорт, синтез белка, липогенез и реакции деацилирования — реацилирования, необходимые для фосфолипидного обмена. Имеется много данных о том, что недостаточность АТФ играет роль в потере целости плазматической мембраны, что характерно для смерти клетки.
4. Ранняя потеря плазматической мембраной избирательной проницаемости — постоянный признак всех видов повреждения клеток. Такие дефекты могут возникать вследствие ряда событий, связанных с потерей АТФ и активацией фосфолипаз. Кроме того, плазматическая мембрана может быть повреждена в результате прямого воздействия некоторых бактериальных токсинов, вирусных белков, компонентов комплемента, веществ из лизированных лимфоцитов (перфоринов), а также ряда физических и химических агентов.
ОСНОВНЫЕ ФОРМЫ ПОВРЕЖДЕНИЯ КЛЕТОКРазличают три формы повреждения клеток: 1) ишемическое и гипоксическое повреждение; 2) повреждение, вызванное свободными радикалами, включая активированный кислород; 3) токсическое повреждение.
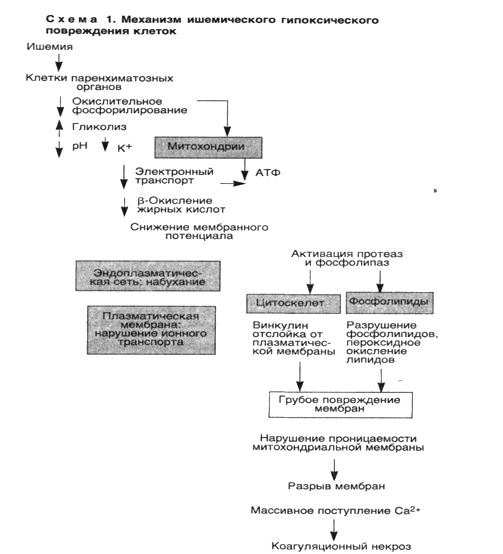
1. Ишемическое и гипоксическое повреждение. Чаще всего оно обусловлено окклюзией артерий. При этом изначально гипоксия воздействует на аэробное дыхание клетки — окислительное фосфорилирование в митохондриях. В связи с тем что напряжение кислорода в клетке снижается, прекращается окислительное фосфорилирование, а образование АТФ уменьшается или останавливается.
Исчезновение АТФ ведет к быстрому набуханию (отеку) клетки — одному из ранних проявлений ишемического повреждения. Отек клетки обусловлен нарушением регуляции объема клетки плазматической мембраной. Баланс между содержимым клетки и окружающей ее средой обеспечивается энергетически зависимым натриевым насосом, который поддерживает концентрацию калия внутри клетки значительно более высокой, чем внеклеточная. Наблюдается отделение рибосом от мембран гранулярной эндоплазматической сети и диссоциация полисом в моносомы. На поверхности клеток могут образовываться "волдыри", а клетки, имеющие на поверхности микроворсинки, их утрачивают (эпителий проксимальных канальцев почек). В цитоплазме и вне клеток появляются "миелиновые фигуры", образующиеся из цитоплазмы и мембран органелл. Митохондрии набухают, а эндоплазматическая сеть остается расширенной.
Необратимые изменения морфологически ассоциируются с выраженной вакуолизацией митохондрий, повреждением плазматических мембран и набуханием лизосом. Вслед за гибелью клетки ее компоненты прогрессивно разрушаются, и происходит выброс ферментов клетки во внеклеточное пространство. Умершие клетки образуют массы, состоящие из фосфолипидов в виде «миелиновых фигур», которые подвергаются фагоцитозу и разрушаются до жирных кислот.
Проникновение ферментов сквозь поврежденную клеточную мембрану, а затем в сыворотку крови позволяет клинически определять параметры смерти клетки. Например, сердечная мышца содержит трансаминазы, лактатдегидрогеназу и креатинкиназу. Повышение содержания этих ферментов в сыворотке крови является клиническим критерием инфаркта миокарда (смерти кардиомиоцитов).
■ Таким образом, основными признаками необратимости повреждения клетки служат невосстановимые повреждения митохондрий, приводящие к потере АТФ, а также развитие глубоких повреждений плазматических мембран, в основе которых лежит ряд биохимических механизмов.
Во-первых, в некоторых ишемизированных тканях, например печени, необратимое ишемическое повреждение сопровождается заметным уменьшением содержания фосфолипидов в клеточной мембране, которое происходит под действием кальцийзависимых фосфолипаз.
Во-вторых, активация протеаз, обусловленная повышением концентрации кальция в цитозоле, ведет к повреждению цитоскелета, выполняющего роль якоря между плазматической мембраной и внутренним содержимым клетки. В результате во время набухания клетки происходит отслойка клеточной мембраны от цитоскелета, что делает мембрану более податливой к растяжению и разрыву.
В-третьих, при ишемии появляется небольшое количество высокотоксичных свободных радикалов кислорода.
Итак, основными механизмами гибели клетки при гипоксии являются нарушение окислительного фосфорилирования, приводящее к недостаточности АТФ, повреждение мембран клетки, а важнейшим медиатором необратимых биохимических и морфологических изменений является кальций (схема 1).
2. Повреждение клетки, вызванное свободными радикалами кислорода. Чаще всего такое повреждение возникает под воздействием химических веществ, лучистой энергии, кислорода и других газов, а также при старении клеток, разрушении опухолей макрофагами и в некоторых иных случаях.
Свободные радикалы представляют собой молекулы кислорода, имеющие один непарный электрон на внешней орбите. В таком состоянии радикал исключительно активен и нестабилен и вступает в реакции с неорганическими и органическими соединениями — белками, липидами и углеводами.
Для повреждения клетки наибольшее значение имеют три реакции, в которые вступают свободные радикалы.
• Свободнорадикальное пероксидное окисление липидов (СПОЛ) мембран. Свободные радикалы в присутствии кислорода могут вызывать пероксидное окисление липидов плазматической мембраны и органелл. Липидно-радикальные взаимодействия приводят к образованию пероксидов, которые сами по себе являются активными соединениями, инициирующими последующее повреждение других жирных кислот; возникает цепь аутокаталитических реакций, обусловливающая обширное повреждение мембран, органелл и самих клеток.
• Окислительное превращение белков. Свободные радикалы вызывают перекрестное связывание таких лабильных аминокислот, как метионин, гистидин, цистин и лизин, а также фрагментацию полипептидных цепей. Окислительное превращение усиливает разрушение ключевых ферментов посредством нейтральных протеаз, содержащихся в цитозоле.
• Повреждение ДНК. Свободные радикалы вступают в реакцию с тимином, входящим в состав ДНК. Такое повреждение ДНК ведет к гибели клетки или ее злокачественному превращению. Митохондриальная ДНК также повреждается.
Свободные радикалы могут разрушаться спонтанно. Например, супероксидный анион-радикал является нестабильным соединением и спонтанно разрушается с образованием кислорода и пероксида водорода. Однако имеется несколько ферментных и неферментных систем, которые способствуют окончанию или инактивации свободнорадикальных реакций. Эндогенными или экзогенными антиоксидантами являются витамин Е; сульфгидрилсодержащие соединения — цистеин и глютатион; белки сыворотки — альбумин, церулоплазмин и трансферрин. Полагают, что трансферрин действует как антиоксидант, связывая свободное железо, которое, как известно, может играть роль катализатора образования свободных радикалов.
Среди ферментов выделяют супероксиддисмутазу, способную превращать супероксидный анион-радикал в пероксид водорода.
Каталаза, сосредоточенная в пероксисомах, разрушает пероксид водорода до кислорода и воды.
При многих патологических процессах конечный результат действия свободных радикалов зависит от баланса между образованием свободных радикалов и их разрушением.
3. Токсическое повреждение. Примером такого повреждения является действие химических веществ, вызывающих повреждение клетки посредством одного из двух механизмов. Во-первых, часть водорастворимых соединений может действовать непосредственно, связываясь с некоторыми молекулами или органеллами. Например, при попадании в организм хлорида ртути ртуть связывает сульфгидрильные группы клеточной мембраны и других белков, вызывая повышение проницаемости клеточной мембраны и торможение АТФаза-зависимого транспорта. В подобных случаях наиболее выраженные изменения наблюдаются в клетках, которые используют, абсорбируют, выделяют или концентрируют эти химические соединения. При попадании в организм хлорида ртути в наибольшей степени страдают клетки желудочно-кишечного тракта и почек. Цианид непосредственно воздействует на ферменты митохондрий. Многие противоопухолевые химиотерапевтические препараты, в том числе антибиотики, также вызывают повреждение клеток посредством цитотоксического действия.
Во-вторых, некоторые другие химические соединения, особенно жирорастворимые токсины, биологически неактивны и вначале превращаются в токсичные метаболиты, которые затем действуют на клетки-мишени. Хотя эти метаболиты могут вызывать повреждение мембран и клеток путем прямого ковалентного связывания с мембранными белками и липидами, наиболее важный механизм повреждения мембран включает образование реактивных свободных радикалов и последующее СПОЛ.
МОРФОЛОГИЯ ПОВРЕЖДЕНИЯ КЛЕТОКВ классической морфологии нелетальное повреждение клеток называется дистрофией (см. лекции 4—7). В большинстве случаев она относится к обратимым повреждениям.
Некроз наряду с апоптозом является одним из двух морфологических выражений смерти клетки (см. лекцию 8).
Апоптоз ответствен за многочисленные физиологические и патологические процессы, происходящие в организме.
1. В результате апоптоза происходят запрограммированное разрушение клеток в процессе эмбриогенеза (включая имплантацию, органогенез и инволюцию) и метаморфоз.
2. Посредством апоптоза развивается гормонозависимая инволюция тканей у взрослых, например атрезия фолликулов в яичниках во время менопаузы, регрессия лактирующей молочной железы после прекращения кормления ребенка.
3. Происходит уничтожение клеток в пролиферирующих клеточных популяциях, таких как эпителий крипт тонкой кишки.
4. Наступает смерть клеток в опухолях, как подвергающихся регрессии, так и с активным ростом клеток.
5. Это механизм смерти иммунных клеток — как В-, так и Т-лимфоцитов, после истощения цитокинов, а также уничтожения аутореактивных Т-клеток в развивающейся вилочковой железе.
6. Это механизм патологической атрофии гормонозависимых тканей, например атрофия предстательной железы после кастрации и исчезновение лимфоцитов в вилочковой железе после введения глюкопротеидов.
7. Это патологическая атрофия паренхиматозных органов после перекрытия протока, например, поджелудочной железы, околоушной слюнной железы, мочеточника.
8. Смерть клетки, вызванная цитотоксическими Т-клетками, например отторжение трансплантата.
9. Это гибель клеток при некоторых вирусных заболеваниях, например при вирусном гепатите (фрагменты клеток при апоптозе известны как тельца Каунсильмена).
10. Клеточная смерть, вызванная различными слабыми повреждающими воздействиями, которые в больших дозах приводят к гибели клетки (радиация, высокие или низкие температуры, цитотоксические противоопухолевые препараты и, возможно, гипоксия), также представляет собой апоптоз.
Механизмы апоптоза. Апоптоз начинается скорее всего в результате действия гормонов и других трофических факторов и является регулятором плотности клеточной популяции. Кроме того, апоптоз может быть одним из механизмов удаления аномальных клеток или клеток, поврежденных токсинами, радиацией и другими воздействиями.
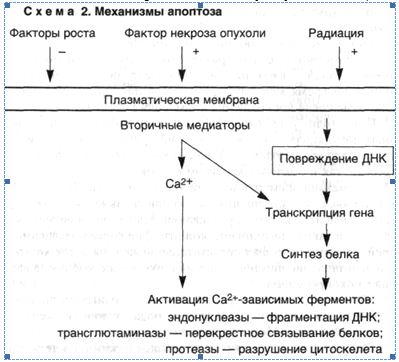
Механизмы возникновения апоптоза различны и зависят от характера воздействия и типа клеток, однако последовательность событий, которые приводят к конечному результату, до конца не выяснена. Вместе с тем ряд механизмов уже установлен (схема 2).
1. Конденсация хроматина. Она обусловлена расщеплением ядерной ДНК в участках связей между нуклеосомами и приводит к образованию фрагментов. Такие фрагменты создают характерную для апоптоза картину ядра, отличную от таковой при некрозе, когда ядро выглядит пятнистым. Эта интернуклеосомальная фрагментация ДНК развивается с участием кальцийчувствительной эндонуклеазы. Эндонуклеаза постоянно присутствует в клетках некоторых типов, например тимоцитах, где она активируется под влиянием цитозольного кальция, тогда как в других клетках фермент образуется перед началом апоптоза.
2. Нарушения объема и формы клеток. Эти процессы связывают с активностью трансглютаминазы, которая вызывает перекрестное связывание цитоплазматических белков, образующих оболочку под плазматической мембраной.
3. Фагоцитоз апоптозных телец макрофагами и клетками других типов. Обеспечивается рецепторами этих клеток, которые связывают и поглощают апоптозные клетки. Одним из таких рецепторов на макрофаге является витронектиновый рецептор δ3- интегрин, который обеспечивает фагоцитоз апоптозных нейтрофилов.
4. Зависимость апоптоза в большинстве случаев от активации гена и синтеза белка является одной из важных его особенностей. При этом индукция генов обеспечивается за счет "стимулов", вызывающих апоптоз, таких как стрессорные белки и протоонкоге-ны. Однако эти гены не связаны напрямую с началом апоптоза. Выявлены апоптозспецифические гены, которые стимулируют или тормозят смерть клетки.
5. Некоторые гены, участвующие в возникновении и росте злокачественной опухоли (онкогены и супрессорные гены), играют регуляторную роль в индукции апоптоза. Например, онкоген р53 в норме стимулирует апоптоз; р53 необходим для развития апоптоза после повреждения ДНК радиацией, хотя апоптоз, вызванный глюкокортикоидами или старением, не зависит от р53. ■ Таким образом, апоптоз является разновидностью смерти клетки, для которой характерна конденсация и фрагментация ДНК. Апоптоз обеспечивает уничтожение клеток при нормальном развитии, тканевом росте, органогенезе и в органах иммунной системы.
Апоптоз может быть также индуцирован под воздействием патологических стимулов. Некоторые из уже известных эффекторных механизмов включают активацию эндонуклеазы, вызванную содержащимся в цитозоле кальцием и приводящую к фрагментации ДНК. Активация трансглютаминазы частично влияет на изменение формы и размеров клеток. Апоптоз заканчивается рецепторнозависимым фагоцитозом апоптозных телец. В некоторых случаях в индукции апоптоза участвуют транскрипция генов и синтез белка, а процесс апоптоза регулируется группой генов, которые участвуют в нормальном росте и дифференцировке клеток.
СУБКЛЕТОЧНЫЕ ИЗМЕНЕНИЯ ПРИ ПОВРЕЖДЕНИИ КЛЕТОКЛизосомы содержат различные гидролитические ферменты — кислую фосфатазу, глюкуронидазу, сульфатазу, рибонуклеазу, коллагеназу и др. Эти ферменты синтезируются в гранулярной (шероховатой) эндоплазматической сети и затем "упаковываются" в пластинчатом комплексе (аппарат Гольджи). На этой стадии их называют первичными лизосомами; они сливаются с окруженными мембраной вакуолями, которые содержат продукты переваривания, и образуют фаголизосомы. Лизосомы участвуют в утилизации фагоцитированного материала посредством гетеро- и аутофагии.
Гетерофагия — феномен, посредством которого материал извне захватывается клеткой с помощью эндоцитоза. Поглощение частиц называется фагоцитозом, а растворенных мелких макромолекул — пиноцитозом. Гетерофагия характерна для фагоцитирующих клеток, таких как нейтрофилы и макрофаги. В качестве примеров гетерофагоцитоза можно привести поглощение бактерий нейтрофильными лейкоцитами и удаление апоптозных клеток и телец макрофагами. Слияние фагоцитозной вакуоли с лизосомой заканчивается растворением захваченного материала.
Аутофагия характеризуется тем, что при ней внутриклеточные органеллы и порции цитозоля вначале отделяются от цитоплазмы в аутофагические вакуоли, образованные из свободных от рибосом мембран гранулярной эндоплазматической сети, которые затем сливаются с первичными лизосомами или элементами пластинчатого комплекса, образуя аутофаголизосому. Аутофагия — распространенный феномен, направленный на удаление разрушенных органелл поврежденной клетки. Он особенно выражен в клетках, атрофирующихся в результате недостаточного питания или гормональной инволюции.
Ферменты лизосом способны разрушать большинство белков и углеводов, но некоторые липиды все равно остаются непереваренными. Лизосомы с непереваренными остатками встречаются в клетках в виде остаточных телец. Например, гранулы пигмента липофусцина представляют собой непереваренный материал, который образовался после внутриклеточного СПОЛ. Некоторые нерастворимые пигменты, такие как частицы угля, попадающие из атмосферы, или пигмент, вводимый при татуировке, могут находиться в фаголизосомах макрофагов десятилетиями.
В лизосомах накапливаются также вещества, которые клетки не могут адекватно метаболизировать. При врожденных лизосомных болезнях накопления, для которых характерен дефицит ферментов, разрушающих различные макромолекулы, происходит ненормальное накопление этих веществ в лизосомах клеток всего тела, особенно в нейронах, приводя к тяжелым аномалиям.
Дисфункция митохондрий играет важную роль при остром повреждении клетки. Различные изменения количества, размеров и формы митохондрий отмечаются в патологических условиях. Например, при гипертрофии и атрофии наблюдается соответственно увеличение и уменьшение количества митохондрий в клетках. Митохондрии могут быть очень крупными и принимать различную форму (мегамитохондрии), например, в случае алкогольной болезни печени. При некоторых врожденных метаболических заболеваниях скелетных мышц — митохондриальных миопатиях — дефекты метаболизма митохондрий сочетаются с увеличением их количества, причем митохондрии часто бывают необычно крупными, имеют аномальные кристы и содержат кристаллоиды. Кроме того, некоторые опухоли (слюнных желез, щитовидной и околощитовидных желез, почки) состоят из клеток с множеством вытянутых митохондрий.
Аномалии цитоскелета встречаются при различных патологических состояниях. Эти аномалии делятся на дефекты функций клетки (локомоторная и движение внутриклеточных органелл) и накопление фибриллярного материала внутри клетки.
Функционирующие миофиламенты и микротрубочки необходимы для различных стадий миграции лейкоцитов и фагоцитоза. Поэтому именно недостаточностью цитоскелета обусловлены некоторые дефекты движения лейкоцитов в ответ на повреждающие стимулы или неспособность таких клеток осуществлять адекватный фагоцитоз. Например, дефект полимеризации микротрубочек при синдроме Чедиака — Хигаси вызывает замедленное слияние лизосом с фагосомами в лейкоцитах, нарушая таким образом фагоцитоз бактерий; в цитоплазме лейкоцитов появляются крупные аномальные лизосомы. Некоторые лекарственные препараты, такие как цитохалазин В, тормозят функцию миофиламентов и таким образом нарушают фагоцитоз. Дефекты в организации микротрубочек могут тормозить подвижность сперматозоидов, вызывая стерильность у мужчин, а также приводить к неподвижности ресничек дыхательного эпителия, что препятствует очищению дыхательных путей от бактерий и способствует развитию бронхоэктазов.
При некоторых типах повреждений клеток наблюдается накопление промежуточных филаментов. Например, тельца Маллори, или алкогольный гиалин, представляют собой эозинофильные интрацитоплазматические включения в клетках печени, которые характерны для алкогольной болезни. Эти включения состоят главным образом из промежуточных филаментов. Нейрофибриллярные включения в мозге при болезни Альцгеймера содержат белки и нейрофиламенты из микротрубочек и отражают повреждение цитоскелета нейронов.
СТАРЕНИЕ КЛЕТОКНесмотря на универсальность, процессу старения трудно дать четкое определение. С возрастом происходят физиологические и структурные изменения почти во всех системах органов. При старении имеют большое значение генетические и социальные факторы, характер питания, а также связанные с возрастом болезни — атеросклероз, сахарный диабет, остеоартроз. Повреждения клеток, обусловленные возрастом, также являются важным компонентом старения организма.
С возрастом прогрессивно страдает ряд функций клеток. Снижается активность окислительного фосфорилирования в митохондриях, синтеза ферментов и рецепторов клеток. Стареющие клетки обладают сниженной способностью к поглощению питательных веществ и восстановлению хромосомных повреждений. К морфологическим изменениям в стареющих клетках относятся неправильные и дольчатые ядра, полиморфные вакуолизированные митохондрии, уменьшение эндоплазматической сети и деформация пластинчатого комплекса. Одновременно происходит накопление пигмента липофусцина.
Старение клеток является многофакторным процессом. Он включает эндогенные молекулярные программы клеточного старения, а также экзогенные влияния, приводящие к прогрессирующему вторжению в процессы выживаемости клеток.
Феномен клеточного старения интенсивно изучается в опытах in vitro. Показано, что в стареющих клетках происходит активация специфических для старения генов, повреждаются гены — регуляторы роста, стимулируются ингибиторы роста, а также включаются и другие генетические механизмы.
Предполагают, что генные дефекты могут быть обусловлены телометрическим укорочением хромосом. Теломеры играют важную роль в стабилизации терминальных порций хромосом и прикреплении их к ядерному матриксу. Например, длина теломеров уменьшается в последних пассажах культуры клеток и в культуре клеток людей старческого возраста. Обнаружена связь между длиной теломера и активностью теломеразы.
Приобретенные повреждения клеток при старении возникают под действием свободных радикалов. Причинами этих повреждений может быть воздействие ионизирующей радиации или прогрессирующее снижение активности антиоксидантных механизмов защиты, например витамина Е, пероксидазы глютатиона. Повреждение клетки свободными радикалами сопровождается накоплением липофусцина, однако сам по себе пигмент не токсичен для клетки. Кроме того, СПОЛ и свободные радикалы вызывают повреждение нуклеиновых кислот как в ядре, так и митохондриях. Мутации и уничтожение митохондриальной ДНК с возрастом становятся просто драматическими. Свободные радикалы кислорода катализируют также образование модификаций белков, включая ферменты, делая их чувствительными к повреждающему действию нейтральных и щелочных протеаз, содержащихся в цитозоле, что ведет к дальнейшему нарушению функций клетки.
Посттрансляционные изменения внутриклеточных и внеклеточных белков также возникают с возрастом. Одна из разновидностей таких изменений — неферментное гликозилирование белков. Например, связанное с возрастом гликозилирование белков хрусталика лежит в основе старческой катаракты.
Наконец, имеются данные о нарушении образования стрессорных белков in vitro у экспериментальных животных при старении. Образование стрессорных белков — важнейший механизм защиты от различных стрессов.
