Лекция 18АУТОИММУНИЗАЦИЯИИММУНОДЕФИЦИТНЫЕ СИНДРОМЫ•
Аутоиммунизация — патологический процесс, в основе которого лежит развитие иммунных реакций на антигены собственных тканей организма.
Развитие иммунной реакции против собственных антигенов является причиной некоторых заболеваний человека, хотя аутоантитела могут быть найдены в сыворотке крови или тканях у многих здоровых людей, особенно в старшей возрастной группе. Безвредные антитела образуются после повреждения ткани и играют физиологическую роль в удалении продуктов разрушения. Кроме того, нормальный иммунный ответ необходим для распознавания собственных антигенов гистосовместимости. •
Аутоиммунные болезни — группа заболеваний, в основе которых лежит развитие иммунных реакций на собственные ткани организма.
Различают три основных признака аутоиммунных заболеваний:
▲ наличие аутоиммунной реакции;
▲ наличие клинических и экспериментальных данных, что такая реакция на повреждение ткани не вторична, а имеет первичное патогенетическое значение;
▲ отсутствие иных определенных причин болезни.
Естественно, что эти признаки наблюдаются лишь при немногих заболеваниях, например при системной красной волчанке.
Встречаются аутоиммунные заболевания, при которых аутоантитела направлены против единственного органа или ткани, тогда эти орган или ткань поражаются. Например, при тиреоидите Хашимото антитела абсолютно специфичны для щитовидной железы. В то же время возможны заболевания с образованием разнообразных антител, что приводит к полиорганным повреждениям. Так, при системной красной волчанке аутоантитела реагируют с составными частями ядер различных клеток, при синдроме Гудпасчера антитела против базальной мембраны легких и почек (перекрестно реагирующие) вызывают повреждения только этих органов. Очевидно, что аутоиммунитет подразумевает потерю самотолерантности.
МЕХАНИЗМЫАУТОИММУННЫХБОЛЕЗНЕЙВ патогенез аутоиммунизации, видимо, вовлечены иммунологические, генетические и вирусные факторы, взаимодействующие с помощью сложных механизмов, которые пока мало известны. Наиболее вероятны следующие.
1.
Обходной путь толерантности Т-лимфоцитов-хелперов. Толерантность к аутоантигену часто обусловлена клональной делецией или анергией специфических Т-лимфоцитов в присутствии полностью компетентных гаптенспецифических В-лимфоцитов. Однако толерантность может быть нарушена одним из двух механизмов.
Модификация молекулы. Если потенциально аутоантигенная детерминанта (гаптен) связывается с новым носителем, то его переносимая часть может быть распознана нетолерантными Т-лимфоцитами как инородная. Затем последняя кооперируется с гаптенспецифическими В-лимфоцитами, образуя аутоантитела. Модификация молекулы может происходить различными способами. Во-первых, возможно образование комплексов аутоантигенов с лекарственными веществами и микроорганизмами. Аутоиммунная гемолитическая анемия, связанная с лекарственными веществами (антигипертензивный препарат метилдофа), может быть обусловлена повреждением поверхности эритроцитов, что способствует формированию нового носителя для Rh-антигена — гаптена. Во-вторых, частичное разрушение аутоантигена приводит к образованию новых антигенных детерминант. Частично разрушенный коллаген и поврежденный ферментами тиреоглобулин или 7-глобулин становятся более иммуногенными, чем нативные образцы. Образование аутоантител к 7-глобулину (ревматоидный фактор), возникающих в процессе бактериальной, вирусной и паразитарной инфекции, может быть связано с повреждением 7-глобулина микроорганизмами или ли-зосомными гидролазами.
Молекулярная мимикрия. Некоторые инфекционные агенты перекрестно реагируют с тканями человека посредством их гаптеновых детерминант. Микроорганизмы могут вызывать антигенный ответ перекрестно реагирующими гаптеновыми детерминантами в ассоциации с их собственным носителем, к которому Т-лимфоциты-хелперы нетолерантны. Антитело, образованное таким образом, может повреждать ткани, которые связаны с перекрестно реагирующими детерминантами. Понятно поэтому, что ревматическое повреждение сердца иногда развивается вслед за стрептококковой инфекцией, так как антитела к стрептококковому белку М перекрестно реагируют с М-протеи-ном в сарколемме сердечной мышцы.
2.
Поликлональная активация лимфоцитов. Некоторые микроорганизмы и продукты их жизнедеятельности способны вызвать поликлональную (антигеннеспецифическую) активацию В-лимфоцитов. Лучше всего исследованы бактериальные липополисахариды (эндотоксин), которые могут индуцировать лимфоциты мышей in vitro к образованию антител против ДНК тимоцитов и эритроцитов. Инфицирование клеток вирусом Эпштейна — Барр может дать те же результаты, так как В-лимфоциты человека имеют рецепторы к этому вирусу.
3.
Дисбаланс функций суп рессорных
и Т-лимфоцитов-хелпе-ров. Снижение функциональной активности супрессорных Т-клеток способствует развитию аутоиммунизации и, наоборот, чрезмерная активность хелперных Т-клеток может вызвать повышение продукции аутоантител В-клетками. Например, при системной красной волчанке человека наблюдается нарушение функционирования или уменьшение содержания (иногда то и другое одновременно) супрессорных Т-клеток и активация хелперных Т-клеток.
4.
Появление секвестрированного антигена. Любой аутоантиген, который полностью секвестрирован в процессе развития, рассматривается как инородный, если попадает в кровоток и на него развивается иммунный ответ. Сперматозоиды, основной белок миелина и кристаллин хрусталика могут попасть в категорию антигенов. Например, травма яичек способствует выбросу спермы в ткани; вслед за этим появляются антитела к сперматозоидам.
5.
Генетические факторы иммунитета. Эти факторы определяют частоту и природу аутоиммунных заболеваний. Во-первых, существует семейная предрасположенность к некоторым аутоиммунным заболеваниям человека, таким как системная красная волчанка, аутоиммунная гемолитическая анемия и аутоиммунный тиреоидит. Во-вторых, имеется связь некоторых аутоиммунных заболеваний с антигенами системы HLA, особенно II классом антигенов. Например, у большинства больных ревматоидным артритом (аутоиммунное заболевание суставов) имеются HLA-DR4 или HLA-DR1 либо оба этих аллеля.
6.
Микробные агенты в аутоиммунитете. Различные микроорганизмы, включая бактерии, микоплазмы и вирусы, могут быть вовлечены в развитие аутоиммунитета. Во-первых, вирусные антигены и аутоантигены могут связываться, образуя иммуногенные единицы. Во-вторых, некоторые вирусы, например вирус Эпштейна — Барр, представляют собой неспецифические, поликлональные В-лимфоцитарные митогены и могут вызывать образование аутоантител. В-третьих, вирусная инфекция может привести к снижению функции супрессорных Т-лимфоцитов.
Вирусы и некоторые другие микроорганизмы, такие как стрептококки и клебсиеллы, могут обладать эпитопами, перекрестно реагирующими с аутоантигенами. Некоторые инфекционные агенты вызывают сильную активацию и пролиферацию СО4+Т-лимфоцитов.
ХАРАКТЕРИСТИКААУТОИММУННЫХБОЛЕЗНЕЙСистемная красная волчанка (СКВ). Это классический прототип мультисистемного заболевания аутоиммунного происхождения. СКВ начинается остро или незаметно, течение хроническое, ремиттирующее и рецидивирующее, часто лихорадочное и характеризуется главным образом поражением
кожи, суставов, почек и серозных оболочек. Фактически может быть поражен любой орган. Клинические проявления СКВ очень вариабельны. Как и большинство аутоиммунных заболеваний, СКВ чаще встречается у женщин в возрасте 20—64 лет.
Этиология и патогенез. Причина СКВ остается неизвестной, однако наличие у этих пациентов аутоантител свидетельствует, что основным дефектом при этом заболевании является недостаточность регуляторных механизмов аутотолерантности (схема 31). Идентифицированы антитела против ядерных и цитоплазматических компонентов клетки, которые не обладают ни органной, ни видовой специфичностью. Эти антитела не только имеют значение для диагностики и лечения, но и играют основную роль в патогенезе, например в развитии иммунокомплексного гломерулонефрита, типичного для этого заболевания.
Антиядерные антитела направлены против некоторых ядерных антигенов и могут быть разделены на четыре группы:
▲антитела к ДНК;
▲ антитела к гистонам;
▲ антитела к негистоновым белкам, связанным с РНК;
▲ антитела к ядерным антигенам.
Кроме того, у больных СКВ выявлено множество других аутоантител, из которых одни направлены против элементов крови (эритроциты, тромбоциты и лимфоциты), другие — против фосфолипидов.
Схема 31. Патогенезсистемнойкраснойволчанки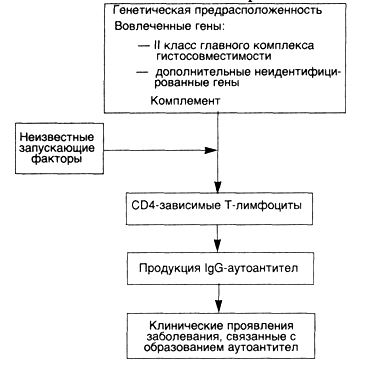 Генетические факторы.
Генетические факторы. Члены семьи больного СКВ имеют повышенный риск развития заболевания. Примерно у 20 % ближайших родственников, не имеющих клинических проявлений СКВ, находят аутоантитела и другие нарушения в иммунной регуляции. Обнаружена более высокая конкордантность (24 %) у монозиготных близнецов по сравнению с дизиготными (1—3 %), причем у монозиготных близнецов, дискордантных по СКВ, виды и титры аутоантител похожи. Видимо, существует генетическая регуляция образования аутоантител, но развитие болезни (тканевые повреждения) зависит от негенетических факторов. Известно, что именно гены главного комплекса гистосовместимости (ГКГС) регулируют продукцию специфических аутоантител. Некоторые больные СКВ имеют врожденный дефицит компонентов комплемента, таких как С2 или С4. Отсутствие компонентов комплемента нарушает элиминацию циркулирующих иммунных комплексов системой мононуклеарных фагоцитов и способствует их осаждению в тканях.
Негенетические факторы. Доказано, что некоторые лекарства (гидролазин, прокаинамид, D-пеницилламин) могут вызывать СКВ-подобный ответ у человека. Ультрафиолетовое облучение обостряет течение заболевания у многих больных, видимо, благодаря способности УФ-лучей влиять на иммунный ответ. Под действием УФ-лучей кератиноциты продуцируют ИЛ-1.
Иммунологические факторы. Полагают, что в основе СКВ лежит гиперактивность В-лимфоцитов. Установлено также, что Т-лимфоциты-хелперы, выделенные из периферической крови больных СКВ, способны индуцировать in vitro секрецию анти-ДНК-антител аутологичными В-лимфоцитами. Эти анти-ДНК-антитела являются катионами и способны осаждаться в почечных клубочках.
Большинство висцеральных повреждений при СКВ обусловлено иммунными комплексами (III тип реакций гиперчувствительности). ДНК-антиДНК-комплексы определяются в клубочках почек и мелких кровеносных сосудах. При появлении аутоантител против эритроцитов, лейкоцитов и тромбоцитов развивайся реакция гиперчувствительности II типа.
Таким образом, системная красная волчанка представляет собой сложное мультифакториальное заболевание, развивающееся
в результате взаимодействия генетических, гуморальных факторов и факторов окружающей среды, которые, действуя совместно, вызывают активацию хелперных Т- и В-лимфоцитов, что способствует секреции различных видов аутоантител.
Морфологические изменения. Исключительно вариабельны. Патогномоничные морфологические изменения практически отсутствуют. При постановке диагноза необходимо учитывать клинические, серологические и морфологические данные. Наиболее характерным повреждением считается выпадение иммунных комплексов, которые находят в кровеносных сосудах, почках, соединительной ткани и коже.
Синдром Шегрена. Характеризуется сухостью глаз (сухой кератоконъюнктивит) и рта (ксеростомия), возникающими в связи с иммунологически обусловленной деструкцией слезных и слюнных желез. Он протекает как изолированное заболевание (первичная форма, или болезнь Шегрена), однако чаще связан с другими аутоиммунными заболеваниями (вторичная форма). Среди этих заболеваний чаще всего встречаются ревматоидный артрит, а также СКВ, полимиозит, склеродермия, васкулит, смешанные заболевания соединительной ткани и тиреоидит.
Этиология и патогенез. Морфологически наблюдаются лимфоцитарная инфильтрация и фиброз серозных и слюнных желез. В инфильтрате содержатся преимущественно активированные СО4
+Т-лимфоциты-хелперы, а также В-лимфоциты, включая плазматические клетки, которые местно секретируют антитела. Остается до конца неясным, опосредованы ли тканевые повреждения только цитотоксическими Т-лимфоцитами, инфильтрирующими железы, или аутоантителами, небольшое количество которых находят в сыворотке крови.
Как и при других аутоиммунных заболеваниях, при синдроме Шегрена имеется ассоциация со II классом аллелей HLA.
В целом развитие синдрома Шегрена связывают с наличием нескольких типов аутоантител, хотя их спектр и не так широк, как при СКВ. Наиболее важными серологическими маркерами этого заболевания являются антитела против двух РНП-антигенов SS-A (Ro) и SS-B (La), которые выявляются у 90 % больных.
Прогрессирующий системный склероз (склеродермия). При этом заболевании чаще всего поражается кожа, хотя нередко страдают желудочно-кишечный тракт, почки, сердце, мышцы и легкие. У некоторых больных основным проявлением патологии длительное время остается поражение кожи, однако у большинства пациентов склеродермия прогрессирует в случае присоединения висцеральных проявлений. Смерть больных наступает от почечной, сердечной, легочной недостаточности или нарушения всасывания в тонкой кишке. Различают две разновидности течения заболевания:
▲ диффузную склеродермию, характеризующуюся широким вовлечением кожи, быстрым прогрессированием и ранними висцеральными проявлениями;
▲ местную склеродермию, сопровождающуюся относительно ограниченным вовлечением кожи (пальцы, предплечье, лицо). Висцеральные проявления присоединяются поздно, а течение заболевания относительно доброкачественное.
Этиология и патогенез. Прогрессирующий системный склероз — заболевание с неизвестной этиологией. Чрезмерное образование коллагена обусловлено взаимодействием многочисленных факторов, которые направлены на продукцию различных факторов роста фибробластов. В фиброгенезе играют роль как иммунологические, так и сосудистые нарушения (схема 32).
В соответствии с иммунологической гипотезой фиброз является следствием
аномальной активации иммунной системы. Предполагают, что Т-лимфоциты, отвечая на какой-то неидентифицированный антиген, накапливаются в коже и выделяют цитокины, которые рекрутируют воспалительные клетки, включая тучные клетки и макрофаги. Некоторые медиаторы, продуцируемые тучными клетками и моноцитами, такие как гистамин, гепарин, ИЛ-1 и ФНО-а, могут усиливать рост фибробластов и увеличивать синтез коллагена. У многих больных склеродермией в коже находят активированные СО4
+Т-лимфоциты-хелперы.
Все больные склеродермией имеют антинуклеарные антитела, которые реагируют с различными внутриядерными мишенями. Два типа антинуклеарных антител более или менее уникальны для прогрессирующего системного склероза. Один из них, направленный против топоизомеразы I ДНК, очень специфичен и присутствует у 28—70
% больных склеродермией. Больные, которые имеют антитела этого типа, чаще страдают легочным фиброзом и заболеваниями периферических сосудов. Антицентромерные антитела другого типа найдены у 22—36 % больных склеродермией и чаще встречаются у пациентов с ограниченным системным склерозом.
Сосудистая гипотеза основывается на наличии предшествующих сосудистых заболеваний у больных прогрессирующим системным склерозом. Фиброз внутренней оболочки пальцевых артерий, например, встречается у всех больных склеродермией. Отмечены также признаки повреждения эндотелия (повышенное содержание фактора Виллебранда) и активация тромбоцитов (увеличение количества циркулирующих тромбоцитов). Повторные повреждения эндотелия сопровождаются агрегацией тромбоцитов, что ведет к выбросу тромбоцитарных факторов, которые вызывают периадвентициальный фиброз. Активированные или поврежденные эндотелиальные клетки сами по себе могут выделять факторы, хемотаксические для фибро-бластов. Наконец, распространенное сужение сосудов микроцир-куляторного русла также приводит к ишемическому повреждению.
Схема 32. Патогенезпрогрессирующегосистемногосклероза (склеродермии)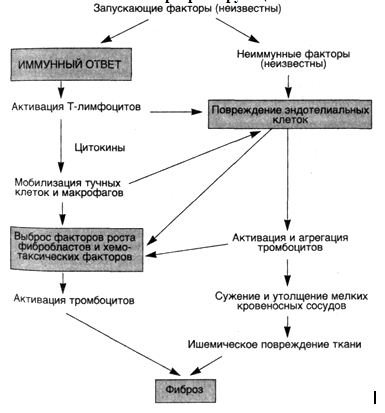
■ Таким образом, в основе прогрессирующего системного склероза лежат различные иммунные нарушения, выраженный фиброз и изменения микроциркуляторного русла. Хотя антигены, запускающие аутоиммунный ответ, и не идентифицированы, установлено, что именно иммунологические механизмы вызывают развитие фиброза с помощью цитокинов, которые активируют фибробласты, или посредством повреждения мелких кровеносных сосудов, либо благодаря обоим механизмам.
Воспалительные миопатии. Это гетерогенная группа заболеваний, характеризующихся иммунологически обусловленным воспалением скелетных мышц. К ним относятся дер-матомиозит и полимиозит, которые могут развиваться сами по себе или сочетаться с другими иммунологически обусловленными болезнями, обычно с прогрессирующим системным склерозом.
Дерматомиозит характеризуется поражением кожи и скелетных мышц, встречается у детей и взрослых. Классическая сыпь при этом заболевании возникает в виде сиреневых или обесцвеченных участков на верхних веках и сопровождается периорбитальным отеком. Нередко появляются шелушащиеся эритематозные высыпания или темно-красные пятна на суставах и локтях. Мышечная слабость развивается медленно, бывает двусторонней симметричной и обычно вначале поражает проксимальные мышцы, поэтому первыми симптомами заболевания бывают затруднения при вставании со стула и ходьбе вверх. Движения, контролируемые дистальными мышцами, страдают позже. Иногда, чаще у детей, возможны внемышечные проявления болезни в виде изъязвлений в желудочно-кишечном тракте и обызвествлений мягких тканей.
При
полимиозите, так же как при дерматомиозите, поражаются симметричные проксимальные мышцы. Однако при полимиозите нет кожных проявлений. Он встречается главным образом у взрослых.
Этиология и патогенез. Этиология воспалительных миопатии неизвестна, но повреждение тканей, видимо, обусловлено иммунными механизмами.
При дерматомиозите основной мишенью служат капилляры. Микроциркуляторное русло атакуют антитела и компоненты комплемента, вызывая появление фокусов некроза миоцитов. При полимиозите, наоборот, возникают повреждения, опосредованные клетками. Около поврежденных мышечных волокон найдены CD8+ цитотоксические Т-лимфоциты и макрофаги, а экспрессия HLA-антигенов I класса увеличена на сарколемме нормальных мышечных волокон.
Как и при других аутоиммунных заболеваниях, при воспалительных миопатиях выявляются антитела.
Диагностика миозита основана на клинических симптомах, Данных электромиографии и биопсии.
Смешанные заболевания соединительной ткани. Описаны у тех больных, у которых сочетаются симптомы СКВ, полимиозита и прогрессирующего системного склероза, а серологически наблюдается высокий титр антител к рибонуклеопротеидам. При этих заболеваниях страдают почки; эффективно лечение кортикостероидами.
Для смешанных заболеваний соединительной ткани характерны артрит, опухание рук, феномен Рейно, аномальная подвижность пищевода, миозит, лейкопения и анемия, лихорадка, лимфаденопатия и гипергаммаглобулинемия.
СИНДРОМЫИММУННОГОДЕФИЦИТА•
Иммунодефицитные заболевания — состояния, обусловленные выпадением одного или нескольких компонентов иммунитета.
Синдром иммунного дефицита по сути представляет собой эксперимент природы, который еще раз убеждает в сложности устройства иммунной системы.
Все иммунодефициты делят на первичные, которые почти всегда детерминированы генетически, и вторичные, связанные с осложнениями инфекционных заболеваний, нарушенным всасыванием, старением, побочными эффектами иммуносупрессии, облучением, химиотерапией и других аутоиммунных болезней.
Первичные иммунодефициты. Это генетически детерминированные заболевания. Они поражают специфический иммунитет (гуморальный и клеточный) или неспецифические механизмы защиты хозяина, обусловленные комплементом и клетками (фагоцитами или естественными киллерами). Хотя большинство иммунодефицитов встречается довольно редко, некоторые из них, например дефицит IgA, довольно-таки распространены, особенно у детей. Обычно первичные иммунодефициты проявляются в детстве в возрасте между 6 мес и 2 годами повышенной чувствительностью и рецидивирующими инфекционными заболеваниями.
Агаммаглобулинемия Брутона, сцепленная с Х-хромосомой. Является одним из самых распространенных первичных иммунодефицитов и характеризуется фактическим отсутствием сывороточных иммуноглобулинов, хотя IgG могут быть обнаружены, в незначительном количестве. Это заболевание связано с Х-хромосомой и встречается у лиц мужского пола. Тяжелые рецидивирующие инфекции начинаются обычно в возрасте 8—9 мес, когда ребенок перестает получать материнские иммуноглобулины. Чаще всего представлены пиогенные микроорганизмы (стафилококки, Haemophilus influenzae). Больные страдают рецидивирующими конъюнктивитом, фарингитом, средним отитом, бронхитом, пневмонией и кожными инфекциями. С большинством вирусных и грибковых инфекций организм больного справляется успешно, так как клеточный иммунитет не нарушен. Вместе с тем существует особый риск развития связанного с вакцинацией полиомиелита и эховирусного энцефалита, а также пневмоцистной пневмонии. Персистирующая лямблиозная инфекция приводит к нарушению всасывания.
При болезни Брутона чаще развиваются аутоиммунные болезни. У половины детей встречаются заболевание типа ревматоидного артрита, а также системная красная волчанка, дерматомиозит и другая аутоиммунная патология.
В костном мозге находят нормальное содержание пре-В-лимфоцитов, представляющих собой крупные лимфоидные клетки с IgM в цитоплазме, но без иммуноглобулинов на поверхности клетки; фактически отсутствуют В-лимфоциты, за исключением редких случаев. Лимфатические узлы и селезенка не имеют герминативных центров, а в лимфатических узлах, селезенке, костном мозге и соединительной ткани отсутствуют плазматические клетки. Небные миндалины особенно плохо развиты или рудиментарны. В то же время количество циркулирующих и тканевых Т-лимфоцитов, функция которых не изменена, остается в норме.
Общий вариабельный иммунодефицит. Представляет собой гетерогенную группу заболеваний. Он может быть врожденным или приобретенным, спорадическим или семейным (с непостоянным типом наследования). Для всех пациентов характерна гипогаммаглобулинемия, обычно связанная с дефектом всех классов антител, но иногда только IgG. Причины иммунодефицита могут быть различными. В противоположность агаммаглобулинемии Брутона у большинства больных содержание В-лимфоцитов в крови и лимфоидной ткани нормальное. Однако эти В-клетки не могут дифференцироваться в плазматические клетки. В большинстве случаев дефект состоит в терминальной дифференцировке В-лимфоцитов, в результате чего они не могут секретировать нормальное количество иммуноглобулинов даже тогда, когда имеются хелперные Т-лимфоциты, а потенциальные супрессорные Т-лимфоциты отсутствуют.
Молекулярная основа аномальной дифференцировки В-лимфоцитов может быть различной. У некоторых больных возникают мутации, которые влияют на экспрессию иммуноглобулиновых генов, у других — дефектные В-лимфоциты, так же как и Функциональные аномалии СD4
+-лимфоцитов (хелперов) или СD8
+Т-лимфоцитов (супрессоров), причем количество CD4+T-лимфоцитов может быть нормальным, но они продуцируют сниженное количество ИЛ-2 и 7-интерферона (ИФН-γ)- В связи с тем что цитокины необходимы для секреции иммуноглобулинов, указанные дефекты Т-лимфоцитов приводят к гипогаммаглобулинемии. У других больных речь идет не об отсутствии Т-лимфоцитов, а скорее об абсолютном увеличении количества CD8+T-лимфоцитов, которые могут подавлять секрецию антител нормальными В-лимфоцитами. Получены данные о генетической предрасположенности к общему вариабельному иммунодефициту.
Клинически заболевание проявляется рецидивирующими инфекциями. Помимо бактериальных инфекций, эти больные страдают тяжелыми энтеровирусными инфекциями, рецидивирующим герпесом и персистирующей диареей, вызванной лямблиями. Гистологически наблюдается гиперплазия В-клеточных участков лимфоидной ткани (лимфоидных фолликулов в лимфатических узлах, селезенке и кишечнике). Расширение этих зон отражает, видимо, дефектную иммунорегуляцию: В-лимфоциты пролиферируют в ответ на антиген, но вследствие нарушенной продукции антител торможение пролиферации посредством IgG отсутствует.
У этих больных высока частота аутоиммунных заболеваний, включая ревматоидный артрит, пернициозную и гемолитическую анемию, и составляет примерно 20 %.
Изолированный дефицит IgA. Широко распространен. Для заболевания характерен очень низкий уровень как сывороточного, так и секреторного IgA. Иммунодефицит может быть семейным или приобретенным после токсоплазмоза, кори либо некоторых других вирусных инфекций. В связи с тем что IgA является основным иммуноглобулином внешней секреции, при его дефиците нарушается защита слизистых оболочек и развиваются инфекции дыхательной, желудочно-кишечной и мочеполовой систем. Больные нередко страдают синопульмональными инфекциями и диареей. У пациентов с дефицитом IgA аллергия респираторного тракта и различные аутоиммунные болезни, особенно системная красная волчанка и ревматоидный артрит, встречаются очень часто. Причина повышенной частоты аутоиммунных и аллергических заболеваний неизвестна.
Основной причиной этого иммунодефицита является дефект дифференцировки В-лимфоцитов, продуцирующих IgA. У большинства больных с селективным дефицитом IgA количество IgA-положительных В-лимфоцитов нормальное, но большинство из них экспрессируют незрелый фенотип, который характеризуется коэкспрессией поверхностных IgD и IgM. Лишь немногие из этих клеток способны in vitro трансформироваться в IgA-плазматические клетки. Сывороточные антитела к IgA обнаружены приблизительно у 40 % больных, что необходимо учитывать при переливании крови, так как при попадании в организм больного крови, содержащей нормальное количество IgA, у него может развиться тяжелая, даже фатальная, анафилактическая реакция.
Синдром Ди Джорджи (гипоплазия вилочковой железы). Это пример селективного Т-лимфоцитарного дефицита, появление которого связано с нарушением развития 3-го и 4-го глоточных карманов, дающих начало вилочковой железе, околощитовидным железам, некоторым светлым клеткам щитовидной железы к ультимобранхиальному телу. Таким образом, у этих больных
отсутствует клеточный иммунный ответ (вследствие гипоплазии
йли отсутствия вилочковой железы), развиваются тетания (отсутствие околощитовидных желез) и врожденные дефекты сердца и крупных сосудов. Кроме того, внешний вид рта, ушей и лица может быть изменен. При отсутствии клеточного иммунитета уровень циркулирующих Т-лимфоцитов низкий и защита против некоторых грибковых и вирусных инфекций слабая. Количество плазматических клеток в лимфоидной ткани нормальное, но тимусзависимые паракортикальные зоны лимфатических узлов и периартериолярных оболочек в селезенке отсутствуют. Содержание иммуноглобулинов в норме.
Синдром Ди Джорджи не относится к числу генетически детерминированных заболеваний, но, по-видимому, является результатом внутриматочного повреждения плода на 8-й неделе беременности.
Тяжелые комбинированные иммунодефицитные заболевания. Характеризуются комбинированным В- и Т-лимфоцитарным дефектом. Больные дети страдают от тяжелых рецидивирующих инфекций, вызываемые Candida albicans, Pneumocystis carinii, Pseudomonas, а также цитомегаловирусом, вирусом ветряной оспы и многими бактериями. Без пересадки костного мозга смерть наступает в первые годы жизни.
В зависимости от локализации мутантного гена и природы генетического дефекта различают два типа наследования: аутосомно-рецессивный и рецессивный, связанный с Х-хромосомой.
Приблизительно у 40 % пациентов с аутосомно-рецессивными формами заболевания отсутствует фермент аденозиндеаминаза, дефицит которого ведет к накоплению деоксиаминазина и его производных, которые особенно токсичны для незрелых лимфоцитов, в первую очередь Т-лимфоцитов. Следовательно, количество Т-лимфоцитов может быть заметно снижено в тяжелых случаях. Реже при аутосомно-рецессивном типе этого заболевания встречается дефект активации Т-лимфоцитов. У этих больных содержание Т-клеток нормальное, однако существует дефицит одного из видов молекул, которые участвуют в активации Т-лимфоцитов.
Рецессивный тип наследования, связанный с Х-хромосомой, встречается приблизительно у 50 % больных. У них происходит мутация, которая воздействует на белок, являющийся рецепторами для ИЛ-2, ИЛ-4 и ИЛ-7.
Характер морфологических изменений зависит от вида генетического дефекта. При двух наиболее распространенных формах иммунодефицита (отсутствие аденозиндеаминазы и мутация рецепторов) вилочковая железа маленькая, лишена лимфоидных клеток. В других случаях лимфоидная ткань гипопластична с заметным уменьшением размеров зон Т-клеток, а в некоторых случаях — как Т-, так и В-зон.
Иммунодефицит с тромбоцитопенией и экземой (синдром Вискотта — Олдрича). Это рецессивное, связанное с Х-хромосомой заболевание, которое характеризуется тромбоцитопенией, экземой, уязвимостью к рецидивирующей инфекции и рано заканчивается смертью. Вилочковая железа морфологически нормальна, однако наблюдается прогрессирующее вторичное истощение Т-лимфоцитов в периферической крови и паракортикальных (тимусзависимых) зонах лимфатических узлов с вариабельным снижением клеточного иммунитета. Ответы на такие белковые антигены, как столбнячный и дифтерийный токсин, могут быть нормальными, однако классически они свидетельствуют о слабом антигенном ответе на полисахаридные антигены. Уровень IgM в сыворотке низкий, a IgG — обычно нормальный. Парадоксально возрастает уровень IgA и IgE. У больных часто развиваются злокачественные лимфомы.
Генетический дефицит системы комплемента. Описан для всех компонентов данной системы и двух ее ингибиторов. Дефицит компонентов комплемента, особенно СЗ, который необходим как для классического, так и альтернативного пути, обусловливает повышенную чувствительность к инфекции, вызываемой патогенными бактериями. Врожденный дефицит Clq, С2 и С4 повышает риск развития иммунокомплексных заболеваний, например системной красной волчанки. При отсутствии ингибитора С1-эстеразы возникает неконтролируемая активация С1-эстера-зы с образованием кинина С2. У этих больных развивается врожденный ангионевротический отек, характеризующийся поражением кожи и слизистых оболочек. Дефицит компонентов классического пути (С5—8) способствует развитию рецидивирующих нейссеровских (гонококковые, менингококковые) инфекций.
